

Significance
The cation channel TRPM3 is highly expressed in the sensory system, where it plays a key role in the detection of noxious heat and the development of inflammatory heat hypersensitivity. Our understanding of the physiological role of TRPM3 in the sensory system and other tissues is hampered by the lack of potent pharmacologic tools, however. This study describes CIM0216, a small-molecule TRPM3 agonist. Our results indicate that CIM0216 is much more potent than established TRPM3 agonists, particularly owing to its ability to open two distinct cation-permeable pores in TRPM3. Using CIM0216 as a pharmacologic tool, we reveal that activation of TRPM3 evokes the release of calcitonin gene-related peptide from sensory nerve terminals and of insulin from pancreatic islets.
Keywords: TRP channel, TRPM3, peptide release, nociceptor
Abstract
Transient receptor potential (TRP) cation channel subfamily M member 3 (TRPM3), a member of the TRP channel superfamily, was recently identified as a nociceptor channel in the somatosensory system, where it is involved in the detection of noxious heat; however, owing to the lack of potent and selective agonists, little is known about other potential physiological consequences of the opening of TRPM3. Here we identify and characterize a synthetic TRPM3 activator, CIM0216, whose potency and apparent affinity greatly exceeds that of the canonical TRPM3 agonist, pregnenolone sulfate (PS). In particular, a single application of CIM0216 causes opening of both the central calcium-conducting pore and the alternative cation permeation pathway in a membrane-delimited manner. CIM0216 evoked robust calcium influx in TRPM3-expressing somatosensory neurons, and intradermal injection of the compound induced a TRPM3-dependent nocifensive behavior. Moreover, CIM0216 elicited the release of the peptides calcitonin gene-related peptide (CGRP) from sensory nerve terminals and insulin from isolated pancreatic islets in a TRPM3-dependent manner. These experiments identify CIM0216 as a powerful tool for use in investigating the physiological roles of TRPM3, and indicate that TRPM3 activation in sensory nerve endings can contribute to neurogenic inflammation.
Transient receptor potential (TRP) channels represent a large and diverse family of nonselective cation channels that respond to a wide range of chemical and physical stimuli and biophysical properties (1). TRP cation channel subfamily M member 3 (TRPM3), a calcium-permeable nonselective cation channel (2), is a typical example of a polymodally gated TRP channel, in that it can be activated by ligands, such as pregnenolone sulfate (PS) and nifedipine, as well as by heat and membrane depolarization (3, 4). Interestingly, recent evidence indicates that combined stimulation with PS and clotrimazole (Clt) leads to the activation of two distinct permeation pathways in TRPM3: the central pore, which is Ca2+-permeable and carries an outwardly rectifying current, and an alternative ion permeation pathway that mediates an inwardly rectifying monovalent cation current (5).
TRPM3 is highly expressed in somatosensory neurons, where it plays decisive roles in the nocifensive response to PS and heat, as well as in the development of heat hyperalgesia during inflammation (3, 6). In these neurons, TRPM3 is frequently coexpressed with TRPA1 and TRPV1, two TRP channels that have emerged as key regulators of neurogenic inflammation by triggering neuropeptide release from sensory nerve endings (7, 8). Whether activation of TRPM3 can also initiate the release of neuropeptides, such as substance P or calcitonin gene-related peptide (CGRP), which elicit vasodilation, vascular leakage, and other responses in peripheral cell types, is unclear, however. In addition, TRPM3 is expressed in pancreatic beta cells, where it is involved in controlling insulin release (4), as well as in various tissues, including brain, pituitary gland, eye, kidney, and adipose tissue (reviewed in ref. 9). The physiological roles of TRPM3 in these tissues remain only poorly understood, owing in part to the lack of potent and specific pharmacologic tools to modulate its action in vitro and in vivo.
Here we describe the identification and characterization of a TRPM3 agonist, CIM0216, with a potency that greatly exceeds that of currently used agonists. This compound has the unique property to open both ion permeation pathways of TRPM3 without the requirement of other channel modulators. We further demonstrate that CIM0216 acts in a TRPM3-dependent manner to induce pain and evoke neuropeptide release from sensory nerve terminals in the skin, and also to release insulin from pancreatic islets. Collectively, these findings provide a novel powerful tool for use in further studies of the physiological functions of TRPM3, and identify TRPM3 as a novel player in neurogenic inflammation.
Results
CIM0216 Selectively Activates TRPM3 in Stably Transfected HEK293 Cells.
A diverse small-molecule library was screened for compounds that modulate TRPM3-mediated Ca2+ responses in HEK293 cells stably expressing murine TRPM3 (HEK-TRPM3 cells), as described previously (3). One such compound, CIM0216, was found to evoke robust increases in intracellular Ca2+ concentration in HEK-TRPM3 cells and was investigated further. The structure of CIM0216 [racemate of 2-(3,4-dihydroquinolin-1(2H)-yl)-N-(5-methylisoxazol-3-yl)-2-phenylacetamide] is presented in Fig. 1A.
Fig. 1.

CIM0216 activates TRPM3 in HEK-TRPM3 cells. (A) Chemical structure of CIM0216. (B) [Ca2+]I was monitored using Fluo-4 AM in HEK-TRPM3 cells before and after the addition of different concentrations of PS (black), CIM0216 (blue), and PS plus CIM0216 (red). Responses were measured as peak increases in fluorescence minus basal, expressed relative to a maximum PS response, and are given as mean ± SEM. n = 2. (C) Time course of Ca2+ imaging measurements in HEK293 cells stably expressing TRPM3 (HEK-TRPM3 cells) and nontransfected (NT) HEK293 cells during application of PS (40 µM) and CIM0216 (1 µM). (D) Time course at ±80 mV of a whole-cell patch-clamp recording on HEK-TRPM3 cells treated with PS (40 µM) and CIM0216 (1 µM), (E) I–V traces corresponding to the time points indicated in D. (F) Time course at ±80 mV of a whole-cell patch-clamp recording showing the application of PS and CIM0216 on nontransfected HEK293 cells. Mean ± SEM values are shown; n = 5. (G) Time course at ±80 mV of whole-cell patch-clamp measurement on HEK-TRPM3 cells presenting the block of CIM0216-induced currents by isosakuranetin (5 µM). (H) Time course at ±80 mV of a patch-clamp recording in whole-cell configuration showing CIM0216 dose-dependent activation of TRPM3 currents in HEK-TRPM3 cells. (I) Dose–response curves of CIM0216 (blue) and PS (black) in HEK-TRPM3. n = 5. Unless indicated otherwise, standard compound concentrations were 40 µM for PS and 1 µM for CIM0216.
