

Significance
Terpene synthases catalyze complex, chain length-specific, electrophilic cyclization reactions that constitute the first committed step in the biosynthesis of structurally diverse terpenoids. (4S)-limonene synthase [(4S)-LS] has emerged as a model enzyme for enhancing our comprehension of the reaction cycle of monoterpene (C10) synthases. While the stereochemistry of the cyclization of geranyl diphosphate to (−)-(4S)-limonene has been the subject of several mechanistic studies, the structural basis for the stabilization of carbocation intermediates and the termination of the reaction sequence have remained enigmatic. We present extensive experimental evidence that the aromatic amino acids W324 and H579 play critical roles in the stabilization of intermediate carbocations. A possible function of these residues as the terminal catalytic base is also discussed.
Keywords: monoterpene synthase, enzyme catalysis, mechanism, carbocation, structure–function relationship
Abstract
Crystal structural data for (4S)-limonene synthase [(4S)-LS] of spearmint (Mentha spicata L.) were used to infer which amino acid residues are in close proximity to the substrate and carbocation intermediates of the enzymatic reaction. Alanine-scanning mutagenesis of 48 amino acids combined with enzyme fidelity analysis [percentage of (−)-limonene produced] indicated which residues are most likely to constitute the active site. Mutation of residues W324 and H579 caused a significant drop in enzyme activity and formation of products (myrcene, linalool, and terpineol) characteristic of a premature termination of the reaction. A double mutant (W324A/H579A) had no detectable enzyme activity, indicating that either substrate binding or the terminating reaction was impaired. Exchanges to other aromatic residues (W324H, W324F, W324Y, H579F, H579Y, and H579W) resulted in enzyme catalysts with significantly reduced activity. Sequence comparisons across the angiosperm lineage provided evidence that W324 is a conserved residue, whereas the position equivalent to H579 is occupied by aromatic residues (H, F, or Y). These results are consistent with a critical role of W324 and H579 in the stabilization of carbocation intermediates. The potential of these residues to serve as the catalytic base facilitating the terminal deprotonation reaction is discussed.
Terpenoids are a structurally diverse group of metabolites with functions in both primary and secondary (or specialized) metabolism. Primary metabolites derived from terpenoid pathway intermediates in plants include sterols, carotenoids, and the side chains of chlorophylls, tocopherols, and quinones of electron transport systems. Many plant hormones are also products of terpenoid metabolism, including abscisic acid, cytokinins, brassinosteroids, and strigolactones (1). Secondary plant metabolites of terpenoid origin can play critical defense-related roles (e.g., sesquiterpene lactones and triterpene saponins serve as antifeedants) and are dominant constituents of essential oils and resins (mono-, sesqui-, and diterpenes) (2). Terpene synthases (TPSs) convert a prenyl diphosphate of a specific chain length to the first pathway-specific (often cyclic) intermediate in the biosynthesis of each class of terpenoids. Whereas some terpene synthases are remarkably specific and only generate one product from a prenyl diphosphate precursor, others release a larger number of products from a common substrate, thus contributing to terpenoid chemical diversity (3). The genomes of plants may only contain one TPS gene [e.g., ent-kaurene (diterpene) synthase in the moss Physcomitrella patens (Hedw.) Bruch & Schimp.], but often harbor sizable families of TPS genes with more than 20 members, which is another source of terpenoid structural variety (4).
All monoterpene synthases (MTSs) use either geranyl diphosphate (GPP) or its 2Z-isomer neryl diphosphate as substrate, but the sequence conservation across species is generally fairly low (2). However, MTSs share a common tertiary structure (the so-called αβ fold), with a C-terminal α-domain containing the active site and an N-terminal β-domain of as yet uncertain function (3). The active site harbors a highly conserved, l-aspartate-rich, DDxxD motif, which is also found in prenyl elongases (5) and a less conserved NSE/DTE motif. The l-aspartate residues bind a trinuclear cluster of divalent metal ions (Mg2+ or Mn2+) involved in the binding and activation of the diphosphate moiety, thereby generating characteristic carbocation intermediates (Fig. 1). Enzymes catalyzing these ionization-initiated cyclization reactions are commonly referred to as class I TPSs (as opposed to class II TPSs that initiate cyclizations by protonation) (3). The remaining course of MTS catalysis is variable and generates acyclic, monocyclic, and/or bicyclic monoterpenes.
Fig. 1.
