

Significance
Small-cell lung cancer (SCLC) is an aggressive carcinoma with few effective treatment options beyond first-line chemotherapy. BH3 mimetics, such as ABT-263, promote apoptosis in SCLC cell lines, but early phase clinical trials demonstrated no significant clinical benefit. Here, we examine the sensitivity of a large panel of cancer cell lines, including SCLC, to ABT-263 and find that high Bcl2-interacting mediator of cell death (BIM) and low myeloid cell leukemia 1 (MCL-1) expression together predict sensitivity. SCLC cells relatively resistant to ABT-263 are sensitized by TORC1/2 inhibition via MCL-1 reduction. Combination of ABT-263 and TORC1/2 inhibition stabilizes or shrinks tumors in xenograft models, in autochthonous SCLC tumors in a genetically engineered mouse model, and in a patient-derived xenograft SCLC model. Collectively, these data support a compelling new therapeutic strategy for treating SCLC.
Keywords: small-cell lung cancer, targeted therapies, BH3 mimetics, apoptosis, BIM
Abstract
BH3 mimetics such as ABT-263 induce apoptosis in a subset of cancer models. However, these drugs have shown limited clinical efficacy as single agents in small-cell lung cancer (SCLC) and other solid tumor malignancies, and rational combination strategies remain underexplored. To develop a novel therapeutic approach, we examined the efficacy of ABT-263 across >500 cancer cell lines, including 311 for which we had matched expression data for select genes. We found that high expression of the proapoptotic gene Bcl2-interacting mediator of cell death (BIM) predicts sensitivity to ABT-263. In particular, SCLC cell lines possessed greater BIM transcript levels than most other solid tumors and are among the most sensitive to ABT-263. However, a subset of relatively resistant SCLC cell lines has concomitant high expression of the antiapoptotic myeloid cell leukemia 1 (MCL-1). Whereas ABT-263 released BIM from complexes with BCL-2 and BCL-XL, high expression of MCL-1 sequestered BIM released from BCL-2 and BCL-XL, thereby abrogating apoptosis. We found that SCLCs were sensitized to ABT-263 via TORC1/2 inhibition, which led to reduced MCL-1 protein levels, thereby facilitating BIM-mediated apoptosis. AZD8055 and ABT-263 together induced marked apoptosis in vitro, as well as tumor regressions in multiple SCLC xenograft models. In a Tp53; Rb1 deletion genetically engineered mouse model of SCLC, the combination of ABT-263 and AZD8055 significantly repressed tumor growth and induced tumor regressions compared with either drug alone. Furthermore, in a SCLC patient-derived xenograft model that was resistant to ABT-263 alone, the addition of AZD8055 induced potent tumor regression. Therefore, addition of a TORC1/2 inhibitor offers a therapeutic strategy to markedly improve ABT-263 activity in SCLC.
Effective cancer-targeted therapies often trigger cell death, commonly via apoptosis, to induce remissions (1–3). For example, we and others previously observed that, among lung cancers with activating EGFR mutations, those with higher expression levels of Bcl2-interacting mediator of cell death (BIM), a key regulator of apoptosis, have a higher response rate and longer progression-free survivals upon treatment with EGFR inhibitors (2, 4). BH3 mimetics are a class of drugs designed to promote apoptosis. These compounds bind to and inhibit antiapoptotic BCL-2 family members, the molecular sentinels of apoptosis. ABT-263 (5) is a BH3 mimetic that directly binds BCL-2 and BCL-XL, which blocks their binding to BIM and thereby enables BIM-mediated induction of apoptosis (6–8). However, ABT-263 does not bind the prosurvival BCL-2 family member myeloid cell leukemia 1 (MCL-1), and high levels of MCL-1 are associated with resistance to BH3 mimetics such as ABT-263 in both the laboratory and the clinic (9–17).
Small-cell lung cancer (SCLC) is a high-grade neuroendocrine carcinoma that accounts for 10–15% of all lung cancers and is commonly associated with a significant tobacco history. Patient outcomes have not improved substantially over the past 30 y, underscoring the need for more effective treatment strategies (18). Recent studies have demonstrated the antitumor activity of BH3 mimetics in laboratory models of SCLC (5, 19–21). These findings spurred clinical trials of the BH3 mimetic ABT-263 (Navitoclax) in SCLC (22). ABT-263 was well tolerated in the clinic with a dose-limiting toxicity of thrombocytopenia (22), an on-target toxicity of BCL-XL inhibition (23). Unfortunately, phase II trials of ABT-263 monotherapy revealed unimpressive activity. Sixteen of 26 evaluable patients had progression of disease, 9 had stable disease, and one had a partial response (24). Consequentially, there has been no further clinical development of ABT-263 as a monotherapy in SCLC.
Despite these findings, several questions remain: which solid tumor malignancies are most sensitive to single-agent ABT-263, and why is SCLC among the most sensitive? In addition, why does SCLC fail to respond to single-agent ABT-263 in the clinic, and how could efficacy be enhanced? Here, we assess the sensitivity of a large panel of cell lines spanning multiple cancer types to ABT-263. We observe that cell lines with high expression of BIM are highly sensitive to ABT-263 and that SCLC expresses higher levels of BIM than other solid tumor malignancies. Although SCLC is among the most sensitive to single-agent ABT-263, efficacy is substantially limited by MCL-1, which also is elevated in SCLC. We find that SCLC can be sensitized to ABT-263 via TORC1/2 inhibition, which leads to reduction of MCL-1 protein levels, thereby permitting BIM-mediated apoptosis. In two SCLC mouse xenograft models, either drug alone has little activity. However, the combination ABT-263 and AZD8055 induces tumor stabilization or regression. Furthermore, we examined the efficacy of this combination in autochthonous lung tumors arising in a genetically engineered mouse model (GEMM) of SCLC, where the combination of ABT-263 and AZD8055 also induces tumor stabilization or regression. By contrast, most tumors progress when treated with either drug alone. Finally, in a patient-derived xenograft model of SCLC in which ABT-263 alone is ineffective, the combination of ABT-263 and AZD8055 causes tumor regression. These studies demonstrate that the combination of ABT-263 and AZD8055 potently suppresses tumor progression across a variety of preclinical SCLC experimental models.
Results
BIM/MCL-1 Ratio Predicts Sensitivity to ABT-263.