In HEK-TRPM3 cells, CIM0216 elicited a dose-dependent Ca2+ response [negative logarithm of the EC50 value (pEC50) = 0.77 ± 0.1 µM], which was not observed in nontransfected HEK293 cells (Fig. S1). The endogenous TRPM3 activator pregnenolone sulfate (PS) also elicited a dose-dependent Ca2+ response, typified by a smaller Ca2+ influx and a fivefold lower potency (pEC50 = 3.0 ± 0.1 µM; n = 2) (see also ref. 4) compared with CIM0216 (Fig. 1B). In the presence of 40 µM PS, the dose dependency of CIM0216 shifted to a lower concentration in HEK-TRPM3 cells (pEC50 = 42 ± 0.6 nM CIM0216 in the presence of PS; Fig. 1B). Single-cell FURA2-ratiometric Ca2+ imaging revealed a robust increase in intracellular Ca2+ concentration (1,145 ± 26 nM; n = 603 from at least three independent measurements) on stimulation with 1 µM CIM0216 in all HEK-TRPM3 cells. These responses were not observed in nontransfected HEK cells (n = 166) or in the absence of extracellular Ca2+ (Fig. 1C and Fig. S1).
In whole-cell patch-clamp experiments, stimulation of HEK-TRPM3 cells evoked robust, doubly rectifying currents (Fig. 1 D and E), which were not observed in nontransfected naïve HEK cells (Fig. 1F) and were blocked by the TRPM3 inhibitor isosakuranetin (5 µM; Fig. 1G) (10). The effect of CIM0216 on TRPM3 currents was concentration-dependent; however, in the patch-clamp experiments, saturating responses were never achieved within the solubility range of CIM0216 and the time period of the experiment. A similar dose-dependent activation for CIM0216 was obtained for HEK293 cells transiently transfected with human TRPM3 (n = 5) (Fig. S2 and Table 1). Remarkably, the PS-induced current increases in HEK-TRPM3 cells at saturating PS concentrations (100 µM) were already obtained at a 200-fold lower concentration of CIM0216 (0.5 µM), indicating that compared with PS, CIM0216 has significantly greater efficacy for activating TRPM3 (Fig. 1I).
Table 1.
CIM0216 selectivity
| Channel | Activation | Block at 10 µM |
| hTRPM1 | − | − |
| hTRPM2 | − | + (17%) |
| mTRPM3 | +++ | − |
| hTRPM3 | +++ | − |
| hTRPM4 | − | − |
| hTRPM5 | − | + (34%) |
| hTRPM6 | − | − |
| hTRPM7 | − | − |
| hTRPM8 | − | ++ (61%) |
| mTRPA1 | + (15 ± 8% of MO response at −80 mV) | − |
This table represents the activating/ blocking activity of CIM0216 toward mouse (m) and human (h) TRPM3 and to other TRPM family members, as well as other sensory TRP channels. In all conditions, the selectivity of CIM0216 was tested by application of 10 µM CIM0216 to HEK293 cells stably expressing hTRPM4 or mTRPM5 or transiently transfected with hTRPM1, hTRPM2, hTRPM6, mTRPM7, hTRPM8, or hTRPV1. The selectivity of CIM0216 toward TRPA1 was tested at a concentration of 10 µM and at −80 mV in CHO cells stably expressing mTRPA1.
To evaluate the specificity of CIM0216 for TRPM3, we examined its effects on all members of the TRPM subfamily. When tested at a concentration of 10 µM, CIM0216 had no stimulating/blocking effect on TRPM1 (11), TRPM4, TRPM6, or TRPM7 currents; however, a small blocking effect of CIM0216 was observed after activation of TRPM2 (16.6% block) and TRPM5 (33.5% block) (Fig. S3 and Table 1). We also tested CIM0216’s specificity toward other sensory TRPs and observed no detectable effect on human TRPV1 and TRPM8 channel activation at a concentration of 10 µM (Fig. S4). In contrast, CIM0216 induced a significant block of 61 ± 5% of the menthol-induced currents in TRPM8-transfected HEK293 cells. In CHO cells stably expressing murine TRPA1, a significant but minor increase in current amplitude (<10% increase at −80 mV relative to the response to stimulation with 100 µM allyl isothiocyanate) was observed after stimulation by 100 µM CIM0216 (Fig. S4 and Table 1).
CIM0216 and Clt Have Common Features for TRPM3.
The activation of TRPM3 currents by CIM0216 developed over time, reaching maximal activation within ∼100 s (Fig. 1 D, G, and H), and then faded out gradually after washout, with a mean ± SEM time constant of 208 ± 12 s (n = 16). This finding contrasts with the rapidly reversible agonistic effect of PS (Fig. 2A). Whereas pretreatment with Clt (10 µM) led to strong potentiation of PS-induced TRPM3 currents (Fig. 2 A, B, G, and H) (5), it had no amplifying effect on the CIM0216-induced TRPM3 current (Fig. 2 C and D). In contrast, the CIM0216-induced TRPM3 outward current was mainly blocked by 10 µM Clt (76.9 ± 3% inhibition at −80 mV and 67.6 ± 5% inhibition at +80 mV; n = 4), possibly suggesting competition between Clt and CIM0216 for a similar binding site (Fig. 2G). Conversely, measurement of intracellular Ca2+ in HEK-TRPM3 cells revealed a dose-dependent potentiation of CIM0216 on PS stimulation (pEC50 = 42 ± 0.6 nM for CIM0216 in the presence of PS) (Fig. 1B).
Fig. 2.

CIM0216s modulation of PS-induced TRPM3 currents shows high similarity to the Clt-induced effects. (A) Time course at ±80 mV of a whole-cell patch-clamp recording showing the effect of Clt (10 µM) on PS (40 µM)-induced HEK-TRPM3 cells. (B) I–V relationship corresponding to the time points indicated in A. (C) Time course at ±80 mV of a whole-cell patch- clamp recording showing the effect of Clt (10 µM) on CIM0216 (1 µM) stimulation in HEK-TRPM3–expressing cells. (D) I–V relationship corresponding to the time points indicated in C. (E) Time course at ±80 mV of whole-cell patch-clamp measurement on HEK-TRPM3 cells after stimulation by PS (40 µM) and CIM0216 (0.1 µM) plus PS (40 µM). (F) I–V relationship corresponding to the time points indicated in E. (G) Mean current increase at +80 mV and −80 mV in HEK-TRPM3 cells after stimulation by PS (40 µM) and CIM0216 (1 µM) in the absence (black bar) and presence (gray bar) of Clt (10 µM). n = 5. (H) Relative potentiation of the PS-induced currents after incubation by Clt (10 µM) and CIM0216 (0.1 and 1 µM) of the inward (−150 mV; black bars) and outward (+150 mV; gray bars) currents. n = 4.
These experiments clearly show that the potentiation of PS-induced Ca2+ influx by CIM0216 occurs at much lower concentrations than in direct activation. In addition, incubation with a low dose of CIM0216 (0.1 µM) in patch-clamp experiments caused a strong potentiation of the currents evoked by PS (37 ± 8-fold potentiation at −150 mV and 10 ± 3-fold potentiation at +150 mV; n = 5), which by itself does not induce appreciable activation of TRPM3 (Fig. 2 E and F). Thus, CIM0216 has greater potency for potentiating PS-induced currents than for activating TRPM3 currents.
A similar potentiation of PS-induced TRPM3 currents was observed for Clt (Fig. 2H). On stimulation by CIM0216, current–voltage (I–V) relationships showed double rectification, with marked inward currents at strongly hyperpolarizing potentials (Figs. 1 and 2), similar to that described for currents activated by the combination of Clt and PS (5). Quantifying the rectification as the ratio of the absolute current amplitude at −150 mV and +150 mV (ratio−150/+150) yielded a rectification score of 0.58 ± 0.02 for currents in the presence of CIM0216, significantly different from the rectification score of PS-induced currents (0.10 ± 0.01; P < 0.001) but comparable to that seen in the combined presence of PS and Clt (Fig. 3A).