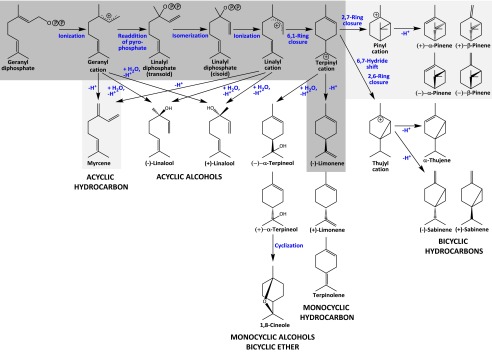
Proposed mechanism for (4S)-limonene synthase catalysis. OPP denotes the diphosphate moiety. The primary pathway in the wild-type enzyme leads to the formation of (−)-limonene (dark gray) and smaller amounts of bicyclic and acyclic products (light gray). Other products shown in this figure are released by mutant enzymes.
Because of the widespread occurrence of (−)-(4S)-limonene throughout the plant kingdom, the relative simplicity of catalysis, the comparatively well-understood mechanism, and the availability of a crystal structure at 2.7-Å resolution (6), (4S)-limonene synthase [(4S)-LS] has become a model for understanding catalysis by class I TPSs. The (4S)-LS gene is translated into a preprotein with an N-terminal targeting sequence for transport to the plastidial envelope membrane (7). Based on results obtained with a series of truncated (4S)-LS mutants, the most likely cleavage site of the preprotein was determined to be at (or near) a tandem pair of arginines (R58 and R59 of the preprotein) (8). If truncated beyond R58, (4S)-LS shows no cyclization activity with GPP but is fully functional with linalyl diphosphate (LPP) as a substrate. Interestingly, the crystal structure of (4S)-LS, complexed with the nonhydrolyzable LPP analog 2-fluorolinalyl diphosphate (FLPP), does not provide evidence for a direct interaction of R58 or R59 with the substrate (9). However, weak interactions, in particular electrostatic interplay of R58 with E363 and hydrogen bonding between R59 and V357/Y435, appear to anchor the N-terminal strand to the outside of the active site, thereby possibly supporting the closure of the active site, while not interfering with the binding of the substrate and intermediates (9). The reaction mechanism of (4S)-LS from spearmint (Mentha spicata L.) has been studied in some detail. The catalytic cascade involves the migration of the diphosphate group to C3 of the geranyl cation (from the original C1) to afford enzyme-bound (3S)-LPP as an intermediate (10) (Fig. 1). Following C2–C3 rotation, the diphosphate is released again to generate a linalyl cation. The proximity of the C6–C7 double bond to the positive charge facilitates an anti-SN′ cyclization to form the (−)-(4S)-terpinyl cation (11, 12) (Fig. 1). Deprotonation from the adjacent methyl group (C8 of the original GPP) yields the monocyclic olefin (−)-(4S)-limonene as the major product (96%). Side products are obtained by either premature deprotonation of the geranyl cation to generate the acyclic olefin myrcene (2%) or additional cyclization of the terpinyl cation (between C2 and C7 of the original GPP) to produce, after deprotonation, a mixture of the bicyclic olefins α- and β-pinene (2%) (10, 13) (Fig. 1).
Using synthetic analogs of GPP with a chiral methyl group at C9 (carrying 1H, 2H, and 3H), Coates et al. demonstrated that the final deprotonation occurs predominantly by re-facial anti-elimination (12). It has been hypothesized that the diphosphate anion released from the prenyl diphosphate substrate may act as the catalytic base for this deprotonation in various prenyl diphosphate synthases and terpene synthases, but no direct evidence is available to date (3, 14–17) and it seems highly unlikely in the present case due to spatial considerations. Here we present a comprehensive dataset to map the active site of spearmint (4S)-LS and evaluate residues with potential roles in stabilizing carbocation intermediates of the reaction cycle. We also discuss the broader implications of our findings for understanding catalysis and reaction termination by this fascinating class of enzymes.
Results
l-Alanine Scanning Mutagenesis Defines Residues Required for Substrate Binding and/or Catalysis.
X-ray data for the crystal structure of the pseudomature form of spearmint (4S)-LS (R58; lacking the plastidial targeting sequence), complexed with FLPP as a nonhydrolyzable analog of the reaction intermediate LPP (9), were used to infer the amino acid residues within closest proximity to the substrate (Dataset S1). Distances were calculated between each of the 10 carbon atoms of the substrate analog and the carbon atoms of all amino acids of the protein (except for those forming peptide bonds). The 48 residues located closest to the substrate analog were exchanged for l-alanine by site-directed mutagenesis (introduced into the pseudomature form of (4S)-LS that is truncated at R58), and these mutant enzymes were expressed in Escherichia coli. Following purification over hydroxyapatite, each recombinant enzyme was incubated with GPP and products of the reaction were analyzed by quantitative chiral phase gas chromatography.
