

Abstract
The ability of small blood vessels to undergo rapid, reversible morphological changes is essential for the adaptive response to tissue injury or local infection. A canonical feature of this response is transient hyperpermeability. However, when leakiness is profound or persistent, adverse consequences accrue to the host, including organ dysfunction and shock. A growing body of literature identifies the Tie2 receptor, a transmembrane tyrosine kinase highly enriched in the endothelium, as an important regulator of vascular barrier function in health and in disease. The principal ligands of Tie2, Angiopoietins 1 and 2, exert opposite effects on this receptor in the context of inflammation. This review will focus on recent studies that have illuminated novel aspects of the exquisitely controlled Tie2 signaling axis while proposing unanswered questions and future directions for this field of study.
Keywords: Angiopoietin, anthrax, ARDS, barrier function, endothelium, inflammation, lung injury, malaria, respiratory distress, sepsis, Tie1, Tie2, vascular leakage, VE-PTP
Abbreviations: Angpt, angiopoietin; PI3K, phosphatidylinositol-3-kinase; MAPK, mitogen-activated protein kinase; VEGF, vascular endothelial growth factor; EC, endothelial cell; siRNA, small interfering RNA; VE-cadherin, vascular endothelial cadherin; VE-PTP, vascular endothelial protein tyrosine phosphatase
Introduction
Cloning of the first Tie family receptor, Tie1, was reported by the laboratory of Kari Alitalo in 1992.1 In the same year, Daniel Dumont, et al., reported the cloning of tek (later renamed Tie2),2 and subsequent knockout studies performed in mice confirmed a critical role for Tie2 in cardiovascular development.3 Localization studies demonstrated specific enrichment of Tie receptors in the endothelium. Using a variation of expression cloning, scientists at Regeneron subsequently identified Angiopoietin-1 (Angpt-1) and Angiopoietin-2 (Angpt-2) as secreted protein ligands of Tie2.4,5 Although there is some evidence that Angiopoietins also binds Tie1,6,7 Tie1 is considered by many an orphan receptor.8 Tie1-Tie2 heterodimers attenuate signaling from Tie2 homodimers (Fig. 1).
Figure 1.
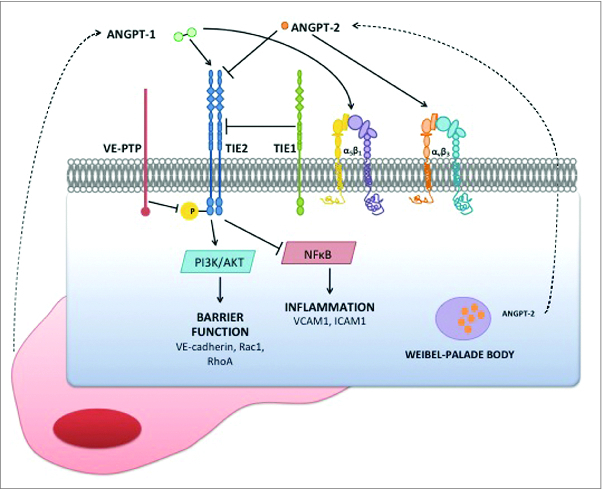
Cell surface components of Angpt-Tie2 axis signaling during inflammation. Endothelial cells are specifically enriched for expression of Tie2, its paralog Tie1, the tyrosine phosphatase VE-PTP, and its ligand Angpt-2. Angpt-1 is secreted by peri-endothelial cells. Several integrins have also been proposed as co- or alternate receptors for the Angpts, examples of which are depicted. In the quiescent vasculature, Tie2 is activated as evident by phosphorylation at tyrosine residues in its intracellular domain. Activated Tie2 promotes barrier function and anti-inflammation. In the context of inflammation, Angpt-2 is both rapidly released from storage granules called Weibel-Palade bodies and is transcriptionally induced. Excess Angpt-2 antagonizes Angpt-1, leading to reduced signaling downstream of Tie2.