Fig. 3.

CIM0216-induced TRPM3 currents show high similarity to PS + Clt-induced TRPM3 currents. (A) Rectification pattern of PS, PS + Clt, and CIM0216-induced TRPM3 currents in HEK-TRPM3 cells, derived by plotting the currents at +150 mV against the currents at −150 mV. (B) Time course at ±80 mV of a whole-cell patch-clamp recording on HEK-TRPM3 cells. At the indicated time points, all Na+ was replaced by NMDG+. (C) Time course at ±80 mV of a patch- clamp recording performed in the inside-out configuration on HEK-TRPM3 cells during application of CIM0216 (1 μM). (D) I–V traces corresponding to indicated time points in C. (E) Current traces obtained with a step protocol in HEK-TRPM3 cells after stimulation by PS (black; 40 µM), PS (40 µM) + Clt (10 µM; red), and CIM0216 (1 µM; blue). (F) G–V plot for CIM0216. The fit was obtained with a Boltzmann function (n = 6).
Plotting the outward vs. inward current amplitudes of the TRPM3 currents showed a linear correlation (R2 = 0.949) between outward and inward current amplitudes over a range of CIM0216 concentrations, indicating that the rectification score is not strongly dependent on CIM0216 concentration (Fig. S5). When N-methyl-d-glucamine (NDMG+) was substituted for Na+ in the extracellular solution, TRPM3 currents evoked by CIM0216 lacked an inward component (Fig. 3B). This indicates that the inward current is carried exclusively by cations, and that NMDG+ is not able to permeate the channel under these circumstances. Inside-out patches of HEK-TRPM3 cells showed a current increase after stimulation by CIM0216, indicating that CIM0216 can activate TRPM3 in a membrane-delimited manner (Fig. 3 C and D).
Conductance–voltage (G–V) curves in the presence of CIM0216 were biphasic, showing a minimal conductance at around −50 mV and increasing conductance at more depolarizing and hyperpolarizing potentials, which saturated at around +200 mV and −250 mV, respectively (Fig. 3 E and F). Identical biphasic G–V curves can be observed in the combined presence of PS and Clt (5). To further investigate the potency of CIM0216 to activate biphasic G–V curves in TRPM3, we analyzed mutant channels with mutations in the S4 domain (W982R) and the TRPM3 pore domain (E1057C) (5). Stimulation by CIM0216 induced G–V currents identical to the PS-induced G–V currents in HEK293 cells overexpressing W982R (Fig. S6). Conversely, HEK293 cells overexpressing the E1057C mutant showed overlapping G–V currents at depolarizing potentials for PS and CIM0216 stimulation. However, at hyperpolarizing potentials, G–V currents were observed only after CIM0216 stimulation (Fig. S6). Taken together, the foregoing data indicate that the effects of a single CIM0216 application on TRPM3 are similar to those of the combined application of PS and Clt.
Given that Clt is able to open the alternative ion permeation pathway in TRPM3 in the presence of PS (5), we considered the possibility that a single application of CIM0216 could open both ionic pores, the central pore and the alternative cation permeation pathway, independent of Clt. Several lines of evidence support this hypothesis. First, the CIM0216-induced inward current is more resistant to Ca2+-dependent desensitization, a process that leads to rapid decay of the outwardly rectifying TRPM3 current (75 ± 7% block at +80 mV), whereas the inward currents showed no Ca2+-dependent desensitization (Fig. 4 A and B). Second, the nonspecific pore blocker La3+ isolated the inwardly rectifying current component. Stimulation of HEK-TRPM3 cells by CIM0216 (1 μM) in the presence of the pore blocker La3+ (100 µM) induced a stable inward rectifying current. An inwardly and outwardly rectifying current component was observed after removal of the La3+ (Fig. 4 C–E). Of note, the G−V curve of the inward current in the presence of La3+ was shifted to more negative voltages (by 13 ± 6 mV) compared with control values (Fig. S7). This slight shift in apparent voltage dependence may reflect a low-affinity block of the alternative ion permeation pathway, as has been described for the gating-pore current in NaV1.4, interactions of La3+ with membrane surface charges, and/or an effect of La3+ on channel gating (12, 13). Third, the La3+-insensitive inward current exhibited identical properties of the alternative permeation pathway activated by PS in the presence of Clt, including (i) strong inward rectification, (ii) inhibition by extracellular Mg2+ and Ca2+ (Fig. 4 F and G), and (iii) increased permeability for guanidinium compared with Na+ when all extracellular Na+ was replaced by guanidinium (Fig. 4H). Taken together, these findings indicate that stimulation with CIM0216 by itself is able to mimic the effects of coapplication of PS plus Clt on TRPM3, namely simultaneous opening of the central pore and the alternative permeation pathway (Fig. S8).
Fig. 4.

Desensitization and permeation properties of the alternative ion permeation pathway. (A) Time course at ±80 mV of a whole-cell patch-clamp recording on HEK-TRPM3 cells during application of CIM0216 (1 µM). At the indicated time points, the extracellular bath concentration was replaced by a solution containing 1 mM [Ca2+]ex. (B) I–V relationship at the time points indicated in A. (C) Time course of a whole-cell patch-clamp recording on HEK-TRPM3 cells at ±150 mV showing CIM0216-induced currents in the presence and absence of the pore blocker La3+. (D) I–V traces corresponding to the indicated time points in C. (E) Current traces obtained with an indicated step protocol from +200 to −200 mV in +20-mV steps under the condition of costimulation with CIM0216 and La3+. (F and G) I–V traces obtained during simultaneous application of CIM0216 and La3+ in the presence of different concentrations of Ca2+ (F) or Mg2+ (G). (Insets) Mean current normalized to the maximum Na+ current in the absence of Ca2+ (F) or Mg2+ (G). n ≥5. (H) I–V traces obtained during simultaneous application of CIM0216 and La3+ in the presence of Na+ and guanidinium (Gua+). Unless indicated otherwise, standard compound concentrations were 40 µM for PS, 100 µM for La3+, and 1 µM for CIM0216.
Synergistic Effects of Heat and CIM0216 on TRPM3.
TRPM3 has been identified as a temperature-sensitive ion channel that is steeply activated by heating (3). Previous work on other thermosensitive TRP channels has demonstrated synergistic effects between chemical agonists and thermal stimuli; for example, PS responses in TRPM3 are potentiated at high temperatures (3). We observed a similar synergism of heat and CIM0216 on TRPM3. We found that increasing the temperature from room temperature (22 °C) to 37 °C strongly potentiated CIM0216 responses in Ca2+ imaging experiments. Interestingly, CIM0216 concentrations as low as 100 nM, which are subthreshold at room temperature, evoked robust responses at 37 °C (Fig. 5 A and B). Taken together, these data demonstrate that the activating effect of CIM0216 on TRPM3 is temperature-dependent.
Fig. 5.