Our initial studies stemmed from the observation that 5 of 11 SCLC human xenografts tested by Shoemaker and colleagues (19) did not respond to ABT-263 and that the majority of patients treated in a phase II study had progression of disease (24). To more broadly identify mediators of response to ABT-263, we examined data collected from a high-throughput drug screen (13) assessing over 500 human cancer cell lines for sensitivity to ABT-263. Because the mechanism underlying ABT-263 activity putatively relies on releasing BIM to promote apoptosis (6), we hypothesized that BIM levels may predict responsiveness to ABT-263. By matching cell line sensitivity to two independent gene expression data sets, we found a modest, but significant, correlation between BIM expression and sensitivity to ABT-263 (Fig. 1A and SI Appendix, Fig. S1A). As we and others have reported (10, 13–17), high MCL-1 expression correlated with resistance to ABT-263 and the related ABT-737, and we also observed MCL-1–based resistance to ABT-263 across both data sets (SI Appendix, Fig. S1 B and C). However, the ratio of BIM to MCL-1 predicted sensitivity to ABT-263 more effectively than the expression of either biomarker alone (Fig. 1B and SI Appendix, Fig. S1D). Moreover, among cell lines, those with the highest BIM levels that also expressed high levels of MCL-1 were not sensitive to ABT-263 nor were those cancers with low expression of both BIM and MCL-1, underscoring the added value of measuring the ratio of BIM to MCL-1 in predicting sensitivity (Fig. 1C and SI Appendix, Fig. S1E). It is notable that the ratio of either BCL-2 or BCL-XL to MCL-1 was inferior to the ratio of BIM to MCL-1 at predicting response to ABT-263 across solid tumor cancers (SI Appendix, Fig. S2). These findings underlie the potential capacity for MCL-1 to mitigate the therapeutic benefit of ABT-263 in cancers with high BIM levels.
Fig. 1.

BIM/MCL-1 expression ratios predict response to ABT-263. (A) Scatter plot of ABT-263 IC50 (μM) values on natural log scale versus relative BIM RNA expression levels measured from the Cancer Cell Line Encyclopedia (CCLE) (45) (n = 331). A linear regression analysis was used to assign a coefficient of determination (R2) of 0.039 and a P value of 0.00043. (B) Scatter plot of ABT-263 IC50 (μM) values on the natural log scale versus the ratio of BIM/MCL-1 expression levels. A linear regression analysis was used to assign a coefficient of determination (R2) of 0.146 and a P value of 2.83e-12. Expression values were taken from the CCLE (45). (C) Scatter plots comparing ABT-263 IC50 (μM) values on the natural log scale between cell lines with high BIM expression (top 10%) and low MCL-1 expression (bottom 75%), cell lines with high BIM expression (top 10%) and high MCL-1 expression (top 25%), and cell lines with low BIM expression and low MCL-1 expression. Expression values were taken from the CCLE (45). An unpaired t test with Welch’s correction was used to assign a P value for differential IC50’s between the BIM high/MCL-1 low group and the two other groups; P = 0.0027 and P = 0.0014, respectively. Red bars are the geometric means of the IC50’s for the groups of cell lines. (D) Box plot showing increased expression of BIM in SCLC cell lines compared with other solid tumors. A Wilcoxon rank-sum test was used to assign a P value for differential expression between the two groups; P value of 7.94e-06. Expression values were taken from the CCLE (45). (E) Cancer cell lines were grouped by cancer type, represented by a numeric value, and correlated to ABT-263 IC50 (μM) values on natural log scale. The key for the different cancer types is in SI Appendix, Fig. S4. A box is drawn around SCLC (#21) for emphasis. Sensitive cell lines were set at IC50’s <1 μM and insensitive cell lines >1 μM. Based on these criteria, a P value of 0.0000033 was assigned from a two-tailed Fisher’s exact test comparing SCLC to other solid tumor types. Of note, there were 10 sensitive SCLC cell lines and 11 insensitive SCLC cell lines (48% sensitive). In contrast, among all other solid tumor types, there were 37 sensitive and 443 resistant cancers (8% sensitive). Note that representative cancer types are shown and not every cancer type analyzed is included.
We found that SCLC lines have increased BIM expression compared with other solid tumor types (Fig. 1D and SI Appendix, Fig. S3) along with enhanced sensitivity to ABT-263 compared with other solid tumor types across a large panel of cancer cell lines (Fig. 1E and SI Appendix, Fig. S4). To determine whether ABT-263 sensitivity in SCLC was mediated by high BIM expression, we knocked down BIM using siRNA. Knockdown with two different siRNAs designed against BIM in SCLC cell lines consistently suppressed ABT-263–induced apoptosis by >50% (Fig. 2 A and B and SI Appendix, Fig. S5 A and B). Next, we engineered H1048 SCLC cells to have either modest (“low”) or marked (“high”) overexpression of MCL-1 (Fig. 2C and SI Appendix, Fig. S5C). Increasing expression of MCL-1 protected cells from ABT-263–induced apoptosis (Fig. 2D). These results support the model that high BIM and low MCL-1 promote sensitivity to ABT-263, whereas high MCL-1 mediates ABT-263 resistance.
Fig. 2.

BIM and MCL-1 mediate ABT-263–induced apoptosis in SCLC, and the ratio of BIM to MCL-1 expression predicts the magnitude of apoptosis in SCLC cell lines. (A) SCLC SW1271 and H1048 cells were treated with either 10 nM scrambled (sc) or BIM siRNA (2), and, the next day, cells were treated with or without ABT-263. Then, cells were (A) prepared and stained with propidium iodide and Annexin-V. Apoptosis was measured by FACS analysis of the percentage of cells positive for Annexin-V 72 h after treatment (the amount of apoptotic cells caused by ABT-263 treatment minus no drug treatment) or (B) lysed and separated by SDS/PAGE, subjected to Western blot, and probed with the indicated antibodies. In A, error bars are SD (n = 3); in B, the asterisk indicates a nonspecific band. (C) H1048 cells transduced with lentiviral particles containing plasmids that express GFP alone (control) or GFP-IRES-MCL-1. MCL-1–overexpressing cells were sorted to isolate cells expressing low and high amounts of MCL-1 based on GFP fluorescence intensity. Protein lysates were prepared and probed with the indicated antibodies. (D) The cells were treated with either no drug (control) or ABT-263 for 48 h. Cells were prepared and stained with propidium iodide and Annexin-V. Apoptosis was measured by FACS analysis of the percentage of cells with Annexin-V positivity. Bars represent mean percentage of apoptotic cells, ABT-263 treatment minus control. Error bars are SD (n = 3). (E) BIM antibody (or IgG control) was added to lysates following treatment with or without ABT-263 for 6 h derived from H1048 cells expressing GFP, low MCL-1, or high MCL-1 (as in C), and BIM-containing complexes were immunoprecipitated and separated by SDS/PAGE, subjected to Western blot, and probed with the indicated antibodies. (F) BIM RNA levels or (G) MCL-1 RNA levels in a panel of 10 human SCLC cell lines were determined by quantitative PCR, and the average of three replicates was plotted versus the amount of apoptosis (n = 3) induced by 1 µM of ABT-263 over 72 h. Each dot represents a unique human SCLC line labeled in red font, with RNA levels relative to GAPDH abundance. A linear regression analysis was used to assign, for F, a coefficient of determination (R2) of 0.142 and a P value of 0.284 (P = not significant), and for G, a coefficient of determination (R2) of 0.135 and a P value of 0.296 (P = not significant). (H) BIM/MCL-1 RNA levels were plotted versus apoptosis in the same lines. A linear regression analysis was used to assign a coefficient of determination (R2) of 0.456 and a P value of 0.032. (I) SCLC H1048 cells were treated with either 50 nM scrambled (sc) or MCL-1 siRNA for 24 h. Cells were reseeded and treated the following day with no drug (control) or ABT-263 and then prepared for FACS analysis of percentage of cells with Annexin-V positivity 24 h after treatment. Error bars are SD (n = 3). (Right) Cell lysates were prepared from the transfected cells and separated by SDS/PAGE, subjected to Western blot, and probed with the indicated antibodies.