Peri-endothelial cells and platelets produce Angpt-1, which binds to and activates Tie2. Cross-phosphorylation of cytoplasmic tyrosine residues close to the C-terminal tail of Tie2 recruits adaptor proteins and initiates signaling through PI3K/Akt and MAP kinases.9-13 Angpt-2 is produced by endothelial cells, and to a lesser degree, macrophages. In endothelial cells, Angpt-2 antagonizes Angpt-1 binding.5 However, in the absence of Angpt-1, Angpt-2 acts as a weak Tie2 agonist.14,15 Both Angpts weakly bind integrins as well,16 the effects of which may be accentuated in situations of low Tie2 expression.17
The opposing roles of Angpt-1 and Angpt-2 on the Tie2 receptor are consistent with results from genetic mouse models. Both the Tie2 and Angpt-1 knockout mouse die in utero of similar defects in cardiac development and blood vessel maturation. In contrast, the Angpt-2 knockout is viable, whereas Angpt-2 overexpression during development phenocopies the Angpt-1 and Tie2 knockouts.4,18 Developmental and pathological angiogenesis remain areas of active inquiry for investigators studying the Angpt-Tie system. In angiogenesis, Tie2 is considered to complement the function of the major mitogenic factor, vascular endothelial growth factor (VEGF). Angpt-2 may destabilize vessels to potentiate the angiogenesis signaled by VEGF whereas Angpt-1 may help stabilize vessels as angiogenesis is completed.
Unlike many other “growth factor” receptor tyrosine kinases, Tie2 is activated throughout the quiescent adult vasculature, suggesting one or more roles in vascular maintenance.19 First using transgenic mice, then adenoviral gene delivery in mice, Thurston, et al., demonstrated that excess Angpt-1 fortifies vascular barrier function against diverse stimuli that promote acute vascular leakage, including VEGF, serotonin, and mustard.20,21 An opposing role for Angpt-2—i.e., barrier weakening—was first reported by Parikh, et al.,22 and independently confirmed in the same year by 2 different laboratories.23,24 Subsequent studies have shown that Angpt-1 counteracts hyperpermeability induced by an array of ligands, each with its own cognate receptor (reviewed in 25)—findings that suggest a downstream mechanism of barrier defense mediated by Tie2 that is discussed below (Fig. 2).
Figure 2.

Intracellular signaling downstream of Tie2 to fortify barrier function. Activated Tie2 signals via PI3K/Akt to the GTPase Rac1. The scaffolding protein IQGAP1 not only maintains Rac1 in its active, GTP-bound form, but may also help localize it to adherens junctions. Active Rac1 signals through p190RhoGAP to inactivate another GTPase, RhoA. The combination of active Rac1 and inactive RhoA promotes cell spreading, cortical distribution of fibrillar actin, and increased accumulation of the barrier effector protein VE-cadherin at the adherens junction.
This review will summarize recent findings that advance our understanding of the Angpt-Tie2 axis by describing how it may be regulated in inflammation and what cellular and tissue responses it modulates.
Vascular leakage in acute inflammation—why and how?
Vascular hyperpermeability is a quintessential feature of acute inflammation, enabling cellular and humoral effectors of immunity access to sites of infection and tissue damage. Venular endothelial cells (ECs) undergo reversible contraction, with the development of transient paracellular gaps that facilitate the egress of humoral contents. This microvascular response is typically localized to the site of injury or infection and is mediated by microbial products (e.g., lipopolysaccharides) and secreted or released host molecules (e.g.,, cytokines, mitochondrial DNA). Hyperpermeability is intimately coupled to enhanced leukocyte adhesion, the first step through which circulating white cells migrate from the blood into tissues. In addition to spatial restriction, the vascular hyperpermeability response tends to be transient as well—the mechanisms of resolution range from downregulation of molecular signaling via receptor endocytosis (e.g., thrombin26) to the elaboration of specific host molecules (e.g.,, resolvins27).