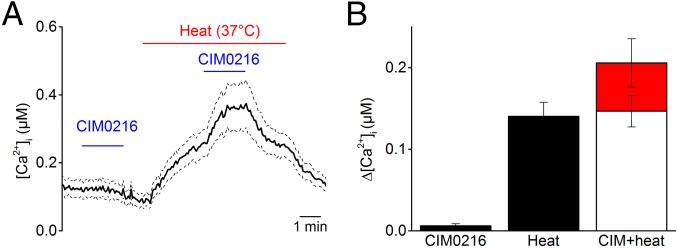
Synergistic effects of heat and CIM0216 on TRPM3. (A) Typical examples of the intracellular calcium increase induced by low doses of CIM0216 (0.1 µM) applied at room temperature (22 °C) and 37 °C in HEK-TRPM3 cells. (B) Bar diagram showing average Ca2+ increases in response to CIM0216 (0.1 µM), heat stimulus (37 °C), and CIM0216 (0.1 µM) at 37 °C. The open bar illustrates the calculated summation of CIM0216 and heat response. The red bar shows the measured value of combined application of heat and CIM0216, illustrating the supra-additive effect of heat on CIM0216 stimulation. n > 42 cells from at least three independent measurements. Data are presented as mean ± SEM.
CIM0216 Activates TRPM3 Channels in Somatosensory Neurons and Evokes Pain.
Previous work revealed that TRPM3 is functionally expressed in mouse dorsal root ganglia (DRG) and trigeminal ganglion neurons (TGN) (3). To test whether CIM0216 is able to specifically activate endogenous TRPM3, we first compared intracellular Ca2+ responses to stimulation with PS and CIM0216 in freshly isolated somatosensory neurons derived from DRG and TGN. Stimulation with CIM0216 evoked robust and reversible calcium signals (ΔCa2+ = 357 ± 14 nM) in 57% of DRG (311 of 543) (Fig. 6 A and D) and 62% of TGN (197 of 317) isolated from wild-type (WT) mice. The majority (96%) of the CIM0216-responsive neurons were responsive to PS as well (Fig. 6 A and E). Importantly, the fraction of CIM0216-sensitive neurons was drastically decreased in DRG and TGN preparations from TRPM3−/− mice, whereas the fraction of neurons that responded to allyl isothiocyanate (MO) (68% in WT and 63% in TRPM3−/−), an activator of TRPA1 (14), or to capsaicin (73% in WT and 77% in TRPM3−/−), an agonist of TRPV1 (15), were not changed significantly (Fig. 6 B and D). The small number of TRPM3−/− neurons that responded to CIM0216 stimulation (11% of all neurons) were always reactive to MO stimulation, and could be blocked in the presence of the TRPA1 inhibitor HC030031 (16) (Fig. S9), possibly suggesting that these responses reflect the very mild agonistic effect of CIM0216 on TRPA1 (Fig. S4). A high fraction of CIM0216 responders (55%; 110 of 200) was found in DRG neurons obtained from Trpa1-deficient mice (Fig. 6 C and D). In 94% of all CIM0216 responders in WT mice, the CIM0216-induced Ca2+ increase could be blocked by application of the TRPM3 blocker isosakuranetin (5 µM). The small subset of neurons that still responded to CIM0216 in the presence of isosakuranetin was responsive to MO as well (Fig. 6 E and F).
Fig. 6.

Reduced CIM0216 responses in TRPM3-deficient sensory neurons. (A–C) Representative traces showing typical patterns of intracellular Ca2+ in DRG and TGN from control animals Trpm3+/+ (A), Trpm3−/− (B), and Trpa1−/− (C) mice in response to PS (20 µM), CIM0216 (1 µM), MO (100 µM), capsaicin (Cap; 1 µM), and K+ (50 mM). (D) Percentage of sensory neurons derived from TRPM3+/+, TRPM3−/−, and TRPA1−/− mice responding to stimulation by PS (20 µM), CIM0216 (1 µM), MO (100 µM), and capsaicin (1 µM). (E) Percentage of sensory neurons responding to CIM0216 in preparations from TRPM3+/+, TRPM3−/−, and TRPA1−/− mice. Different colors correspond to the different subtypes of CIM0216 responders based on PS and MO sensitivity. (F) Representative traces showing typical response patterns of intracellular Ca2+ in DRG and TGN from control animals in response to CIM0216 (1 µM), MO, and high K+. At the indicated time bar, the TRPM3 blocker isosakuranetin (5 µM) was added.
In whole-cell current recordings in DRG and TGN from WT mice, CIM0216 activated a significant current increase (13.0 ± 3 pA/pF at –80 mV and 17.3 ± 5 pA/pF at +80 mV) in 62% of the neurons (n = 13) (Fig. 7 A and B). CIM0216-induced currents showed a doubly rectifying I–V relationship (ratio−120/+120 = 0.47 ± 0.06; n = 8). Application of La3+ induced 100% blockage of the outward current, but only partial blockage of the inward current (Fig. 7 A and B). These properties are identical to those observed for CIM0216 stimulation in a TRPM3-overexpression system (Fig. 4C) and for coapplication of PS plus Clt (5). TRPM3-deficient neurons showed no detectable current response to CIM0216 stimulation (Fig. 7C) (n = 12). Note that in both Trpm3+/+- and Trpm3−/−-derived neurons, application of CIM0216 induced an inhibition of outward current of varying amplitude. This blockage was never observed in Ca2+ imaging experiments, however, suggesting that this small inhibitory effect is not sufficient to induce excitation of sensory neurons.
Fig. 7.

Neuronal and nocifensive responses to CIM0216. (A) Amplitude of currents at ±80 mV of a patch-clamp recording in whole-cell configuration during application of CIM0216 (1 µM) and La3+ (100 µM) in a TRPM3+/+ TGN. (B) I–V relationship of the CIM0216-induced current at time points as indicated in A. (Inset) CIM0216-induced current increase in the presence and absence of La3+, obtained as the difference between two traces in B. (C) Amplitude of currents at ±80 mV of a patch-clamp recording in whole-cell configuration during application of CIM0216 (1 µM) in a TRPM3−/− TGN. (D) Total number of behavioral responses (paw licks and lifts within 2 min) in response to intraplantar injection of vehicle (vhc; 0.5% DMSO), PS (15 nmol/paw), CIM0216 (2.5 nmol/paw), or capsaicin (Caps; 0.5 nmol/paw) in WT mice (black bars) and TRPM3−/− mice (red bars). n = 9 animals for each genotype. Data are presented as mean ± SEM. The black line represents the unpaired t test; the blue bar, the paired t test. *P < 0.05; **P < 0.01.
To investigate whether CIM0216 can activate TRPM3 in vivo, we tested for nocifensive behavior in WT and TRPM3−/− mice following injection of CIM0216 into the plantar skin of the hind paw. Injection of CIM0216 (2.5 nmol) in the hind paw evoked strong nocifensive behavior (i.e., paw licking and lifting) in control mice (Fig. 7D). The nocifensive behavior to CIM0216 was remarkably more exhaustive compared with intraplantar injection of a sixfold-higher dose of PS (15 nmol). TRPM3−/− mice completely lacked this nocifensive response to CIM0216 and PS, whereas injection of the TRPV1-agonist capsaicin (0.5 nmol) evoked a normal nocifensive behavior in both genotypes (Fig. 7D). Taken together, these findings indicate that CIM0216 induced pain in a TRPM3-dependent manner, with a pain response more robust than that to PS.