To further assess the mechanism of ABT-263 response, we performed immunoprecipitation of BIM complexes from whole-cell lysates. Please note that the BIM immunoprecipitations successfully depleted >90% of the cellular BIM (SI Appendix, Fig. S5D). In control cells, ABT-263 treatment led to loss of BIM binding to BCL-XL and BCL-2, whereas it induced BIM binding to MCL-1 (Fig. 2E). This indicates that MCL-1 binds the BIM that is released from complexes with BCL-2 and BCL-XL upon treatment with ABT-263. This likely contributes to the mechanism whereby MCL-1 expression mitigates the apoptotic response induced by ABT-263. Incremental overexpression of MCL-1 resulted in more BIM/MCL-1 complexes (Fig. 2E) and fewer BIM/BCL-2 complexes, consistent with the abrogation of apoptosis induced by MCL-1 overexpression. Consistent with these results and similar to the analyses of the large cell line panel described above, BIM or MCL-1 expression alone was not significantly predictive of apoptosis following ABT-263 therapy in a subset of SCLC cell lines (Fig. 2 F and G). However, the ratio of BIM/MCL-1 correlated significantly with apoptotic response in SCLC cell lines (Fig. 2H). Thus, BIM and MCL-1 levels substantially impact sensitivity to ABT-263–mediated apoptosis in SCLC cell lines, with high BIM and low MCL-1 expression associated with enhanced sensitivity to ABT-263.
TORC1/2 Inhibition Suppresses MCL-1 and Sensitizes SCLC Cells to ABT-263.
Despite some of the preclinical promise of ABT-263 against SCLC (Fig. 1E) (5, 19), ABT-263 has demonstrated minimal clinical activity in SCLC as monotherapy (22, 24). BIM levels are higher in SCLC relative to other solid tumor cell lines (Fig. 1D and SI Appendix, Fig. S3), but MCL-1 levels are also relatively high in SCLC (SI Appendix, Fig. S5E), suggesting that high MCL-1 levels may be mitigating the efficacy of ABT-263 in SCLC. Consistent with this notion, knockdown of MCL-1 using two different siRNAs sensitized the H1048 SCLC cell line to ABT-263 (Fig. 2I and SI Appendix, Fig. S6A). We therefore sought to identify pharmacological strategies that could suppress MCL-1 levels and increase sensitivity to ABT-263. We did not pursue obatoclax, which does target MCL-1 in addition to other BCL-2 family members, because early clinical trials suggest that obatoclax causes toxicity independent of its effects on BCL-2 family proteins, potentially limiting its clinical utility (25–27). Rather, we considered TORC1/2 inhibitors because they suppress MCL-1 protein levels in some cancers (7, 28–31). We observed that the TORC1/2 catalytic inhibitor AZD8055 (32) potently suppressed protein levels of MCL-1 in SCLC cells (Fig. 3A) and markedly enhanced ABT-263–induced apoptosis in four different SCLC cell lines examined (Fig. 3B). BIM immunoprecipitations confirmed that AZD8055 abrogated the formation of MCL-1/BIM complexes normally induced by ABT-263 treatment (Fig. 3C and SI Appendix, Fig. S6B; compare the combination to ABT-263). Consistent with the proposed mechanism of apoptosis induced by this combination, overexpression of MCL-1 blocked apoptosis induced by the combination (Fig. 3D and SI Appendix, Fig. S6 C and D). Importantly, AZD8055 and ABT-263 interfered with cell cycle progression in all SCLC cell lines (SI Appendix, Fig. S7). Thus, the combination of ABT-263 and AZD8055 both increased apoptosis and induced growth arrest of SCLCs, suggesting that this combination could be of superior efficacy compared with treatment uniquely targeting BCL-2 family members. Interestingly, we found that treatment of SCLC cells with rapamycin, an allosteric mTOR inhibitor, diminished expression of pS6, a downstream target of TORC1, but, unlike AZD8055 therapy, failed to suppress p4E-BP1 signaling, lower MCL-1 levels, or sensitize to ABT-263 as well as AZD8055 (SI Appendix, Fig. S8 A and B). These data are consistent with other studies demonstrating that TORC1/2 catalytic inhibitors more effectively suppress 4E-BP1 phosphorylation and cap-dependent translation than rapamycin or rapalogs (31, 33). In fact, resistance to single-agent AZD8055 has been reported to emerge through reactivation of cap-dependent translation, including that of MCL-1, in the presence of the drug (34). Of note, although others found that BCL2-associated X protein (BAX) plays a role in rapamycin sensitization of ABT-737 (35), we did not observe an increase in BAX expression in the ABT-263/AZD8055 combination treatment compared with ABT-263 treatment (SI Appendix, Fig. S8C) despite the induction of substantial apoptosis (∼20%) at the same time point (SI Appendix, Fig. S8D).
Fig. 3.

Combination treatment with ABT-263 and the TORC1/2 inhibitor AZD8055 leads to robust apoptosis and antitumor activity in vivo. (A and B) The indicated SCLC cell lines were treated with no drug (–), 500 nM AZD8055, 1 μM ABT-263, or a combination of 500 nM AZD8055 and 1 μM ABT-263. (A) After 16 h of treatment, lysates were probed with the indicated antibodies. (B) After 72 h of treatment, cells were prepared and stained with propidium iodide and Annexin-V, and apoptosis was measured by FACS analysis of the percentage of cells with Annexin-V positivity. Bars represent mean the amount of apoptosis induced by each treatment minus control. Error bars are SD (n = 3). **P < 0.05 by Student's t test, ABT-263 versus combination treatment group and AZD8055 versus combination group. (C) H1048 cells were treated with no drug (−), 1 μM ABT-263, 500 nM AZD8055, or 500 nM AZD8055/1 μM ABT-263 for 2 h, and lysates were immunoprecipitated with BIM antibody or IgG negative control. Precipitates were analyzed by Western blot analyses with the indicated antibodies. (D) H1048 cells transduced with lentiviral particles expressing GFP alone (GFP) or GFP-IRES-MCL-1 (the “high”-expressing MCL-1 cells from Fig. 2C) were treated with either no drug (–) or 500 nM AZD8055/1 μM ABT-263. Following 72 h of treatment, cells were prepared and stained with propidium iodide and Annexin-V, and apoptosis was measured by FACS analysis of the percentage of cells with Annexin-V positivity. Bars represent mean percentage of apoptotic cells following combination treatment over control. Error bars are SD (n = 3). (E) Human SCLC H1048 and H82 cells were grown as xenograft tumors in Nu/Nu mice, and, when tumors were ∼100–200 mm3, mice were randomized into treatment cohorts: control (no drug), 16 mg/kg/qd AZD8055, 80 mg/kg/qd ABT-263, or the combination of AZD8055 and ABT-263. AZD8055 was given ∼1.5 h before ABT-263 for combination treatments. Tumor measurements were performed approximately three times per week by calipers, and the average tumor volume + SEM for each cohort is displayed. (F) Tumors were harvested from H1048 tumor-bearing mice approximately 3 h after drug administration at the end of treatment and tumor lysates were subjected to Western blot analyses and probed with the indicated antibodies. (G) Slides were prepared from formalin-fixed tissue of H1048-tumor bearing mice approximately 3 h after drug administration and stained with CC3 to quantify apoptosis. Quantification of CC3 is shown in SI Appendix, Fig. S10A.