Rare individuals who suffer leukocyte adhesion deficiency vividly illustrate the adaptive aspects of the localized inflammatory response. The most well-characterized of this family of syndromes involves ITGB2, a gene encoding the β2 integrin chain. The β2-integrin chain contributes to 3 major leukocyte cell surface complexes, including the receptor LFA1 that binds the endothelial adhesion molecule ICAM1. Loss-of-function mutations in ITGB2 disrupt leukocyte-endothelial interactions, resulting in recurrent and chronic bacterial infections.28 Mutations in FERMT3, encoding a protein called Kindlin3, produce a similar immune deficiency that is believed to arise from intracellular destabilization of β2-integrin-mediated focal adhesions.29
In certain settings, vascular leakage becomes diffuse and persists. Systemic bacterial infections, also referred to as “sepsis,” constitute a common and deadly example of this phenomenon. When localized infection spreads into the blood, the initially constrained leakage response that helps the host eliminate invasive pathogens appears to affect multiple, distant vascular beds simultaneously. Common clinical manifestations include soft-tissue swelling when skin and skeletal muscle microvessels become excessively leaky; acute respiratory distress syndrome when lung microvessels leak to the point of flooding the airspaces with fluid, thus impairing efficient gas exchange; and poor perfusion of tissues (shock) due to inadequate intravascular blood volume. For reasons that are incompletely understood, certain vascular beds—e.g., those in the brain and heart—are typically spared this systemic leakage.
The biomechanical basis of vascular leakage in acute inflammation is still debated, but seminal ultrastructural studies from Guido Majno and George Palade in the 1960s provided powerful visual evidence of 2 linked processes—(1) the contraction of endothelial cells in venules and (2) the development of gaps between adjacent cells through which water and large molecules could exit the bloodstream.30,31 In the ensuing decades, investigators have delineated the molecular apparatus that governs the remodeling of the actin cytoskeleton to promote cellular contraction (reviewed in32). RhoA and Rac1 are representative members of a large family of GTPases that interact with each other and signal to enzymes that directly modify actin-myosin links to remodel the cytoskeleton. RhoA is activated downstream of canonical permeability mediators, including thrombin and bacterial lipopolysaccharide. Through an associated kinase called ROCK, RhoA signals the reorganization of actin-myosin complexes into a “stress-fiber” arrangement that exerts centripetal force on cell membranes and favors endothelial contraction. Rac1 directly opposes RhoA and drives endothelial cell spreading characterized by a redistribution of actin into a peripheral or “cortical” arrangement. Activation of Rac1 thereby favors endothelial barrier fortification.
The formation of gaps between cells also implies the disassembly of intercellular junctions. Depending upon the vascular bed under consideration, distinct junctional complexes have been identified that form molecular and anatomical units. These include adherens junctions, tight junctions, and gap junctions (reviewed in33). Studies addressing Tie2-dependent regulation of vascular permeability have focused on VE-cadherin, an essential component of adherens junctions.34 The extracellular domain of VE-cadherin homophilically engages VE-cadherin of an adjacent cell in a calcium-dependent fashion. This trans interaction is essential for maintenance of the endothelial barrier.34 The cytoplasmic domain of VE-cadherin non-covalently interacts with a family of catenins, which, in turn, bind actin. Through these internal interactions, VE-cadherin is structurally linked to the actin cytoskeleton, thus facilitating 2-way communication. As a result, barrier dysfunction can arise from signaling events that first pass through the actin cytoskeleton or conversely, from the outside inwards.34
Several endothelial signaling pathways have been proposed as modulators of vascular leakage during inflammation, including the VEGF pathway, the Slit-Robo system, and sphingosine-1-phosphate receptors.35-37 A review of the interconnections among these (and other) cascades and the Angpt-Tie2 system is beyond the scope of this review except to note that results from independent laboratories suggest convergence of downstream signaling from these membrane receptors to VE-cadherin and the actin cytoskeleton.11,38
Angpt-1 and Angpt-2
The ability of excess Angpt-1 to counteract vascular leakage was originally described by Thurston, et al., and subsequently verified in endotoxemic mice.20,21,39 Direct evidence linking the Angpt-Tie2 pathway to pathologic vascular leakage came in 2006 through cellular, mouse, and human studies showing induction of Angpt-2 in the context of sepsis, sufficiency of excess Angpt-2 to promote leakage in otherwise healthy animals, and amelioration of experimental inflammation and organ injury by genetically depleting Angpt-2.22-24
Intriguingly, 2 of the original 3 Angpt-2 mouse studies in inflammation focused on the lung.22,23 The lung may be a prominent site for observing sequelae of impaired Tie2 signaling for several potentially inter-related reasons: (1) Tie2 and its ligands are heavily expressed in the lung; (2) due to its rich vascular network, the lung possesses the highest abundance of ECs relative to total cell population; (3) finally, since intact vascular barrier function is essential for normal lung function, derangements in the vascular barrier are readily detectable and often provoke profound consequences.