Neuropeptide Release by CIM0216 Depends on TRPM3 Activity.
The sensory TRP channels TRPA1 and TRPV1 participate in neurogenic inflammation and vasodilation, by initiating the release of calcitonin gene-related peptide (CGRP) and substance P (7, 8, 17). Previous results have shown that PS responsiveness, like capsaicin responsiveness (18), is restricted almost exclusively to small-diameter (<25 µm) DRG neurons, which are known to include unmyelinated nociceptor neurons (3). In addition, a large subpopulation of PS-responsive DRG neurons was also responsive to capsaicin and MO, illustrating a functional coexpression of TRPM3 and TRPV1/ TRPA1.
To test whether activation of TRPM3 in nerve terminals also leads to neuropeptide release and neurogenic inflammation, we performed release experiments using isolated mouse hind paw skin. The skin is densely innervated with peptidergic CGRP containing nerve endings. A 5-min incubation of the skin with PS produced a dose-dependent release of CGRP, which was absent in skin preparations derived from TRPM3−/− knockout mice (Fig. 8A). A similar PS-induced CGRP release was observed in TRPV1−/− and TRPA1−/− mice (Fig. S10). In addition, the novel TRPM3 agonist CIM0216 caused a significant, dose-dependent increase of cutaneous CGRP release in WT animals, which was largely reduced in the presence of the antagonist isosakuranetin (5 µM) and was absent in skin preparations of Trpm3-deficient mice (Fig. 8 B and C), suggesting a selective action of the compound on TRPM3. In addition, CIM0216 evoked comparable CGRP release in TRPV1−/− TRPA1−/− double-knockout mice as in WT mice (Fig. S10). Collectively, these results indicate that chemical ligands such as CIM0216 can activate TRPM3 in sensory nerve terminals, leading to neuropeptide release and acute neurogenic inflammation.
Fig. 8.

TRPM3 agonists release CGRP from skin nerve terminals. (A) PS increased the outflow of CGRP from skin preparations in a concentration-dependent manner. No significant increase of CGRP release was detected in TRPM3−/− tissue. Asterisks refer to P < 0.01 between genotypes (n = 10 vs. 7, ANOVA and LSD statistical tests). (B) CGRP release from WT skin during a 5-min incubation with CIM0216 at different concentrations (1, 5, 10, 50, and 100 µM) and capsaicin (0.3 µM) for comparison. (C) CGRP release during consecutive 5-min periods (5 min baseline, next 5 min, etc.). CIM0216 (50 µM) was applied during the period tagged 5–10 min to hind paw skin derived from WT mice (black; n = 7) and Trpm3-deficient mice (red; n = 4). In the presence of the TRPM3 blocker isosakuranetin (5 µM), no significant CIM0216-induced CGRP release was observed in WT skin (blue). n = 7; same animals as above. *P < 0.01 between the WT groups, ANOVA and Fisher’s least significant differences test.
CIM0216 Activates TRPM3 Channels in Pancreatic Beta Cells and Releases Insulin.
TRPM3 expression has been described in the pancreas, and stimulation of pancreatic islets with PS evokes insulin secretion (4). To evaluate the effect of CIM0216 in pancreatic islets, we performed Ca2+ imaging on isolated pancreatic islets and used PS and CIM0216 as specific TRPM3 activators. Stimulation by PS and CIM0216 induced an increase in fluorescence in all islets derived from control mice (n = 5) (Fig. 9 A and C). In contrast, no Ca2+ increase was observed in pancreatic islets derived from Trpm3-deficient mice (n = 13) after stimulation by CIM0216 (Fig. 9B). As a positive control, a high-K+ solution (30 mM) was applied at the end of every experiment, and no difference in fluorescence increase was observed in islets derived from control and Trpm3-deficient mice (Fig. 9C).
Fig. 9.

CIM0216 induces TRPM3-dependent calcium and current increases and enhances insulin release from pancreatic islets. (A and B) Effect of CIM0216 on intracellular [Ca2+] in islets derived from WT mice (A) and Trpm3−/− mice (B). The islets were bathed in a solution containing 3 mM glucose. PS (40 µM)-, CIM0216 (1 µM)-, 10 mM glucose-, and high-K+ (50 mM)-containing solutions were added at the indicated time points. (C) Average increase in fluorescence ratio (F350/F380) after stimulation with PS (40 µM), CIM0216 (1 µM), and 50 mM K+ in islets from WT and Trpm3−/− mice. n = 9 from three mice; ***P < 0.001, unpaired t test. (D) Amplitude of currents at ±80 mV of a patch-clamp recording in whole-cell configuration during application of PS (40 µM), CIM0216 (1 µM), and La3+ (100 µM) in a Trpm3+/+ isolated pancreatic islet cell. (E and F) I–V relationship of the PS-induced (E) and CIM0216-induced (F) currents at time points as indicated in D. (G) Mean currents at ±80 mV of a patch-clamp recording in whole-cell configuration during application of PS (40 µM) and CIM0216 (1 µM) in Trpm3−/− isolated pancreatic islet cells (n = 5). (H) Mean current increases at ±80 mV of a patch-clamp recording in whole-cell configuration during application of PS (40 µM) or CIM0216 (1 µM) on Trpm3+/+ and Trpm3−/− pancreatic islet cells (n ≥ 5). (I) Insulin release after a 15-min incubation with vehicle (vhc, DMSO), PS (100 µM), CIM0216 (5 µM and 20 µM), high K+ (50 mM), and glucose (Gluc; 20 mM) in WT mice (black bars) and TRPM3−/− mice (red bars). n = 4 from four independent measurements; **P < 0.01, unpaired t test.
In whole-cell patch-clamp experiments on isolated WT pancreatic cells, stimulation by PS induced an outwardly rectifying current (1 ± 1 pA at –80 mV and 47.3 ± 14 pA at +80 mV) in 100% of the pancreatic cells (n = 6) that could be blocked in the presence of La3+ (Fig. 9 D, E, and H); however, stimulation by CIM0216 induced a doubly rectifying current (81.9 ± 19 pA at –80 mV and 539.6 ± 159 pA at +80 mV) in all of the pancreatic cells (n = 8). Incubation by La3+ induced a block of the outward current (78% inhibition), whereas the inward current remained mainly unaffected (37% inhibition) (Fig. 9 D and F). In contrast, PS and CIM0216 stimulation did not induce a current increase in Trpm3−/−-derived pancreatic cells (Fig. 9 G and H).
An increase in intracellular Ca2+ causes exocytosis of large vesicles and thereby releases insulin (19). Islets incubated with PS or CIM0216 showed a dose-dependent increase in insulin release compared with vehicle-incubated islets (Fig. 9I; n ≥ 3 independent experiments). In contrast, in Trpm3-deficient islets, no additional insulin release was observed after incubation with the TRPM3 agonists (Fig. 9I). In addition, no significant differences between WT and Trpm3 knockout islets were observed following incubation with a high-glucose– or high-K+–containing solution (Fig. 9I). These data demonstrate that stimulation of pancreatic islets by CIM0216 induces the release of insulin in a TRPM3-specific manner.