Previous studies have suggested that mTOR is necessary for efficient cap-dependent translation of MCL-1, and thus mTOR inhibitors may decrease MCL-1 expression by decreasing translation. To determine if AZD8055 affected the half-life of MCL-1 protein, we treated H1048 cells with the cytoplasmic protein synthesis inhibitor, cycloheximide (CHX), alone or in combination with AZD8055. We found the rate of MCL-1 protein degradation was comparable over time in the CHX-treated cells versus the CHX+AZD8055 combination-treated cells, suggesting a similar mode of MCL-1 inhibition between CHX and AZD8055 (SI Appendix, Fig. S9A). Treatment at 6 h with AZD8055 alone left a nearly equivalent amount of cellular MCL-1 as CHX alone or the combination of CHX and AZD8055 (SI Appendix, Fig. S9 A and B). These data are consistent with translational inhibition being the major mechanism underlying suppression of MCL-1 protein levels following mTORC inhibition.
Combination of ABT-263 and AZD8055 Causes Tumor Regression in Xenograft Models.
We next assessed the efficacy of AZD8055 and ABT-263 in vivo using H1048 and H82 SCLC cell line xenograft tumor models. ABT-263 has been shown to be only modestly effective in the H1048 model and minimally effective in the H82 model (19) and therefore may be more representative of SCLC clinical outcomes with single-agent BH3 mimetics such as ABT-263 (22, 24). Consistent with previous data (19), ABT-263 (80 mg/kg/qd) was partially effective in established H1048 xenografts, whereas single-agent AZD8055 (16 mg/kg/qd) modestly slowed tumor growth (Fig. 3E). However, the combination induced marked tumor regressions to nearly undetectable sizes (Fig. 3E). In the faster-growing H82 xenografts, ABT-263 was ineffective as previously reported (19). Single-agent AZD8055 had a modest effect on tumor growth (Fig. 3E). However, the combination almost completely blocked growth of these tumors (Fig. 3E). Pharmacodynamic studies confirmed that AZD8055 suppressed phosphorylation of TORC1 targets S6 and 4E-BP1 and decreased MCL-1 protein levels in vivo (Fig. 3F). Moreover, cleaved caspase 3, a marker of apoptosis, was significantly increased following combination treatment (Fig. 3G and SI Appendix, Fig. S10A), consistent with the in vitro studies. No overt toxicities were observed in the tumor bearing mice treated with the combination (SI Appendix, Fig. S10B).
Combination of ABT-263 and AZD8055 Is Superior to Either Drug Alone in a SCLC Genetically Engineered Mouse Model.
Although cell line xenograft models of SCLC are valuable tools for assessing drug activity in vivo, they may not fully model the tissue environment and heterogeneity of autochthonous tumors (those growing in their native tissue context). To rigorously assess the activity of the combination of ABT-263 and AZD8055 in vivo, we used a GEMM of SCLC (36). Tumors are initiated in this model via conditional inactivation of both alleles of Tp53 and Rb1 in pulmonary neuroendocrine cells, resulting in the development of tumors that model the histology, metastatic spread, and acquired genetic alterations observed in human SCLC (37–39). Tumor-bearing SCLC GEMMs were randomized to receive no treatment, AZD8055 alone (16mg/kg/qd), ABT-263 alone (80 mg/kg/qd), or both ABT-263 and AZD8055 and were treated for 21 d (Fig. 4A). Magnetic resonance imaging (MRI) of the thorax was performed 1 day before starting treatment and on day 21 of treatment, and lung tumor volumes pre- and posttreatment were quantified. Most animals had exactly one measurable lung tumor. If more than one tumor was observed in separate lobes of the lungs, the larger tumor was measured. Only tumors that were histologically confirmed to be high-grade neuroendocrine tumors or had clear evidence of distant metastatic spread on MRI were included in the analysis. Tumors progressed in all untreated animals (n = 7), although we observed significant variability in the rate of progression over the 21-d period (Fig. 4 B–D and SI Appendix, Table S1). All tumors treated with AZD8055 alone progressed (n = 5) (Fig. 4 B, E, and F and SI Appendix, Table S1). Of six tumors treated with ABT-263, five progressed and one regressed (Fig. 4 B, G, and H and SI Appendix, Table S1). By contrast, in six animals treated with the combination of ABT-263 and AZD8055, three tumors regressed and three others showed relatively limited tumor progression (Fig. 4 B, I, and J and SI Appendix, Table S1). The responses of tumors to the combination of ABT-263 and AZD8055 were significantly superior to responses either to the drug alone or to no treatment. Consistent with these findings and those in the human xenograft models (Fig. 3 E–G), CC3 staining in allografted SCLC GEMM tumors was markedly apparent in the combination treatment at the 3-d time point (Fig. 4 K–N), again indicating a strong apoptotic response following administration of this regimen. Ex vivo cell lines derived from SCLC GEMMs treated with AZD8055 had reduction of MCL-1 protein levels and were sensitized to ABT-263 (SI Appendix, Fig. S11 A and B), consistent with the mechanism delineated in human cell lines and the activity observed in vivo.
Fig. 4.