Mutations affecting the conserved l-aspartate residues known to be of crucial importance for complexing divalent active site metal ions (Mg2+ or Mn2+) in monoterpene synthases and related enzymes (5, 18), D352A, D353A, and D496A, predictably resulted in inactive enzymes (Dataset S2). The majority of the remaining mutants generated product profiles similar to those of the wild-type enzyme [(−)-limonene (96.6%), myrcene (1.0%), (−)-α-pinene (0.6%), (+)-α-pinene (0.7%), (−)-β-pinene (0.5%), (+)-β-pinene (0.5%), and (+)-sabinene (0.3%)]. However, some single residue exchanges led to a significantly reduced rate of (−)-limonene production, with increases in the formation of side products and novel monoterpenes not detectable in assays with the wild-type enzyme (Dataset S2). The acyclic monoterpene myrcene was produced in substantial amounts by M458A (10.1%), I348A (9.7%), and T349A (8.3%). Acyclic monoterpene alcohols were released at fairly high levels from mutant enzymes W324A [equal amounts (26.7% each) of (−)- and (+)-linalool], M458A [equal amounts (14.3% each) of (−)-and (+)-linalool], and H579A [8.6% (−)-linalool] (Dataset S2). A novel product derived from an altered reaction stereochemistry was (4R)-(+)-limonene generated by M458A (8.6%). Monocyclic alcohols were found to be novel products of M458A [27.3% (+)-α-terpineol], H579A [25.0% (−)-α-terpineol], and L492A [8.0% (-)-α-terpineol] (Dataset S2). Increased amounts of bicyclic products were formed by N345A [36.6% (−)-sabinene, 14.8% (+)-α-pinene, and 13.9% (+)-β-pinene]. Additional novel products generated in smaller amounts by mutant enzymes were α-thujene (N345A) and 1,8-cineole (T349A, L492A, and M458A) (Dataset S2).
To further assess the observed loss in fidelity in mutant enzymes, we plotted the distance between each amino acid and the substrate analog in the (4S)-LS crystal structure against the percentage of (−)-limonene released by the enzyme after this residue was mutated to l-alanine (Fig. 2). Interestingly, mutations affecting the nine amino acid residues positioned in closest proximity to the substrate analog (i.e., those with a carbon atom at a distance of less than 5 Å to a carbon atom of the substrate analog) released (−)-limonene at less than 80% of the products, indicating a possible direct role in substrate (or intermediate) binding and/or catalysis. The only exceptions were D352A and D353A (with minimum C–C distances of 5.1 and 8.1 Å, respectively), which are known to be required for the divalent metal ion-mediated binding of the pyrophosphate after its release from the prenyl diphosphate, and thus do not interact with the prenyl or emerging terpenoid moiety (3) (Fig. 2).
Fig. 2.

Mutant enzymes lose fidelity when residues that likely function in substrate binding and/or catalysis are exchanged. The closest distance of a carbon atom of each amino acid in (4S)-limonene synthase is plotted against the percentage of (−)-limonene formation when the residue is exchanged with l-alanine.
W324A and H579A Mutants Are Catalytically Impaired and Preferentially Generate an Acyclic Hydrocarbon and Monoterpene Alcohols.
Under routine end-point assay conditions (200 µg purified protein), all available substrate (GPP concentration at 5 mM) was turned over by the wild-type and most mutant enzymes. However, two mutants, W324A and H579A, converted only small amounts of substrate during the 2-h time frame of the assay. Under kinetic assay conditions, when the R58 wild-type enzyme converts ≤50% of the substrate, the specific activity of R58 was 0.19 nmol (h × mg protein)−1, and the catalytic constants determined in our assays were similar to those reported in the literature (Km = 12.6 µM; kcat = 0.037 s−1) (8). The specific activities of the W324A and H579A mutant enzymes were significantly reduced and, therefore, kinetic values could not be determined with accuracy.