Discussion
In this paper, we have presented a newly identified and potent activator of TRPM3, CIM0216, which has the unique property to open distinct cation permeation pathways by itself and represents, to our knowledge, the most potent activator of TRPM3 identified so far. The use of this agonist has allowed us to further investigate the physiological consequences of the opening of TRPM3 in vivo. In particular, we demonstrate that activation of TRPM3 can evoke the release of CGRP from sensory nerve terminals in the skin, implying a potential role in the initiation of neurogenic inflammation. Furthermore, our present results show that potent TRPM3 agonists can act as secretagogues triggering insulin release from pancreatic islets, even at low extracellular glucose levels.
Our results demonstrate that CIM0216 activates TRPM3 in a membrane-delimited manner, because CIM0216 is capable of activating the channel in the inside-out patch-clamp configuration. We and other investigators have reported that PS acts on TRPM3 only when applied at the extracellular side of the membrane (4, 5, 20), whereas CIM0216 is active from the intracellular side as well. Of note, a similar intracellular interaction side on TRPM3 has been suggested for Clt (5). The Clt-induced block of TRPM3 currents after stimulation by CIM0216 might point to a possible competition between Clt and CIM0216 for the same interaction side; however, further investigation is needed to identify the CIM0216 interaction side on TRPM3.
We identify CIM0216 as a very potent activator of TRPM3. At first sight, there is very little structural analogy between CIM0216 and known TRPM3 ligands, such as PS or nifedipine. A previous study reported that a negative charge at position C3 of the steroid PS is required for TRPM3 activation (20); however, no acidic substituents are present on CIM0216, and as such, the compound does not have a negative charge. Moreover, we show evidence for a role of CIM0216 as a modulator of PS-induced TRPM3 responses, as is described for Clt. The structure of Clt is a basic compound comprising four aromatic rings centered around a carbon atom. CIM0216 has the same basic properties and comprises three aromatic ring systems around a central linking moiety.
We found that activation of TRPM3 by CIM0216 stimulation induced an inwardly and outwardly rectifying current in both TRPM3-overexpressing cells and cells that endogenously express TRPM3. This I–V relationship differs from the outwardly rectifying TRPM3 current described for other TRPM3 agonists like PS and nifedipine and for heat (3, 4, 20), but is similar to that measured on combined application of endogenous neurosteroids and exogenous chemicals such as Clt (5). The properties of the inwardly rectifying component indicate that CIM0216 itself is able to open the same alternative pathway observed after the combined application of PS and Clt. The large inward current, which under physiological conditions is carried mainly by Na+, makes CIM0216 an ultrapotent TRPM3 agonist, especially in excitable cells, such as sensory neurons and pancreatic β cells. In these cells, robust Na+ influx may facilitate membrane depolarization and concomitant Ca2+ influx via voltage-gated Ca2+ channels, thereby augmenting intracellular Ca2+ signals and vesicular release.
Using CIM0216, we have described a novel physiological function of TRPM3 as a mediator of neuropeptide release and neurogenic inflammation. In particular, we have shown that incubation of mouse hind paw skin with different TRPM3 agonists induces a dose-dependent CGRP release that was absent in the presence of a specific TRPM3 blocker or in hind paw skin derived from Trpm3-deficient mice. This finding suggests that opening of TRPM3 can lead to CGRP-induced vasodilation and vascular leakage, similar to what has been described for TRPV1 and TRPA1 (7, 21, 22). We note that the functional expression of a Ca2+-permeable ion channel in a small subpopulation of C and Aδ fibers does not constitute evidence that such channels are involved in CGRP release, given that, for instance, TRPM8-expressing neurons show no coexpression with typical markers of nociceptor neurons, such as CGRP, substance P, and isolectin B4, and TRPM8 activation is not known to cause neuropeptide release (23). Previous studies have shown that application of PS induces vasoconstriction, an effect blocked by pre-exposure to a TRPM3-blocking antibody (24, 25). It should be noted that these studies used relatively high PS doses and were performed on aorta, which is poorly braided with CGRP fibers. Further investigation of Trpm3-deficient mice will clarify the role of TRPM3 in vascular smooth muscle contractility.
Several lines of evidence indicate that CIM0216-induced nociceptive behavior and CGRP release is due to the activation of TRPM3, and not to activation of other nociceptive transducers, such as TRPV1 and TRPA1. First, no or very little stimulating effects were observed in cells overexpressing TRPV1 and TRPA1 after stimulation by at least 10 µM CIM0216. Second, the majority of the sensory neurons that responded to CIM0216 were also positive for PS stimulation, known as a selective TRPM3 activator (4). Only a very limited fraction of neurons (∼10%) were unresponsive to PS stimulation but positive for CIM0216 and MO stimulation. Third, the number of CIM0216-sensitive nociceptors was strongly reduced in TRPM3-deficient mice, and the remaining responders could be blocked by the TRPA1 inhibitor HC030031. Fourth, a similar high percentage of neurons (55% of all neurons) responded to PS and CIM0216 stimulation in neurons derived from Trpa1-deficient mice. Fifth, in contrast to WT mice, TRPM3-deficient mice did not react to CIM0216 injection into the hind paw, and incubation with CIM0216 did not induce further CGRP release in hind paw skin. Sixth, a similar CIM0216-induced CGRP release was observed in hind paw skin of WT, TRPV1−/−, and TRPA1−/− knockout mice.
We also have illustrated the functional role of TRPM3 in pancreatic cells and the involvement of TRPM3 in insulin release. Stimulation of isolated pancreatic islets by TRPM3 activators PS and CIM0216 induced a strong increase in intracellular Ca2+ and current density and further insulin release, which was fully lacking in TRPM3−/− cells. The first indications of the involvement of TRPM3 in insulin release were reported by previous studies showing that PS led to a rapid Ca2+ influx and enhanced insulin secretion from pancreatic islets (4). Remarkably, no difference in calcium influx or insulin release was detected between control and Trpm3-deficient mice after stimulation with high glucose concentrations. In addition, TRPM3−/− mice and TRPM3+/+ controls exhibited no obvious differences in basal blood glucose levels (3). Taken together, these results suggest only a minor role for TRPM3 in normal glucose-induced insulin release, although the possibility that TRPM3 modulates insulin release under specific (patho)physiological conditions cannot be excluded.
In conclusion, our observations identify CIM0216 as the most potent known agonist of TRPM3, with the unique property of opening different ion permeation pathways of TRPM3 independent of other channel modulators. This novel agonist has led to the identification of a role of TRPM3 in the release of CGRP and insulin.
Materials and Methods
Cell Culture and Transfection.
HEK293 cells stably expressing murine TRPM3 and nontransfected HEK293 were designed and cultured as described previously (3). DRG and TGN were isolated as described previously (26). Because the RNA expression of TRPM3 varies very little between different ganglia (27), DRG were isolated from along the entire vertebral column of the mouse. Isolation of pancreatic islets and dissociation into single cells were performed as described previously (28).
Animals.