Combination ABT-263 and AZD8055 induce tumor regressions in a GEMM of SCLC. (A) Schematic of GEMM experiment. Gray space in mouse drawing indicates lung; red circle indicates lung tumor. (B) SCLC GEMMs harboring radiographically measurable lung tumors were randomized to receive treatment for 21 d with 80 mg/kg/day ABT-263, 16 mg/kg/day AZD8055, combination ABT-263/AZD8055, or no treatment. Chest and upper abdomen were imaged by MRI on the day before starting treatment and on day 21 of treatment. For each animal, the largest single lung tumor that was histologically confirmed to be a high-grade neuroendocrine tumor was measured radiographically, and volume was computed. The percentage of tumor volume change for each animal is plotted as a single circle, with black bars indicating mean and SEM. Also included for comparison is the percentage of tumor volume change by MRI when equivalent GEMMs were treated with cisplatin (7 mg/kg IP on days 1 and 8) and etoposide (10 mg/kg on days 2 and 9). Notably, the follow-up imaging time point for the chemotherapy-treated animals was 28 d. Unpaired two-tailed t tests for statistical significance comparing tumor volume percentage change between pairs of datasets were performed. Untreated versus ABT-263/AZD8055: P = 0.0222. AZD8055 versus ABT-263/AZD8055: P = 0.00350. ABT-263 versus ABT-263/AZD8055: P = 0.0333. Cisplatin/etoposide versus ABT-263/AZD8055: 0.1589 (not significant). (C–J) Representative examples of MRI images (with tumors outlined in green) on day 1 and day 21 of a 21-d treatment period, with the treatment indicated. The asterisk in G and H indicates additional tumor in a separate lobe, which is not included in measurements. (Scale bar: 5.0 mm.) (K–N) A GEMM SCLC tumor was dissected from the lung and implanted s.c. into a NSG mouse. An established tumor in the NSG mouse was then divided and s.c. implanted directly into four nu/nu NSG mice. There was no in vitro intermediate. Once tumors were established, nu/nu mice were treated for 3 d with (K) vehicle for both drugs, (L) AZD8055 (16 mg/kg/qd), (M) ABT-263 (80 mg/kg/qd), (N) ABT-263 (80 mg/kg PO daily), and AZD8055 (16 mg/kg PO daily). Tumors were collected and fixed 3 h after the final treatment. Sections from tumor were stained to detect CC3, shown as brown stain. (Scale bar: 100 μm.)
SCLC tumors often respond to first-line chemotherapy in patients, although these responses are almost invariably transient. To provide context for the responsiveness to the AZD8055/ABT-263 combination in the SCLC GEMM model, we treated mice with a combination of cisplatin (7 mg/kg IP on days 1 and 8) and etoposide (10 mg/kg on days 2 and 9), the standard chemotherapies used to treat SCLC in patients. When we compared responses to the combination chemotherapy at day 28 to responses to the targeted therapy combination at day 21, the regimen of ABT-263 and AZD8055 improved responses, although the differences did not reach statistical significance (Fig. 4B). These data show an encouraging efficacy of the ABT-263/AZD8055 combination therapy in reference to standard of care chemotherapy in this model.
AZD8055 Sensitizes a SCLC Patient-Derived Xenograft Model to ABT-263.
To further gain insight into the translational potential of our findings, we next determined the efficacy of the combination in a third type of in vivo model, a patient-derived xenograft (PDX). This type of in vivo model may more faithfully recapitulate the high degree of genomic complexity of the human disease. Patient-derived xenografts derived from a histologically confirmed SCLC case were implanted into NOD scid gamma (NSG) mice, and mice were monitored for tumor growth (Fig. 5A and SI Appendix, Fig. S12). Growing tumors were subsequently treated with either single-agent ABT-263 (80 mg/kg/qd) (n = 4) or the combination of ABT-263 (80 mg/kg/qd) and AZD8055 (16 mg/kg/qd) (n = 4) (there were not enough tumor-bearing mice to treat with single-agent AZD8055). Consistent with a recent report highlighting overall modest activity of ABT-737 against PDXs of SCLC (35), the tumors in all mice treated with single-agent ABT-263 grew fairly rapidly despite drug treatment (Fig. 5A). In contrast, the addition of AZD8055 to ABT-263 led to consistent tumor regressions and showed no sign of regrowth when the experiment was ended ∼50 d after drug treatments began (Fig. 5A). Additionally, pharmacodynamic analyses of the tumors demonstrated marked decrease in MCL-1 expression in the combination cohort (Fig. 5B). These data indicate substantial activity of this combination in a third type of SCLC preclinical model and demonstrate that, in a SCLC PDX model, AZD8055 down-regulates MCL-1 to sensitize SCLC to ABT-263.
Fig. 5.

Addition of AZD8055 to ABT-263 sensitizes a PDX model of SCLC. (A) Pieces of a patient-derived SCLC tumor were implanted into NSG mice and monitored for subsequent growth. Upon growing to ∼150–300 mm3, tumors were treated with ABT-263 (80 mg/kg/qd) or ABT-263 (80 mg/kg/qd) plus AZD8055 (16 mg/kg/qd). AZD8055 was given ∼1.5 h before ABT-263 for combination treatments. Tumor measurements were performed approximately three times per week by calipers, and the average tumor volume + SEM for each cohort is displayed. Treatment start is indicated by an arrow. (B) Approximately 3 h after final drug administration, tumors were surgically excised and tumor lysates were prepared and subjected to Western blot analyses and probed with the indicated antibodies. ABT-263–treated and ABT-263 and AZD8055 combination–treated (ABT+AZD) tumors were compared to two untreated tumors (No Rx). Each number represents a tumor from a unique mouse.
Efficacy of Low-Dose ABT-263 Plus AZD8055 in Vitro and in Vivo.
Therapeutic dosing in humans is sometimes limited by drug toxicities that often preclude sufficient target inhibition in patients. The synergistic effect of the ABT-263/AZD8055 combination prompted us to test whether lower doses of either AZD8055 or ABT-263 could still efficiently suppress cell viability and demonstrate in vivo efficacy. In human SCLC cell lines, we found that even low-nanomolar doses of AZD8055 (5 or 16.66 nM) were sufficient to sensitize SCLCs to ABT-263 (100 nM) (Fig. 6A). This sensitization to low-dose AZD8055 was also observed in the ex vivo cell lines derived from SCLC GEMMs (SI Appendix, Fig. S11B). Similarly, a low dose of ABT-263 (3.3 nM) was sufficient to strongly suppress cell viability in combination with 50 nM AZD8055 (SI Appendix, Fig. S13). In the H211 cell line, which is sensitive to single-agent ABT-263 in vitro and in vivo (19), addition of AZD8055 further sensitized cells to ABT-263 (Fig. 6A). Furthermore, in the presence of ABT-263, p4E-BP1 was almost fully suppressed and MCL-1 was significantly down-regulated by low-nanomolar doses of AZD8055 (Fig. 6B). In the dose–response analyses, the amount of apoptosis induced by the combination was inversely related with the amount of MCL-1 protein remaining in response to increasing doses of AZD8055 (Fig. 6C). These in vitro findings prompted us to assess lower doses of AZD8055 in vivo. Strikingly, a low dose of 2 mg/kg of AZD8055 (Fig. 6D, compared with 16 mg/kg, Fig. 3E) in combination with ABT-263 (80 mg/kg) was sufficient to induce regressions in the H1048 SCLC xenograft model.
Fig. 6.