To further investigate the potential roles of W324 and H579 in (4S)-LS catalysis, additional mutants were generated. W324 and H579 were exchanged with another aromatic residue (W324H, W324F, W324Y, H579F, H579Y, and H579W), a base (W324H, W324K, and H579K), or a randomly selected amino acid. The majority of W3234 mutant enzymes had extremely low specific activities [≤0.02 nmol (h × mg protein)−1; 3–10% of wild type], the only exception being W324F and W324Y [0.08 and 0.06 nmol (h × mg protein)−1; 50% and 32% of wild-type activity, respectively]. The H579 mutants had specific activities ranging between 40% and 50% of those of the wild-type enzyme. The product profiles were significantly altered in all 12 W324 mutants (W324A, W324C, W324F, W324H, W324I, W324K, W324L, W324P, W324Q, W324S, W324T, and W324Y) and five of nine H579 mutants (H579A, H579D, H579K, H579N, and H579W) (Dataset S3). The majority of W324 and H579 mutants generated notable amounts of the acyclic hydrocarbon myrcene (Fig. 3), which is released by premature deprotonation from the geranyl or linalyl cation (Fig. 1). The most prominent products of W324 mutants were acyclic alcohols, the formation of which is caused by premature quenching of an early carbocation intermediate (linalyl cation) by a water molecule (Fig. 1). An example is W324P, which forms 39.6% (−)-linalool and 40.5% (+)-linalool (80.1% acyclic alcohols) (Fig. 3A). Another notable class of products was monocyclic alcohols [(−)-α- and (+)-α-terpineol; detectable in 11 W324 mutants], which are generated by quenching of the terpinyl cation by a water molecule (Fig. 1) (19). The highest relative amounts were produced by W324A [11.6% (−)-α-terpineol and 5.7% (+)-α-terpineol; 17.3% moncyclic alcohols] (Fig. 3A). An exception was the W324H mutant which, in addition to monoterpene alcohols, produced high amounts of bicyclic monoterpenes [42.0% (+)-sabinene and 7.8% (−)-sabinene]. The catalytically impaired H579 mutants, in agreement with W324 mutant data, were also characterized by the production of considerable amounts of acyclic and monocyclic alcohols. A representative example is H579K, which released 14.9% (−)-linalool, 14.9% (+)-linalool, 22.1% (−)-α-terpineol, and 4.1% (+)-α-terpineol (56.0% alcohols) (Fig. 3B). A W324A/H579A double mutant had no detectable enzyme activity.
Fig. 3.

Mutant enzymes produce acyclic monoterpene olefins, and acyclic and monocyclic monoterpene alcohols, when amino acid exchanges are made in positions occupied by residues required for stabilizing the α-terpinyl cation. (A) W324 and (B) H579 mutants.
Discussion
(4S)-LS Mutants with Substituted Active-Site Residues Lose Fidelity.
An analysis of the X-ray crystallographic structure of (4S)-LS (8), complexed with a substrate analog, enabled us to hypothesize which amino acid residues are likely to form the active site of the enzyme. We further hypothesized that exchanges of residues involved in substrate binding and catalysis would generate mutant enzymes with lower specific activity and/or result in altered product profiles. A total of 48 amino acid residues were found to be arranged within a radius of about 25 Å from any carbon atom of the C10 substrate analog. When these residues were substituted with l-alanine, the mutant enzymes were mostly unaffected in their specific activity and fidelity. However, 11 mutants released (−)-limonene as less than 80% of the total monoterpene products. In these fidelity-impaired enzymes, a residue containing a carbon atom within close proximity to a substrate carbon atom was mutated (Fig. 4A). Strong hydrogen bonding below 3 Å can thus occur between donor oxygens or nitrogens of active site amino acids (e.g., –OH in T349, or S454; –NH– in W324 or H579) and acceptor sites of the substrate or intermediates. By analogy to an earlier mechanistic proposal for 5-epi-aristolochene synthase (TEAS) (20), we propose that the carbocation reaction intermediates of (4S)-LS might move deeper into a hydrophobic pocket [composed of W324, N345, T349, S454, M458, and H579 in (4S)-LS], which would enhance the stabilization of the positive charge by aromatic residues situated at the bottom of the active site. The region of positive charge around the divalent metal ions interacts directly with the diphosphate leaving group, thereby immobilizing this anion and preventing a recapture of the allylic carbocation (which would regenerate GPP and terminate the reaction) (20, 21).
Fig. 4.

Possible roles of W324 and H579. The carbon skeletons of the substrate analog (FLPP) and the α-terpinyl cation intermediate are depicted in green. (A) Residues lining the active site of (4S)-limonene synthase with a bound substrate analog based on crystal structural data (9). (B) Proposed stabilization of the α-terpinyl cation intermediate by carbocation-π interactions with H579 and/or W324. (C) Orientation of α-terpinyl cation so that H579 can act as a catalytic base. (D) Orientation of α-terpinyl cation so that W324 can act as a catalytic base. Note that the deprotonation reactions in C and D would both result in a product with the correct stereochemistry [(−)-limonene].
W324 and H579 Appear to Play Roles in Stabilizing the α-Terpinyl Cation.