The TRPM3−/−, TRPV1−/−, and TRPA1−/− mice have been described previously (3, 18, 29). The knockout strains were backcrossed at least six times into the C57BL/6J background, and C57BL/6J mice served as WT controls. Mice of all genotypes were housed under identical conditions, with a maximum of five animals per cage, on a 12-h light–dark cycle and food and water ad libitum. All experiments were performed using 8- to 12-wk-old mice. Only male mice were used in the behavioral experiments.
Small-Molecule Library Screening.
Small molecules were present in black 384-well assay plates (Greiner) with buffer containing 138 mM NaCl, 5.4 mM KCl, 4 mM CaCl2, 2 mM MgCl2, 10 mM glucose, and 10 mM Hepes; pH 7.2 with NaOH). HEK-TRPM3 and nontransfected HEK293 cells were loaded with 2 µM Fluo-4 AM (Invitrogen) for 30 min, detached using trypsine, collected in calcium-free medium (138 mM NaCl, 5.4 mM KCl, 2 mM MgCl2, 10 mM glucose, and mM 10 Hepes; pH 7.2 with NaOH), and added to the assay plate at ± 2,000 cells per well. The final CaCl2 concentration in the assay solution was 2.4 mM. Fluorescence (excitation at 485 nm; emission at 535 nm) was measured with an Envision fluorescence reader before and after the addition of 50 µM PS. All responses were normalized to the response to 50 µM PS in the absence of small molecules.
Fluorescence Imaging and Electrophysiology.
The protocol and imaging system for standard Ca2+ measurements have been described elsewhere (26). The standard imaging solution contained 150 mM NaCl, 10 mM Hepes, 2 mM CaCl2, and 1 mM MgCl2 (pH 7.4 with NaOH). For measurements on sensory neurons, cells were considered responders if the fluorescence ratio increased by 0.05 above the basal value on compound application. Nonresponsive cells that did not respond to application of 50 mM KCl were excluded from the analysis.
For [Ca2+]i measurements in isolated pancreatic islets, islets were loaded with 1 μM Fura-2 for 1 h at 37 °C in culture medium. [Ca2+]i from the Fura-2–loaded islets was measured by monitoring the fluorescence ratio (F350/F380) every 2 s (after correction for background fluorescence) using a monochromator-based system consisting of a Polychrome IV monochromator (FEI Munich) with an additional TILL Photonics photomultiplier, both controlled by Pulse software (HEKA Elektronik). Standard extracellular solution for Ca2+ imaging measurements contained 120 mM NaCl, 4.8 mM KCl, 2.5 mM CaCl2, 1.2 mM MgCl2, and 10 mM Hepes (pH 7.4 with NaOH), with various concentrations of glucose added as indicated.
Whole-cell membrane currents and inside-out currents were measured with an EPC-10 patch-clamp amplifier and PatchMasterPro Software (HEKA Elektronik). Current measurements were performed at a sampling rate of 20 kHz, and currents were digitally filtered at 2.9 kHz. For whole-cell recordings on HEK293 cells, the standard extracellular solution contained 150 mM NaCl, 1 mM MgCl2, and 10 mM Hepes (pH 7.4 with NaOH), and the standard internal solution contained 100 mM CsAsp, 45 mM CsCl, 10 mM EGTA, 10 mM Hepes, and 1 mM MgCl2 (pH 7.2 with CsOH). In experiments allowing Ca2+-dependent desensitization of TRPM3, Mg2+ was replaced by Ca2+ in the extracellular solution, and Cs+ was replaced by Na+ in the pipette solution. Whole-cell current recordings on primary cells were performed using similar solutions, supplemented with 10 mM glucose in the external solution for pancreatic islets and 10 mM glucose and 100 nM TTX in the external solution and 5 mM TEA in the internal solution for sensory neurons. The standard patch pipette resistance was between 2.5 MΩ and 5 MΩ when filled with pipette solution. Between 50% and 70% of the series resistance was compensated.
Behavior.
PS (250 µM), CIM0216 (50 µM), and capsaicin (10 µM) were dissolved in PBS + 0.1% DMSO. An intraplantar injection of 20 μL was performed using a 30G needle coupled to a Hamilton syringe, and behavior was recorded. Experiments were performed during the light cycle. Nocifensive behavior was quantified during the first 2 min after injection (3). Execution and analysis of behavioral experiments were done blinded, that is, without knowledge of the mouse genotype (3). All animal experiments were carried out in accordance with the European Union Community Council guidelines and approved by the local Ethics Committee.
CGRP Release.
Skin flaps of both mouse hairy hind paws were excised from the lower leg and foot and wrapped around acrylic rods, with the corium side exposed as described previously (30). Samples were placed in carbogen-gassed synthetic interstitial fluid (SIF) inside a shaking bath set to 32 °C for a washout period of 30 min. Skin flaps were then passed consecutively through a set of four glass tubes containing 700 μL of SIF, with each incubation step lasting 5 min. The first two incubation steps were performed to determine basal CGRP release and variability at 32 °C. The third incubation step assessed heat (47°, 50°, or 52 °C) or chemically induced CGRP release. In the case of chemical treatment, one hind paw skin was stimulated with the TRPM3 agonist CIM0216 (50 µM in SIF) and the other was exposed to CIM0216 together with the TRPM3 blocker isosakuranetin (5 µM). The fourth incubation step assessed recovery from the response in SIF at 32 °C. The CGRP content of the incubation fluid was measured using commercial enzyme immunoassay kits with a detection threshold of 5 pg/mL (Bertin Pharma). The EIA plates were photometrically analyzed using a microplate reader (Dynatech). All results are presented as measured by the EIA in pg CGRP/mL SIF. To reduce interindividual variability and day-to-day baseline variability, the data were normalized to the second individual baseline value (before stimulation). This value was subtracted from all four data points of a typical experiment, so that only the absolute change in CGRP release (∆ pg/mL) is displayed in the figures.
Insulin Release.
The isolated islets were used immediately after isolation. Insulin release was measured as described previously (28) using a commercially available ELISA kit (Crystal Chem).
Data Analysis.
Electrophysiological data were analyzed using WinASCD software (Guy Droogmans, Catholic University of Leuven) and Origin 8.6 (OriginLab). Origin 8.6 was also used for statistical analysis and data display. Pooled data of continuous parameters are expressed as mean ± SEM from n biological replicates. The Student paired or unpaired two-tailed t test was used for statistical comparisons of two datasets. For CGRP measurements, statistical comparisons were performed using Statistica 7 software (Statsoft). All-time series of experimental values were first analyzed for the effect of stimulation (heat, chemical) compared with baseline, using the nonparametric Wilcoxon matched-pairs test. The baseline-normalized (i.e., ∆) CGRP values were entered into a one-way ANOVA, followed by Fisher’s least significant difference test, focusing on the peak values of stimulated CGRP release. P < 0.05 was considered statistically significant. P < 0.01 is indicated by double asterisks; P < 0.001, by triple asterisks. Data points represent mean ± SEM of the given number (n) of identical experiments. Unless indicated otherwise, experiments were performed at room temperature (23 ± 1 °C).