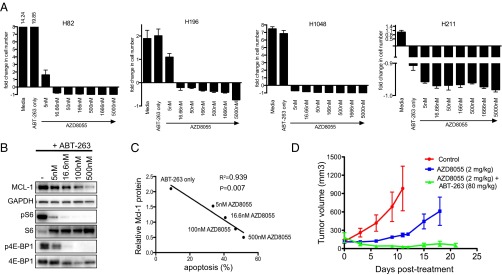
Low doses of AZD8055 sensitize SCLC to ABT-263. (A) SCLC cell lines were treated with no drug (Media), 100 nM ABT-263 (ABT-263 only), or 100 nM ABT-263 in combination with the indicated dose of AZD8055 (ranging from 5 to 5,000 nM AZD8055) for 7 d. The number of viable cells was determined by CellTiter-Glo and presented as fold-change of cells compared with day 0 (i.e., negative values represent loss of cells from day 0). Error bars are SD of cells treated in triplicate. (B) H1048 cells were treated with the indicated dose of AZD8055 with 100 nM ABT-263 or 100 nM ABT-263 alone (–) for 16 h, and equal amounts of lysates were subjected to Western blot analyses and probed with the indicated antibodies. (C) The amount of MCL-1 protein relative to GAPDH as determined by quantification on a SynGene GBox was plotted against the amount of apoptosis induced by the corresponding 72 h drug treatments (as determined by FACS analysis of the percentage of Annexin-V–positive cells, treatment minus no-treatment control). Note: AZD8055 treatments were in the presence of 100 nM ABT-263, as in (B). For the linear regression analysis, the R2 value was 0.939 and the P value was 0.007. (D) H1048 cells were grown as xenograft tumors in Nu/Nu mice. When tumors reached ∼100–200 mm3, mice were randomized into treatment cohorts: control (no drug), 2 mg/kg AZD8055, or the combination of AZD8055 (2 mg/kg) and ABT-263 (80 mg/kg). AZD8055 was given ∼1.5 h before ABT-263 for combination treatments. Tumor measurements were performed approximately three times per week by calipers, and the average tumor volume ± SEM for each cohort is displayed.
Discussion
Direct targeting of apoptotic regulators has emerged as an effective therapeutic approach in cancer. One such class of compounds includes BH3 mimetics, such as ABT-263, which block the binding of BCL-2 and BCL-XL to BIM and other proapoptotic proteins. In this study, we examined the efficacy of ABT-263 across a panel of cancer cell lines and observed that the ratio of BIM to MCL-1 predicted sensitivity (Fig. 1 and SI Appendix, Fig. S1). These data agree with a recent report by Roberts et al. showing that a high BIM to MCL-1 ratio indicated a favorable response to ABT-263 in nine patients with chronic lymphocytic leukemia (11).
There is significant interest in further developing BH3 mimetics to treat SCLC, and thus we specifically sought to improve the efficacy of ABT-263 in these cancers. We found that, although levels of BIM were relatively high in SCLC lines (Fig. 1D), MCL-1 levels were also relatively high, contributing to ABT-263 resistance (SI Appendix, Fig. S5E). Thus, high BIM levels may “prime” SCLC for apoptosis and in this way contribute to the sensitivity of SCLC to ABT-263. However, the lack of sensitivity of SCLC to ABT-263 in the clinic may be a result of high MCL-1 levels in these cancers. Therefore, we used these data as a rationale to develop a novel targeted therapy for SCLC based on the intrinsic high levels of BIM and the need to suppress MCL-1 (via TORC inhibition) to sensitize to ABT-263. The ABT-263/TORC1/2 inhibitor combination strategy was more effective than either agent alone, not only in increasing the induction of apoptosis, but also in suppressing proliferation. Inducing both apoptosis and growth arrest recapitulates the effects of other successful targeted therapy paradigms (2), including EGFR inhibitors for EGFR mutant lung cancers and ALK inhibitors for ALK-positive lung cancers.
Notably, the enhanced induction of apoptosis and tumor regression by the combination of ABT-263 and AZD8055 was achieved even with low concentrations of AZD8055 in both cell lines and mouse xenografts and was directly correlated with the relative reduction in MCL-1 protein level (Fig. 6). This suggests that AZD8055 may be effectively combined with ABT-263 even at doses that would otherwise be subtherapeutic as a single agent, widening the potential therapeutic window of this combination. As other BH3 mimetics are being currently developed in the clinic, it will be interesting to determine the differential activity of each to build on the promising concept of combining TORC inhibitors with BH3 mimetics.
A recent report by Gardner et al. demonstrated that rapamycin sensitized several SCLC human cell lines and PDXs to the structurally related BH3-mimetic ABT-737 (35). However, our study reveals significant differences between the two approaches. Unlike the TORC catalytic site inhibitor, rapamycin fails to significantly down-regulate MCL-1 (SI Appendix, Fig. S8A). This finding is consistent with results demonstrating that TORC1/2 catalytic inhibitors more effectively suppress 4E-BP1 phosphorylation and cap-dependent translation than rapamycin or rapalogs (31, 33). Thus, the TORC catalytic inhibitor is uniquely capable of down-regulating MCL-1 protein expression, which is key for the combination to induce apoptosis. Indeed, we found that addition of rapamycin did not consistently increase the amount of apoptosis induced by ABT-263 (SI Appendix, Fig. S8B). Another major difference between these therapeutic approaches is that the AZD8055/ABT-263 combination appears effective in low-BCL-2–expressing SCLCs (NCI-H82 and NCI-H446, Figs. 3 B and E and 6A), which Gardner et al. (35) found were resistant to the combination of rapamycin with ABT-737. In contrast to the findings reported by Gardner et al. (35), we did not observe that ABT-263, rapamycin, or AZD8055 consistently affected BAX levels (Figs. 3F and 5B and SI Appendix, Fig. S8C). One possibility for this difference is that Gardner et al. (35) assessed for changes in BAX expression after more prolonged treatment (1 d to 1 w). Although we did not detect BAX changes after >14 d of treatments in the H1048 xenograft model (Fig. 3F) or in the PDX model of SCLC (∼50 d of treatment), we assayed for BAX expression only at shorter time points in vitro (16 h). Thus, BAX regulation through TORC1 inhibition may be a later event in some models of SCLC. Importantly, Gardner et al. (35) observed that the mTOR pathway was active in all of the PDX models that they studied, further supporting the concept that combining mTORC catalytic site inhibitors with BH3 mimetics in SCLC could be beneficial.
Interestingly, we observed that the BIM to MCL-1 ratio predicted sensitivity to ABT-263 not only in SCLC, but also across a large panel of cancer cells encompassing a wide range of malignancies (Fig. 1B and SI Appendix, Fig. S1D). We indeed recently reported that combination ABT-263 and AZD8055 also showed efficacy in KRAS and BRAF mutant colorectal cancers (7). In that study, we demonstrated that AZD8055 led to loss of MCL-1 in those cancers, thereby sensitizing to ABT-263. However, this combination therapy was not effective in KRAS and BRAF wild-type colorectal cancers, where TORC1/2 inhibition failed to suppress MCL-1. Thus, it is likely that this combination therapy may specifically be effective for cancers in which TORC1/2 inhibition suppresses MCL-1 expression.