Following the cyclization step, the positive charge of the α-terpinyl cation could be stabilized by cation-π interactions with the electron-rich heteronuclear aromatic rings of W324 and H579, which would be particularly strong if the carbocation were situated, as hypothesized, in the hydrophobic active site pocket (Fig. 4B). Analogous stabilizing roles had been proposed for aromatic residues in sesquiterpene synthases (20, 21). The importance of l-histidine in TPS catalysis was already recognized by Rajaonarivony et al. (22) based on experiments with histidine-directed inhibitors, but the data presented here provide to our knowledge the first evidence for the involvement of a specific residue (H579). One would predict that, if a charge-stabilizing residue was exchanged with a nonaromatic residue, the reaction outcome would reflect early termination products. Indeed, almost all W324 mutants released significant amounts of acyclic myrcene and (+/−)-linalool (up to 83% of total products), which is indicative of a reaction that terminates before the cyclization to the α-terpinyl cation (Fig. 1). Interestingly, W324F and W324Y also formed mostly early termination products (77% and 81%, respectively), indicating that other aromatic amino acids could not effectively substitute for W324. It is thus not surprising that the W324 residue is conserved in angiosperm MTSs (Dataset S4). H579 mutants generally released a mixture of myrcene, (+/−)-linalool and (+/−)-α-terpineol, with the exception of H579F and H579Y (aromatic substitutions), which, similar to the wild-type enzyme, generated (−)-limonene as the only major product (>85%). Interestingly, angiosperm MTSs that generate cyclized products carry an aromatic residue (H, F, or Y) in the position corresponding to H579 in (4S)-LS. The formation of cyclic alcohols by W324 and H579 mutants provides evidence that these residues are also required for avoiding quenching of the reaction by water, thereby further stabilizing the α-terpinyl cation for subsequent deprotonation. The high-resolution structure of (+)-bornyl diphosphate synthase (at 2.0 Å) indicated the presence of water molecules in the active site of this MTS (19), which could not be discerned in the lower resolution structure (at 2.7 Å) of (4S)-LS (8). (+/−)-Linalool (formed by W324 mutants) would be expected to be generated by water capture of the linalyl cation, where the charge is closer to the metal ion-bound pyrophosphate at the top of the active site. (+/−)-α-Terpineol (formed in H579 mutants) would be predicted to be produced by reaction with water approaching from the bottom of the active site. It is presently unknown whether water molecules are admitted to the active site in W324 and H579 mutants or, if already present in wild-type (4S)-LS, are permitted to react with carbocation intermediates in these mutants.
Which Residues Are Involved in the Final Deprotonation Reaction?
The ultimate steps of the (4S)-LS reaction cascade are the deprotonation of a carbocation and release of the olefin final product. Several authors have previously proposed that the initially released pyrophosphate may act as the catalytic base (3, 14–17). This is likely correct for bornyl diphosphate synthase (BPPS) from culinary sage (Salvia officinalis L.), where the pyrophosphate is recaptured in the terminating step of the reaction (19, 23). However, such a recapture does not occur in other characterized MTSs and, if the carbocation was situated and stabilized, as hypothesized, in a hydrophobic pocket of the active site, then the metal ion-bound diphosphate would be too distant to act as a base (Fig. 4). Based on quantum mechanical calculations, Hong and Tantillo (24) proposed pathways for the formation of BPP by BPPS. In all models, the bicyclic bornyl cation assumes a conformation that positions C2 for a recapture of the diphosphate anion, and we propose that, in other MTSs, the α-terpinyl carbocation interacts with the charge-stabilizing aromatic residues W324 (conserved) and H579 (or other aromatic residues in the same position) at the bottom of the active site cavity (separated from the diphosphate anion, which is bound by residues forming the top of the active site).
H579 has the properties of a catalytic base and, in one energy-minimized docking orientation of the α-terpinyl cation, is positioned close to the proton to be removed (Fig. 4C). W324 is situated close to the leaving proton in an alternative docking orientation of the α-terpinyl cation (Fig. 4D). Whereas H579 could be the primary base in wild-type (4S)-LS, it can be exchanged with other, nonbasic, residues with only minor effects on enzyme activity. If W324 were to act as a catalytic base (resulting in a protonated indole with an approximate pKa of −2) (25), the protonation would occur preferentially at Cγ, which is a possibility because the α-terpinyl cation is a strong conjugate acid (pKa ∼ −10). The significant reduction of specific enzyme activity in all W324 mutants could be interpreted as evidence that the termination reaction (rather than the initial isomerization) (10) has become rate limiting. The fact that the W324A/H579A double mutant had no measurable enzyme activity is consistent with a function of these residues as potential catalytic bases. An alternative explanation would be that W324 (and even more so W324/H579) mutations cause an aberrant binding of GPP. This issue can only be resolved with a series of more detailed kinetic evaluations outside the scope of the current study. The work presented here constitutes the most extensive combination of mutagenesis and product analysis in a model MTS, and it can be concluded that very subtle modifications in active site structure can result not only in alteration of termination chemistry, but also in the regiochemistry and stereochemistry of the cyclization reaction itself. The native (4S)-LS, an enzyme of very high fidelity, must therefore guide the reaction course very precisely to avoid such digression. Our data are the foundation for future efforts to better understand the as yet unpredictable outcome of reactions catalyzed by MTSs.