Supplementary Material
Acknowledgments
We thank all members of the Laboratory of Experimental Gynecology and Obstetrics and of the Laboratory of Ion Channel Research for helpful discussions. We are very grateful to B. Colsoul and R. Vennekens for introducing the isolation technique for pancreatic islets. This work was supported by grants from the Belgian Federal Government (IUAP P7/13, to T.V.), the Research Foundation Flanders (G.0565.07 and G.0825.11, to T.V. and J.V.), and the Research Council of Catholic University of Leuven (IOF- HB/12/023, to J.V. and T.V., and PF-TRPLe, to T.V.) and by the Planckaert-De Waele fund (to J.V.). K.H. and K.D.C. are funded by the Fonds Wetenschappelijk Onderzoek Belgium.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1419845112/-/DCSupplemental.
References
- 1.Voets T, Talavera K, Owsianik G, Nilius B. Sensing with TRP channels. Nat Chem Biol. 2005;1(2):85–92. doi: 10.1038/nchembio0705-85. [DOI] [PubMed] [Google Scholar]
- 2.Montell C, et al. A unified nomenclature for the superfamily of TRP cation channels. Mol Cell. 2002;9(2):229–231. doi: 10.1016/s1097-2765(02)00448-3. [DOI] [PubMed] [Google Scholar]
- 3.Vriens J, et al. TRPM3 is a nociceptor channel involved in the detection of noxious heat. Neuron. 2011;70(3):482–494. doi: 10.1016/j.neuron.2011.02.051. [DOI] [PubMed] [Google Scholar]
- 4.Wagner TF, et al. Transient receptor potential M3 channels are ionotropic steroid receptors in pancreatic beta cells. Nat Cell Biol. 2008;10(12):1421–1430. doi: 10.1038/ncb1801. [DOI] [PubMed] [Google Scholar]
- 5.Vriens J, et al. Opening of an alternative ion permeation pathway in a nociceptor TRP channel. Nat Chem Biol. 2014;10(3):188–195. doi: 10.1038/nchembio.1428. [DOI] [PubMed] [Google Scholar]
- 6.Vriens J, Nilius B, Voets T. Peripheral thermosensation in mammals. Nat Rev Neurosci. 2014;15(9):573–589. doi: 10.1038/nrn3784. [DOI] [PubMed] [Google Scholar]
- 7.Bautista DM, et al. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell. 2006;124(6):1269–1282. doi: 10.1016/j.cell.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 8.Bautista DM, et al. Pungent products from garlic activate the sensory ion channel TRPA1. Proc Natl Acad Sci USA. 2005;102(34):12248–12252. doi: 10.1073/pnas.0505356102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oberwinkler J, Philipp SE. Trpm3. Handbook Exp Pharmacol. 2014;222:427–459. doi: 10.1007/978-3-642-54215-2_17. [DOI] [PubMed] [Google Scholar]
- 10.Straub I, et al. Flavanones that selectively inhibit TRPM3 attenuate thermal nociception in vivo. Mol Pharmacol. 2013;84(5):736–750. doi: 10.1124/mol.113.086843. [DOI] [PubMed] [Google Scholar]
- 11.Oancea E, et al. TRPM1 forms ion channels associated with melanin content in melanocytes. Sci Signal. 2009;2(70):ra21. doi: 10.1126/scisignal.2000146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Armstrong CM, Cota G. Modification of sodium channel gating by lanthanum: Some effects that cannot be explained by surface charge theory. J Gen Physiol. 1990;96(6):1129–1140. doi: 10.1085/jgp.96.6.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sokolov S, Scheuer T, Catterall WA. Ion permeation and block of the gating pore in the voltage sensor of NaV1.4 channels with hypokalemic periodic paralysis mutations. J Gen Physiol. 2010;136(2):225–236. doi: 10.1085/jgp.201010414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bandell M, et al. Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron. 2004;41(6):849–857. doi: 10.1016/s0896-6273(04)00150-3. [DOI] [PubMed] [Google Scholar]
- 15.Caterina MJ, et al. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature. 1997;389(6653):816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- 16.Eid SR, et al. HC-030031, a TRPA1 selective antagonist, attenuates inflammatory- and neuropathy-induced mechanical hypersensitivity. Mol Pain. 2008;4:48. doi: 10.1186/1744-8069-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Earley S. TRPA1 channels in the vasculature. Br J Pharmacol. 2012;167(1):13–22. doi: 10.1111/j.1476-5381.2012.02018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caterina MJ, et al. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288(5464):306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- 19.Gillis KD, Misler S. Single cell assay of exocytosis from pancreatic islet B cells. Pflugers Arch. 1992;420(1):121–123. doi: 10.1007/BF00378654. [DOI] [PubMed] [Google Scholar]
- 20.Drews A, et al. Structural requirements of steroidal agonists of transient receptor potential melastatin 3 (TRPM3) cation channels. Br J Pharmacol. 2014;171(4):1019–1032. doi: 10.1111/bph.12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kunkler PE, Ballard CJ, Oxford GS, Hurley JH. TRPA1 receptors mediate environmental irritant-induced meningeal vasodilatation. Pain. 2011;152(1):38–44. doi: 10.1016/j.pain.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nassini R, et al. The “headache tree” via umbellulone and TRPA1 activates the trigeminovascular system. Brain. 2012;135(Pt 2):376–390. doi: 10.1093/brain/awr272. [DOI] [PubMed] [Google Scholar]
- 23.Peier AM, et al. A TRP channel that senses cold stimuli and menthol. Cell. 2002;108(5):705–715. doi: 10.1016/s0092-8674(02)00652-9. [DOI] [PubMed] [Google Scholar]
- 24.Inoue R, et al. Transient receptor potential channels in cardiovascular function and disease. Circ Res. 2006;99(2):119–131. doi: 10.1161/01.RES.0000233356.10630.8a. [DOI] [PubMed] [Google Scholar]
- 25.Naylor J, et al. Pregnenolone sulphate- and cholesterol-regulated TRPM3 channels coupled to vascular smooth muscle secretion and contraction. Circ Res. 2010;106(9):1507–1515. doi: 10.1161/CIRCRESAHA.110.219329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alpizar YA, et al. Allyl isothiocyanate sensitizes TRPV1 to heat stimulation. Pflugers Arch. 2014;466(3):507–515. doi: 10.1007/s00424-013-1334-9. [DOI] [PubMed] [Google Scholar]
- 27.Vandewauw I, Owsianik G, Voets T. Systematic and quantitative mRNA expression analysis of TRP channel genes at the single trigeminal and dorsal root ganglion level in mouse. BMC Neurosci. 2013;14:21. doi: 10.1186/1471-2202-14-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colsoul B, et al. Loss of high-frequency glucose-induced Ca2+ oscillations in pancreatic islets correlates with impaired glucose tolerance in Trpm5−/− mice. Proc Natl Acad Sci USA. 2010;107(11):5208–5213. doi: 10.1073/pnas.0913107107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kwan KY, et al. TRPA1 contributes to cold, mechanical, and chemical nociception but is not essential for hair-cell transduction. Neuron. 2006;50(2):277–289. doi: 10.1016/j.neuron.2006.03.042. [DOI] [PubMed] [Google Scholar]
- 30.Kichko TI, Reeh PW. Why cooling is beneficial: Non-linear temperature-dependency of stimulated iCGRP release from isolated rat skin. Pain. 2004;110(1-2):215–219. doi: 10.1016/j.pain.2004.03.033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.