Angpt-1 mediated barrier defense is orchestrated by a cascade of signaling that triggers cell-spreading characterized by a cortical distribution of fibrillar actin and increased junctional accumulation of VE-cadherin.12,13,22,39,40 Activated Tie2 signals through PI3K and Akt to activate the small GTPase Rac1. Rac1 is stabilized in the active, GTP-bound form by the scaffolding protein IQGAP1.40 IQGAP1 may also be important for localizing the barrier-protective signaling downstream of Tie2 to the adherens junction. Rac1 activation leads to phosphorylation of p190RhoGAP, a GTPase-activating protein that shifts another GTPase, RhoA, toward the inactive, GDP-bound form. Thus, Tie2 activates Rac1 and deactivates RhoA. The former event favors cell spreading and junctional VE-cadherin accumulation whereas the latter event inhibits actin stress fiber formation and actin-myosin complexes that would retract cell membranes centripetally.
Integrins have been proposed to be involved in Angpt-1 signaling in 2 different ways—either as part of a physical complex with Tie2 in ECs (e.g., αvβ3)16 or even as alternate receptors in non-ECs (e.g., α5β1 and αvβ5).41 Many integrins directly bind components of the extracellular matrix, and since Angpt-1 adheres to the matrix,42 a direct integrin-Angpt-1 interaction is highly plausible. Integrin binding (e.g., to αvβ3) has also been proposed as a mechanism for Angpt-2-mediated sprouting angiogenesis.17 The interaction of integrins with the extracellular matrix helps root endothelial cells via focal adhesions to the basement membrane—as a result, integrins that serve as “receptors” for components of the ECM such as fibronectin have a critical role in the maintenance of endothelial barrier integrity.43 However, a direct link between Angpt binding to integrins and modulation of endothelial barrier function has not been shown. And the mechanisms by which Angpt-Tie signaling impacts integrin-mediated focal adhesions requires further elucidation.