Materials and Methods
Calculating C–C Distances from (4S)-LS Crystal Structure.
Coordinates of the carbon atoms of all amino acids were obtained from the published crystal structure of (4S)-LS (PDB accession identifier 2ONH; complexed with the C10 substrate analog FLPP). A perl script was used to calculate each distance using the following formula:
Site-Directed Mutagenesis.
The spearmint (4S)-LS cDNA, truncated at R58 to eliminate the plastidial targeting sequence, was cloned into the pSBET vector (26) as described before (8). Point mutations were introduced by PCR using a modified overlap extension strategy as outlined previously (27). A complete list of primers used in these reactions is provided in Dataset S5. PCR amplicons were digested with BamHI and NdeI, gel purified, and ligated into a similarly digested and purified vector. The products of the ligation reactions were transformed into XL-1 blue competent cells and plated onto LB-agar plates containing 50 µg/mL kanamycin. Individual colonies were picked and bacteria grown under selection pressure. Plasmid DNA was then isolated using a commercial kit (GeneJet Mini Prep kit, Fermentas–Thermo Fisher Scientific) and the inserted gene sequence was confirmed by a commercial service provider (Eurofins Genomics).
Recombinant Protein Expression and Purification.
Vectors containing (4S)-LS wild-type (R58) and mutant genes were transformed into E. coli BL21 DE3 cells, which were then grown overnight to an OD of ∼1.0 at 37 °C in LB medium containing 50 µg/mL kanamycin. The cultures were induced with 0.5 mM isopropyl-1-thio-β-d-galactopyranoside and grown for another 24 h at 16 °C. Cells were harvested by centrifugation at 2,500 × g for 10 min, suspended in a cell disruption buffer [50 mM MOPSO, 10 mM DTT, 10% (vol/vol) glycerol; pH 7.0], and sonicated on ice three times for 15 s each. The supernatant (400 µL) obtained after centrifugation at 15,000 × g was mixed with 100 mg hydroxyapatite [preequilibrated with 10 mM sodium phosphate (pH 7.0)] in an Eppendorf tube and gently mixed by tube inversion for 1 h at 4 °C. The hydroxyapatite was then allowed to settle by gravity and the supernatant was removed with a Pasteur pipette. Weakly bound proteins were removed by washing with 50 mM MOPSO (pH 7.0). (4S)-LS was eluted with 100 mM sodium phosphate (pH 7.0). SDS gel electrophoresis indicated a >90% purity of the recombinant enzymes.
Enzyme Assays.
Purified enzyme (200 µg) was reacted with 0.5 mM GPP (obtained synthetically according to ref. 28) in 400 µL MOPSO buffer (pH 7.0) overlayed with 100 µL hexane for 16 h at 31 °C (gentle agitation). Enzymatic reactions were terminated by vigorous mixing of the aqueous and organic phases, and phase separation was achieved by placing samples in a freezer (−20 °C) for 2 h. The upper hexane layer was removed for analysis by gas chromatography with flame ionization detection (model 7890, Agilent Technologies) under the following conditions: injector at 250 °C, 20:1 split injection mode (1 µL); detector at 250 °C (H2 flow at 30 mL/min, airflow at 400 mL/min, makeup flow (He) at 25 mL/min); Cyclodex-B chiral column (J&W Scientific 112–2532; 30 m × 0.25 mm, 0.25 µm film thickness) operated at 2 mL/min flow rate with He as carrier gas; oven heating program starting at 40 °C with a ramp to 120 °C at 2°/min, a second ramp to 200 °C at 50 °C/min, and a final hold at 200 °C for 2 min. Peaks were identified based on comparisons of retention indices with those of authentic standards and verified where necessary by standard GC-MS methods (29). For kinetic assays, enzymatic assay times were adjusted to 2 h to ensure that no more than 20% of the available substrate was consumed. Kinetic parameters were determined by varying substrate concentrations while maintaining other reactants at saturation. Kinetic constants (Km and Kcat) were calculated by nonlinear regression analysis (Origin 8; OriginLab).