We extended our study beyond the more traditional preclinical strategies by using both a SCLC GEMM model and a PDX from a patient with SCLC. Importantly, each autochthonous tumor in the SCLC GEMM model, although initiated by inactivation of Tp53 and Rb1, develops a unique set of additional genetic alterations (39). Furthermore, tumors themselves are heterogeneous, harboring multiple distinct subclones (39). Autochthonous GEMM tumors may therefore more closely recapitulate the genetic heterogeneity found in human SCLC tumors (24, 40, 41) and may be a more rigorous assay for therapeutic efficacy than in vitro studies or mouse xenograft models. They also model the tumor microenvironmental features and immune interactions of human disease more fully than s.c. xenografts in immune-compromised hosts. The fact that we consistently observed stabilization or regression of tumors in response to combination of ABT-263 and AZD8055 in the GEMM, compared with either drug alone, further supports the notion that this combination may be more broadly active in patients than ABT-263 monotherapy. Notably, of six autochthonous tumors treated with ABT-263 alone, one showed significant regression whereas the other five significantly progressed. This range of sensitivities also mirrors the range of sensitivities to ABT-263 in human cell lines and in patients (19, 24).
There was a trend toward GEMM tumors responding more to ABT-263 and AZD8055 than to the standard of care chemotherapies used in patients, cisplatin and etoposide. The relative lack of response of the GEMM tumors to cisplatin and etoposide is interesting because, by contrast, human SCLC tumors have a response rate of ∼50% to combination platinum and etoposide (42). Consistent with our findings, Singh and colleagues (43) showed that Tp53/Rb1-deleted SCLC GEMMs were only modestly sensitive to combination carboplatin and irinotecan, which have similar clinical efficacy as cisplatin and etoposide in patients (42, 44). The SCLC GEMM may therefore more closely resemble a chemotherapy-resistant subset of human SCLC. If this is true, then our study supports the use of the combination of ABT-263 and TORC1/2 inhibition even in second line setting in patients. Alternatively, it is possible that we did not achieve sufficient levels of cisplatin and etoposide in the mice to induce the extent of tumor regression often seen in patients.
Our PDX study demonstrated lack of efficacy of single-agent ABT-263, consistent with the lack of efficacy observed with single-agent ABT-737 by Gardner et al. (35). The combination, however, induced regressions of all of the combination-treated tumors (Fig. 5A). Taken together with the potent efficacy demonstrated in the human xenograft mouse models of SCLC (Fig. 3) and the GEMM model (Fig. 4), the effect of the combination of AZD8055 and ABT-263 was consistent and profound across several mouse models, even at low concentrations of AZD8055 (Fig. 6D).
Overall, we found that high BIM levels helped promote sensitivity to BCL-2/BCL-XL inhibitors in SCLC, but efficacy was mitigated by MCL-1. The addition of TORC1/2 inhibitors to BCL-2/BCL-XL inhibitors in SCLCs can lead to marked tumor responses in an array of complementary mouse models and therefore may improve the efficacy of BH3 mimetics for the treatment of SCLC.
Materials and Methods
All materials and methods are described in SI Appendix. These include information on cell lines, Western blotting, immunoprecipitation, plasmid preparation, siRNA transfections and shRNA transductions, viability assays, FACS death and cell cycle assays, mouse experiments, and statistical methods.
All studies in GEMM and allograft models were performed under an Institutional Animal Care and Use Committee- and Massachusetts Institute of Technology Committee on Animal Care-approved animal protocol.
The mouse xenografts and patient-derived xenografts were performed in accordance with the Massachusetts General Hospital Subcommittee on Research Animal Care.
Supplementary Material
Acknowledgments
We thank the Koch Institute Swanson Biotechnology Center for technical support, specifically the Hope Babette Tang (1983) Histology Facility and the Applied Therapeutics and Live Animal Imaging facility, and Alex Shoemaker (AbbVie) for helpful suggestions. T.J. is a Howard Hughes Medical Institute Investigator and a Daniel K. Ludwig Scholar. This work was supported by Dana Farber/Harvard Cancer Center (DF/HCC) Lung Cancer Specialized Program in Research Excellence NIH–National Cancer Institute (NCI) Grant 2P50 CA09057 (to J.A.E.); NIH Grants R01CA140594 (to J.A.E.), R01CA137008-01 (to J.A.E.), and 1U54HG006097-01 (to C.H.B.); Department of Defense Grants W81-XWH-13-1-0323 (to T.J.) and W81-XWH-13-1-0325 (to J.A.E.); U.S. Army Medical Research and Material Command Acquisition Activity is the awarding and administering acquisition office (T.J. and J.A.E.); Cancer Center Support Grant P30-CA14051 from the NCI (to T.J.); the David H. Koch Institute for Integrative Cancer Research at the Massachusetts Institute of Technology (Koch Institute)–DF/HCC Bridge Project (T.J. and J.A.E.); Grant 086357 from the Wellcome Trust (to C.H.B.); awards from the Burroughs Wellcome Fund and the Howard Hughes Medical Institute (to M.N.R.); a Lung Cancer Research Foundation grant (to A.C.F.); an American Cancer Society Postdoctoral Fellowship (to A.C.F.); and a grant from Uniting Against Lung Cancer (to A.F.F.).
Footnotes
Conflict of interest statement: J.A.E. receives sponsored research from Novartis and AstraZeneca and consults for Novartis and AstraZeneca. H.E. receives sponsored research from AstraZeneca.
This article is a PNAS Direct Submission. J.D.M. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1411848112/-/DCSupplemental.