In the context of acute inflammation, experiments with a non-homologous peptide mimetic of Angpt-1 provide unique evidence that Tie2 itself, rather than integrins, is likely to be the primary transducer of Angpt-1-mediated barrier defense. The peptide mimetic, termed vasculotide, is composed of His-His-His-Arg-His-Ser-Phe oligomers tethered to a polyethylene backbone. The HHHRHSF peptide was identified through an unbiased screen for Tie2 binders and shares no homology with Angpt-1.44 When administered to septic mice, vasculotide phenocopied the ability of Angpt-1 to prevent vascular leakage and death in experimental sepsis.45,46 Further, David, et al., showed that the protection conferred by vasculotide was abrogated with partial deletion of Tie2.45
Compared to Angpt-2, the unique adhesiveness of Angpt-1 to the extracellular matrix may have ramifications beyond integrin binding.42 First, as a secreted product of pericytes, Angpt-1 may accumulate in the matrix near the endothelium and be “available” for Tie2 ligation, thus providing a potential explanation for the sustained Tie2 phosphorylation observed in the quiescent vasculature.19 However, a recent report of a postnatal Angpt-1 knockout mouse showed no baseline defects in the vasculature.47 This could suggest that basal Tie2 signaling is dispensable in the postnatal vasculature or that Tie2 is activated in other ways—e.g., ligand-independent or even via Angpt-2. To evaluate these possibilities, both the local extent of Angpt-1 gene excision and the activation state of Tie2 will require careful investigation. Second, Angpt-1 adhesiveness to the matrix may influence which signaling cascades are activated downstream of Tie2 (vide infra).48,49 Lastly, while many human observational studies in critical illness have described a disease-associated rise in circulating Angpt-2 level, the circulating concentration of Angpt-1 is consistently depressed as well (reviewed in25), albeit with a lower relative magnitude. Inferring that the matrix binding of Angpt-1 increases its volume of distribution relative to Angpt-2, the statistically significant but relatively minor falls in circulating Angpt-1 concentration during sepsis may, in fact, reflect a more profound downregulation of local Angpt-1 available for Tie2 binding. Such a hypothesis is in agreement with a recent study of kidney biopsies from septic humans.50 The mechanisms underlying the fall in Angpt-1 have not been defined.
For Angpt-2, both loss- and gain-of-function experiments suggest an antagonist function that potentiates vascular leakage in sepsis.22,51 The human studies in sepsis show that individuals with poor outcomes not only develop early elevation of circulating Angpt-2, but suffer a further rise as clinical status deteriorates toward multi-organ dysfunction and death.22,51 While pre-formed Angpt-2 protein is stored in the Weibel-Palade bodies of ECs and rapidly released upon exposure to inflammatory cytokines,52 the clinical results suggest that the most severe disease is associated with de novo biosynthesis of Angpt-2. In line with this, Angpt-2 mRNA is elevated in experimental sepsis and in vivo siRNA against Angpt-2, whether delivered before or after the cecal ligation and puncture surgery to create sepsis, improves survival.51,53 The mechanisms underlying Angpt-2 induction in sepsis may relate to Tie2 inhibition as evidence for a Tie2-Angpt-2 feedback loop has been shown in cultured ECs.54 The relevance of this mechanism to endothelial inflammation requires further clarification.
Although some studies suggest a protective role of Angpt-2 in the context of infection, the majority of the literature supports the concept that Angpt-2 is deleterious in sepsis (reviewed in55). Recently, Western analysis of lung homogenates from septic mice has shown that Angpt-2 siRNA-knockdown restores Tie2 phosphorylation, providing in vivo evidence that the induction of Angpt-2 in sepsis leads to Tie2 deactivation and that Angpt-2 worsens outcomes in sepsis.53 The human results are equally impressive. Circulating Angpt concentrations have been reported in approximately 4000 subjects with critical illnesses since the initial studies in 2006 (reviewed in25). High Angpt-2 (or an elevated Angpt-2/Angpt-1 ratio or a depressed Angpt-1) concentration has consistently been associated with adverse outcomes. The degree of circulating Angpt-2 elevation observed within the first hour of emergency room admission in patients suspected to harbor infection may even help predict the future development of shock and the probability of death.51 Studies from van der Heijden, et al., show a robust association of Angpt-2 levels with direct measurements of vascular leakage among patients in the intensive care unit.56,57 Finally, a genomic screen conducted in 2 stages among nearly 1,000 subjects with acute respiratory distress syndrome (ARDS) found a powerful association between common polymorphisms in the ANGPT2 locus and adverse outcomes in the ICU, arguing that changes in Angpt-2 expression may fundamentally impact the course and outcome of pulmonary vascular leakage incited by inflammation.58