Carbocation Structure and Docking.
The molecular geometry of the α-terpinyl cation was optimized by ab initio electronic structure calculation and minimization (using default values for bond length, bond angles, and dihedral angles) with the Gaussian03 program at the HF/6–31G level (Gaussian Inc.). The optimized geometry output file was converted into the standard format for the Protein Data Bank. Molecular docking of the α-terpinyl cation in the (4S)-LS active site was performed with AutoDock Vina (vina.scripps.edu/) using default settings.
Supplementary Material
Acknowledgments
The authors thank Ms. Lisa Washburn and Ms. Deanna Heidorn for technical assistance. This work was supported by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences, US Department of Energy Grant DE-FG02-09ER16054 (to B.M.L.) for the biochemical characterization of mutant enzymes and the National Institutes of Health Grant GM-31354 (to R.B.C.) for the generation of site-directed mutants.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1501203112/-/DCSupplemental.
References
- 1.Bouvier F, Rahier A, Camara B. Biogenesis, molecular regulation and function of plant isoprenoids. Prog Lipid Res. 2005;44(6):357–429. doi: 10.1016/j.plipres.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 2.Lange BM, Ahkami A. Metabolic engineering of plant monoterpenes, sesquiterpenes and diterpenes—current status and future opportunities. Plant Biotechnol J. 2013;11(2):169–196. doi: 10.1111/pbi.12022. [DOI] [PubMed] [Google Scholar]
- 3.Gao Y, Honzatko RB, Peters RJ. Terpenoid synthase structures: A so far incomplete view of complex catalysis. Nat Prod Rep. 2012;29(10):1153–1175. doi: 10.1039/c2np20059g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen F, Tholl D, Bohlmann J, Pichersky E. The family of terpene synthases in plants: A mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 2011;66(1):212–229. doi: 10.1111/j.1365-313X.2011.04520.x. [DOI] [PubMed] [Google Scholar]
- 5.Tarshis LC, Proteau PJ, Kellogg BA, Sacchettini JC, Poulter CD. Regulation of product chain length by isoprenyl diphosphate synthases. Proc Natl Acad Sci USA. 1996;93(26):15018–15023. doi: 10.1073/pnas.93.26.15018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Degenhardt J, Köllner TG, Gershenzon J. Monoterpene and sesquiterpene synthases and the origin of terpene skeletal diversity in plants. Phytochemistry. 2009;70(15-16):1621–1637. doi: 10.1016/j.phytochem.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 7.Colby SM, Alonso WR, Katahira EJ, McGarvey DJ, Croteau R. 4S-limonene synthase from the oil glands of spearmint (Mentha spicata). cDNA isolation, characterization, and bacterial expression of the catalytically active monoterpene cyclase. J Biol Chem. 1993;268(31):23016–23024. [PubMed] [Google Scholar]
- 8.Williams DC, McGarvey DJ, Katahira EJ, Croteau R. Truncation of limonene synthase preprotein provides a fully active ‘pseudomature’ form of this monoterpene cyclase and reveals the function of the amino-terminal arginine pair. Biochemistry. 1998;37(35):12213–12220. doi: 10.1021/bi980854k. [DOI] [PubMed] [Google Scholar]
- 9.Hyatt DC, et al. Structure of limonene synthase, a simple model for terpenoid cyclase catalysis. Proc Natl Acad Sci USA. 2007;104(13):5360–5365. doi: 10.1073/pnas.0700915104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rajaonarivony JI, Gershenzon J, Croteau R. Characterization and mechanism of (4S)-limonene synthase, a monoterpene cyclase from the glandular trichomes of peppermint (Mentha x piperita) Arch Biochem Biophys. 1992;296(1):49–57. doi: 10.1016/0003-9861(92)90543-6. [DOI] [PubMed] [Google Scholar]
- 11.Pyun HJ, et al. Regiospecificity and isotope effects associated with the methyl-methylene eliminations in the enzyme-catalyzed biosynthesis of (R) and (S)-limonene. J Org Chem. 1993;58(15):3998–4009. [Google Scholar]
- 12.Coates RM, et al. Stereochemistry of the methyl → methylene elimination in the enzyme-catalyzed cyclization of geranyl diphosphate to (4S)-limonene. Chem Commun (Camb) 1997;21:2079–2080. [Google Scholar]