References
- 1.Faber AC, Ebi H, Costa C, Engelman JA. Apoptosis in targeted therapy responses: The role of BIM. Adv Pharmacol. 2012;65:519–542. doi: 10.1016/B978-0-12-397927-8.00016-6. [DOI] [PubMed] [Google Scholar]
- 2.Faber AC, et al. BIM expression in treatment-naive cancers predicts responsiveness to kinase inhibitors. Cancer Discov. 2011;1(4):352–365. doi: 10.1158/2159-8290.CD-11-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Faber AC, et al. Differential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition. Proc Natl Acad Sci USA. 2009;106(46):19503–19508. doi: 10.1073/pnas.0905056106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Costa C, et al. The impact of EGFR T790M mutations and BIM mRNA expression on outcome in patients with EGFR-mutant NSCLC treated with erlotinib or chemotherapy in the randomized phase III EURTAC trial. Clin Cancer Res. 2014;20(7):2001–2010. doi: 10.1158/1078-0432.CCR-13-2233. [DOI] [PubMed] [Google Scholar]
- 5.Tse C, et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68(9):3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 6.Del Gaizo Moore V, et al. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest. 2007;117(1):112–121. doi: 10.1172/JCI28281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Faber AC, et al. mTOR inhibition specifically sensitizes colorectal cancers with KRAS or BRAF mutations to BCL-2/BCL-XL inhibition by suppressing MCL-1. Cancer Discov. 2014;4(1):42–52. doi: 10.1158/2159-8290.CD-13-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sale MJ, Cook SJ. The BH3 mimetic ABT-263 synergizes with the MEK1/2 inhibitor selumetinib/AZD6244 to promote BIM-dependent tumour cell death and inhibit acquired resistance. Biochem J. 2013;450(2):285–294. doi: 10.1042/BJ20121212. [DOI] [PubMed] [Google Scholar]
- 9.Chen S, Dai Y, Harada H, Dent P, Grant S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 2007;67(2):782–791. doi: 10.1158/0008-5472.CAN-06-3964. [DOI] [PubMed] [Google Scholar]
- 10.Lin X, et al. ‘Seed’ analysis of off-target siRNAs reveals an essential role of Mcl-1 in resistance to the small-molecule Bcl-2/Bcl-XL inhibitor ABT-737. Oncogene. 2007;26(27):3972–3979. doi: 10.1038/sj.onc.1210166. [DOI] [PubMed] [Google Scholar]
- 11.Roberts AW, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: Results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol. 2012;30(5):488–496. doi: 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Al-Harbi S, et al. An antiapoptotic BCL-2 family expression index predicts the response of chronic lymphocytic leukemia to ABT-737. Blood. 2011;118(13):3579–3590. doi: 10.1182/blood-2011-03-340364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garnett MJ, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483(7391):570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adams JM, et al. Subversion of the Bcl-2 life/death switch in cancer development and therapy. Cold Spring Harb Symp Quant Biol. 2005;70:469–477. doi: 10.1101/sqb.2005.70.009. [DOI] [PubMed] [Google Scholar]
- 15.van Delft MF, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10(5):389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mason KD, et al. In vivo efficacy of the Bcl-2 antagonist ABT-737 against aggressive Myc-driven lymphomas. Proc Natl Acad Sci USA. 2008;105(46):17961–17966. doi: 10.1073/pnas.0809957105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nguyen M, et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci USA. 2007;104(49):19512–19517. doi: 10.1073/pnas.0709443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodriguez E, Lilenbaum RC. Small cell lung cancer: Past, present, and future. Curr Oncol Rep. 2010;12(5):327–334. doi: 10.1007/s11912-010-0120-5. [DOI] [PubMed] [Google Scholar]
- 19.Shoemaker AR, et al. Activity of the Bcl-2 family inhibitor ABT-263 in a panel of small cell lung cancer xenograft models. Clin Cancer Res. 2008;14(11):3268–3277. doi: 10.1158/1078-0432.CCR-07-4622. [DOI] [PubMed] [Google Scholar]
- 20.Sartorius UA, Krammer PH. Upregulation of Bcl-2 is involved in the mediation of chemotherapy resistance in human small cell lung cancer cell lines. Int J Cancer. 2002;97(5):584–592. doi: 10.1002/ijc.10096. [DOI] [PubMed] [Google Scholar]
- 21.Hann CL, et al. Therapeutic efficacy of ABT-737, a selective inhibitor of BCL-2, in small cell lung cancer. Cancer Res. 2008;68(7):2321–2328. doi: 10.1158/0008-5472.CAN-07-5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gandhi L, et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol. 2011;29(7):909–916. doi: 10.1200/JCO.2010.31.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mason KD, et al. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128(6):1173–1186. doi: 10.1016/j.cell.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 24.Rudin CM, et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res. 2012;18(11):3163–3169. doi: 10.1158/1078-0432.CCR-11-3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCoy F, et al. Obatoclax induces Atg7-dependent autophagy independent of beclin-1 and BAX/BAK. Cell Death Dis. 2010;1:e108. doi: 10.1038/cddis.2010.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hwang JJ, et al. Phase I dose finding studies of obatoclax (GX15-070), a small molecule pan-BCL-2 family antagonist, in patients with advanced solid tumors or lymphoma. Clin Cancer Res. 2010;16(15):4038–4045. doi: 10.1158/1078-0432.CCR-10-0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paik PK, et al. A phase II study of obatoclax mesylate, a Bcl-2 antagonist, plus topotecan in relapsed small cell lung cancer. Lung Cancer. 2011;74(3):481–485. doi: 10.1016/j.lungcan.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wendel HG, et al. Dissecting eIF4E action in tumorigenesis. Genes Dev. 2007;21(24):3232–3237. doi: 10.1101/gad.1604407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schatz JH, et al. Targeting cap-dependent translation blocks converging survival signals by AKT and PIM kinases in lymphoma. J Exp Med. 2011;208(9):1799–1807. doi: 10.1084/jem.20110846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mills JR, et al. mTORC1 promotes survival through translational control of Mcl-1. Proc Natl Acad Sci USA. 2008;105(31):10853–10858. doi: 10.1073/pnas.0804821105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsieh AC, et al. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell. 2010;17(3):249–261. doi: 10.1016/j.ccr.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chresta CM, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010;70(1):288–298. doi: 10.1158/0008-5472.CAN-09-1751. [DOI] [PubMed] [Google Scholar]
- 33.Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci USA. 2008;105(45):17414–17419. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cope CL, et al. Adaptation to mTOR kinase inhibitors by amplification of eIF4E to maintain cap-dependent translation. J Cell Sci. 2014;127(Pt 4):788–800. doi: 10.1242/jcs.137588. [DOI] [PubMed] [Google Scholar]
- 35.Gardner EE, et al. Rapamycin rescues ABT-737 efficacy in small cell lung cancer. Cancer Res. 2014;74(10):2846–2856. doi: 10.1158/0008-5472.CAN-13-3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meuwissen R, et al. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell. 2003;4(3):181–189. doi: 10.1016/s1535-6108(03)00220-4. [DOI] [PubMed] [Google Scholar]
- 37.Sutherland KD, et al. Cell of origin of small cell lung cancer: Inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell. 2011;19(6):754–764. doi: 10.1016/j.ccr.2011.04.019. [DOI] [PubMed] [Google Scholar]
- 38.Dooley AL, et al. Nuclear factor I/B is an oncogene in small cell lung cancer. Genes Dev. 2011;25(14):1470–1475. doi: 10.1101/gad.2046711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McFadden DG, et al. Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell. 2014;156(6):1298–1311. doi: 10.1016/j.cell.2014.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peifer M, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012;44(10):1104–1110. doi: 10.1038/ng.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rudin CM, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet. 2012;44(10):1111–1116. doi: 10.1038/ng.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hanna N, et al. Randomized phase III trial comparing irinotecan/cisplatin with etoposide/cisplatin in patients with previously untreated extensive-stage disease small-cell lung cancer. J Clin Oncol. 2006;24(13):2038–2043. doi: 10.1200/JCO.2005.04.8595. [DOI] [PubMed] [Google Scholar]
- 43.Singh M, et al. Anti-VEGF antibody therapy does not promote metastasis in genetically engineered mouse tumour models. J Pathol. 2012;227(4):417–430. doi: 10.1002/path.4053. [DOI] [PubMed] [Google Scholar]
- 44.Rossi A, et al. Carboplatin- or cisplatin-based chemotherapy in first-line treatment of small-cell lung cancer: The COCIS meta-analysis of individual patient data. J Clin Oncol. 2012;30(14):1692–1698. doi: 10.1200/JCO.2011.40.4905. [DOI] [PubMed] [Google Scholar]
- 45.Barretina J, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.