The exact mechanism for Tie2 antagonism by Angpt-2 remains incompletely understood. For example, although multimerization of Angpt-1 driven by unique N-terminal residues is critical for its ability to activate Tie2,59 domain-swap experiments between Angpt-1 and Angpt-2 have shown that the ability of Angpt-2 to antagonize Tie2 does not depend upon its oligomerization state.60 Studies of the Angpt-2-Tie2-complex crystal structure from Barton, et al., also support the hypothesis that all Angpts interact with Tie2 in a structurally similar fashion.61
More recently, these authors compared their original results to new crystallographic analysis of the Angpt-1-Tie2 complex.62 Superposition revealed no structural shifts in Tie2 that would account for altered signaling, leading the authors to investigate Angpt residues outside the receptor-binding domain. By swapping residues 463–465 of Angpt-1 (T-A-G) with 461–463 of Angpt-2 (P-Q-R), Yu, et al., found that chimeric Angpt-2—i.e., Angpt-2 bearing T-A-G at positions 461–463 in place of P-Q-R—clustered Tie2 monomers overexpressed in the endothelioma cell line EA.hy926. The chimeric Angpt—T-A-G also favored formation of Tie2 homodimers rather than Tie1-Tie2 heterodimers and activated downstream Akt similarly to native recombinant Angpt-1.62 Conversely, the mutant version of Angpt-1, Angpt-1-P-Q-R, exhi-bited attenuated function in these assays (Fig. 3). These results reinforce the importance of Tie1 as a co-receptor that helps tune Tie2 signaling.
Figure 3.

Angpt-1 vs. Angpt-2 at the Tie2 interface. (upper) Angpt-1 and Angpt-2 bind the 2nd immunoglobulin domain of Tie2. Both Angpts share a modular structure consisting of an N-terminal superclustering domain, a coiled-coil domain, a linker region, and finally, a C-terminal fibrinogen-like fold that binds Tie2 (illustrated as ball). (lower) An alignment of fibrinogen-like-folds of Angpt-1 vs. Angpt-2 shows extensive homology. Using structure-based mutagenesis, Yu, et al., have presented evidence that amino acids 461–463 of Angpt-2 (P-Q-R) and 463–465 of Angpt-1 (T-A-G) confer the antagonist vs. agonist property of each ligand. For example, chimeric Angpt-2-TAG functions as a Tie2 agonist whereas chimeric Angpt-1-PQR exhibits antagonist effects.
In the setting of chronic inflammation or vascular remodeling, the pericyte also appears to contribute to the barrier function of small blood vessels. Using a DNA-based aptamer to block the pericyte mitogen platelet-derived growth factor (PDGF)-B, Fuxe, et al., demonstrated that Angpt-1 relies on pericyte coverage to achieve antileakage during chronic infection with the respiratory pathogen Mycoplasma pulmonis.63 In agreement with the established endothelium-centric model of acute inflammation, the authors observed no PDGF-B dependency for Angpt-1-mediated barrier protection against bradykinin challenge. The involvement of pericytes in chronic vascular inflammation has also been demonstrated for Angpt-2. More recently, Ziegler and colleagues showed that long-term vascular overexpression of Angpt-2 was associated with vascular hyperpermeability and loss of capillary-associated pericytes, changes that were reversed by ectopic Angpt-1 or PDGF-B.64
Tie1 and Tie2
Expression of Tie1 is increased at sites of inflammation and vascular remodeling, such as atherosclerotic plaques, healing wounds, tumor-perfusing blood vessels, and maturing ovarian follicles.65-67 Tie1 is critical during embryonic development as global knockout mice die in utero between E13.5–14.5 with severe edema and ruptured microvessels.68 In contrast, postnatal or adult-specific Tie1 deletion appears to have no overt effects on the normal vasculature, but instead, retards tumor angiogenesis and attenuates experimental atherosclerosis.65,69 Studies using fluorescence resonance energy transfer (FRET), structural modeling, and directed mutagenesis suggest that Tie1 and Tie2 directly interact at the cell surface in the absence of any unidentified binding mediator.7 The structural basis of this interaction appears to be a large protein-protein interface over which multiple electrostatic contacts occur between the ectodomains of Tie1 and Tie2. In their study, Seegar, et al., showed that Angpt-1, but not Angpt-2, could dissociate Tie1-Tie2 heterodimers and promote Tie2 clustering.7
Compared to VEGF and other growth factors receptors, the activation of Tie2 proceeds by a unique physical mechanism in which stimulation of ECs with ligand—here, Angpt-1—leads to a rapid translocation of Tie2 receptor molecules to the inter-endothelial junctions where they form homophilic contacts in trans.48,49 Compared to basolaterally distributed Tie2, junctionally localized receptor molecules preferentially activate the PI3K/Akt pathway, which promotes EC survival and barrier function.12 In contrast, activation of Tie2 molecules localized to the interface with extracellular matrix leads to downstream MAP kinase signaling and cell migration.