- 13.Alonso WR, Rajaonarivony JI, Gershenzon J, Croteau R. Purification of 4S-limonene synthase, a monoterpene cyclase from the glandular trichomes of peppermint (Mentha x piperita) and spearmint (Mentha spicata) J Biol Chem. 1992;267(11):7582–7587. [PubMed] [Google Scholar]
- 14.Hosfield DJ, et al. Structural basis for bisphosphonate-mediated inhibition of isoprenoid biosynthesis. J Biol Chem. 2004;279(10):8526–8529. doi: 10.1074/jbc.C300511200. [DOI] [PubMed] [Google Scholar]
- 15.Seemann M, et al. Pentalenene synthase. Analysis of active site residues by site-directed mutagenesis. J Am Chem Soc. 2002;124(26):7681–7689. doi: 10.1021/ja026058q. [DOI] [PubMed] [Google Scholar]
- 16.Köksal M, Jin Y, Coates RM, Croteau R, Christianson DW. Taxadiene synthase structure and evolution of modular architecture in terpene biosynthesis. Nature. 2011;469(7328):116–120. doi: 10.1038/nature09628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Köksal M, Hu H, Coates RM, Peters RJ, Christianson DW. Structure and mechanism of the diterpene cyclase ent-copalyl diphosphate synthase. Nat Chem Biol. 2011;7(7):431–433. doi: 10.1038/nchembio.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aaron JA, Christianson DW. Trinuclear metal clusters in catalysis by terpenoid synthases. Pure Appl Chem. 2010;82(8):1585–1597. doi: 10.1351/PAC-CON-09-09-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whittington DA, et al. Bornyl diphosphate synthase: Structure and strategy for carbocation manipulation by a terpenoid cyclase. Proc Natl Acad Sci USA. 2002;99(24):15375–15380. doi: 10.1073/pnas.232591099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Starks CM, Back K, Chappell J, Noel JP. Structural basis for cyclic terpene biosynthesis by tobacco 5-epi-aristolochene synthase. Science. 1997;277(5333):1815–1820. doi: 10.1126/science.277.5333.1815. [DOI] [PubMed] [Google Scholar]
- 21.Lesburg CA, Caruthers JM, Paschall CM, Christianson DW. Managing and manipulating carbocations in biology: Terpenoid cyclase structure and mechanism. Curr Opin Struct Biol. 1998;8(6):695–703. doi: 10.1016/s0959-440x(98)80088-2. [DOI] [PubMed] [Google Scholar]
- 22.Rajaonarivony JI, Gershenzon J, Miyazaki J, Croteau R. Evidence for an essential histidine residue in 4S-limonene synthase and other terpene cyclases. Arch Biochem Biophys. 1992;299(1):77–82. doi: 10.1016/0003-9861(92)90246-s. [DOI] [PubMed] [Google Scholar]
- 23.Wise ML, Savage TJ, Katahira E, Croteau R. Monoterpene synthases from common sage (Salvia officinalis). cDNA isolation, characterization, and functional expression of (+)-sabinene synthase, 1,8-cineole synthase, and (+)-bornyl diphosphate synthase. J Biol Chem. 1998;273(24):14891–14899. doi: 10.1074/jbc.273.24.14891. [DOI] [PubMed] [Google Scholar]
- 24.Hong YJ, Tantillo DJ. Quantum chemical dissection of the classic terpinyl/pinyl/bornyl/camphyl cation conundrum-the role of pyrophosphate in manipulating pathways to monoterpenes. Org Biomol Chem. 2010;8(20):4589–4600. doi: 10.1039/c0ob00167h. [DOI] [PubMed] [Google Scholar]
- 25.Balón M, Carmora MC, Muñoz MA, Hidalgo J. The acid-base properties of pyrrole and its benzologs indole and carbazole. A reexamination from the excel acidity method. Tetrahedron. 1989;45(23):7501–7504. [Google Scholar]
- 26.Schenk PM, Baumann S, Mattes R, Steinbiss HH. Improved high-level expression system for eukaryotic genes in Escherichia coli using T7 RNA polymerase and rare ArgtRNAs. Biotechniques. 1995;19(2):196–198, 200. [PubMed] [Google Scholar]
- 27.Katoh S, Hyatt D, Croteau R. Altering product outcome in Abies grandis (-)-limonene synthase and (-)-limonene/(-)-alpha-pinene synthase by domain swapping and directed mutagenesis. Arch Biochem Biophys. 2004;425(1):65–76. doi: 10.1016/j.abb.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 28.Davisson VJ, Woodside AB, Poulter CD. Synthesis of allylic and homoallylic isoprenoid pyrophosphates. Methods Enzymol. 1985;110:130–144. doi: 10.1016/s0076-6879(85)10068-6. [DOI] [PubMed] [Google Scholar]
- 29.Adams RP. In: Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry. Kozlowski AC, editor. Allured Books; Carol Stream, IL: 2007. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.