In multiple conditions associated with acute vascular leakage, not only does the activation of Tie2 fall, but the abundance of receptor molecules also declines.13,45,51,70 As a result, there appears to be a fold2- impairment in Tie2 signaling—a smaller pool of receptor molecules and a lesser fraction of which is in the activated state. While most studies involving this pathway have focused on the ligands, it is likely that the fall in Tie2 expression itself is also important. The mechanisms underlying this change may relate to transcriptional regulation as both protein and mRNA abundance fall. Indeed, microRNAs may be important as studies by Suarez, et al., have shown that artificial reduction of the microRNA-processing enzyme Dicer leads to an increase in Tie2 mRNA and protein in ECs.71 Endothelial sensors of blood flow may be upstream regulators of Tie2 expression as a model of abrupt flow cessation over cultured ECs has demonstrated reduced Tie2 expression.72
Mechanisms of Tie2 protein downregulation may also be involved, but are less well-studied. Receptor ubiquitination has been described as a key step for ligand-induced internalization and degradation of cell-surface Tie2 protein.73 Depending on which Tie2 ligand is bound and the degree of receptor activation, the rate and extent of endocytic internalization also differs, at least in cultured ECs.15
VE-PTP
Phosphatase enzymes counteract kinase-mediated signal transduction. Vascular-endothelial protein tyrosine phosphatase (VE-PTP, mouse ortholog of the human PTP-β) is the only known tyrosine phosphatase whose expression is limited to vascular endothelium during development. In 1999, Risau's group reported that VE-PTP binds and de-activates Tie2.74 Knockout studies from Vestweber's group and then scientists at Regeneron in 2006 demonstrated that VE-PTP is essential for maturation of primitive blood vessels during development and that the enzyme is expressed in the adult vasculature.75,76
The biology of VE-PTP is complex, and the reader is referred to an excellent review.77 Part of the complexity with respect to barrier function stems from the observation that VE-PTP also targets phosphorylated tyrosines on VEGFR2 and VE-cadherin.78,79 Whereas tyrosine phosphorylation of VEGFR2 and Tie2 promote downstream signaling from each receptor, tyrosine phosphorylation of VE-cadherin leads to internalization via endocytosis and weakening of the endothelial barrier.
Depending upon the target considered, VE-PTP could be predicted either to support barrier function (by dephosphorylating VE-cadherin or VEGFR2) or weaken barrier function (by dephosphorylating Tie2). Indeed, activated VEGFR2 loosens inter-endothelial contacts by inducing phosphorylation of VE-cadherin,80 whereas activated Tie2 not only counteracts the permeability effect of VEGF on VEGFR2, but increases barrier stability by enhancing junctional localization of VE-cadherin.12,21 This event requires de-phosphorylation of VE-cadherin at critical tyrosine residues.81 Downregulation of VE-PTP expression has been shown to reduce VE-cadherin adhesiveness, leading to increased transendothelial permeability. Morevoer, artificial stabilization of the VE-PTP-VE-cadherin interaction has been reported to attenuate the permeability response to VEGF or bacterial lipopolysaccharide.82
In vivo experiments are helping to resolve whether VE-PTP is primarily barrier-protective or -disruptive. In mice, Vestweber's group has shown that VE-PTP maintains the endothelial barrier by forming a close association with VE-cadherin that favors the dephosphorylated, membrane-localized form of VE-cadherin.82 How VE-PTP's effects on Tie2 fit into this scheme requires further experiments. One recent study suggests that an antibody targeting VE-PTP reduces its interaction with Tie2, but not its interaction with VE-cadherin.83 This result raises the possibility that VE-PTP inhibition may improve barrier function in vivo.
Future directions
Each component of the Tie2 signaling axis merits further investigation. Noted above, Tie1 is induced during states of inflammation and vascular remodeling. No ligand has been conclusively identified for this receptor. While it is possible that Tie1 is a transmembrane tyrosine kinase that lacks a high-affinity ligand, the discovery of such a molecule could have implications far beyond inflammation and vascular leakage. Second, although integrins have been identified as important low-affinity Angpt receptors—particularly in states of reduced Tie2 expression—the mechanistic basis that distinguishes Angpt-2 as a Tie2 antagonist during inflammation still remains incompletely understood. The mutagenesis studies from Yu, et al., would imply that Angpt-2 is always an antagonist when Tie2 and Tie1 are present, never an agonist. Third, experiments showing that the non-homologous Angpt-1-mimetic, vasculotide, recapitulates Angpt-1s protective effects in experimental sepsis independently implicate Tie2 in Angpt-dependent barrier regulation. Further effort is needed to understand why Tie2 expression falls in several states of vascular leakage. Tie2 and Angpt-2 are highly enriched in endothelial cells, but are notably also expressed by small populations of haematopoietic -lineage cells. While much of the biology pertaining to Tie2-expressing macrophages has been described in the context of cancer, more work is needed to test whether haematopoietic cells contribute to vascular leakage in inflammation in Tie2-dependent ways.84 Finally, a number of therapeutic commercialization efforts are underway, both in the US and abroad. As Tie2-axis drugs penetrate the clinical market, there will be a need for careful trials to define the utility of these compounds in settings of vascular leakage.
Conclusions
The stereotypical morphological response of microvascular ECs and the rapid reversibility of associated vascular leakage suggest that the endothelium executes an organized and highly regulated response to acute inflammatory stimuli. The Angiopoietin-Tie2 system is one of several endothelial pathways proposed to be prominently involved in this response. In the settings of severe infection and critical illness, a combination of genetic, biochemical, and experimental evidence argue that the disruption of normal Tie2 signaling critically contributes to vascular leakage and its injurious sequelae. A growing body of literature has elucidated key events to account for the disruption of Tie2 signaling during acute inflammation and to explain the downstream consequences of the switch in Tie2 signaling. While important questions remain unanswered, ongoing efforts to apply knowledge of the Angiopoietins and Tie2 in the context of human diseases herald an exciting era of discovery and translation in this field.
Disclosure of Potential Conflicts of Interest
S.M.P. advises Vasomune Therapeutics and Eunoia Biotech and is listed as an inventor on disclosures and patents related to Angiopoietins held by Beth Israel Deaconess Medical Center.
Acknowledgments
The authors would like to thank Drs. Chandra Ghosh (Boston, USA) and Sascha David (Hannover, Germany) for critical review and helpful advice with this manuscript.
Funding
Research in S.M.P's laboratory is supported by the National Institutes of Health (R01-HL093234 and R01-DK095072) and the American Diabetes Association.
References
- 1.Partanen J, Armstrong E, Makela TP, Korhonen J, Sandberg M, Renkonen R, Knuutila S, Huebner K, Alitalo K. A novel endothelial cell surface receptor tyrosine kinase with extracellular epidermal growth factor homology domains. Mol Cell Biol 1992; 12:1698-707; PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dumont DJ, Yamaguchi TP, Conlon RA, Rossant J, Breitman ML. Tek, a novel tyrosine kinase gene located on mouse chromosome 4, is expressed in endothelial cells and their presumptive precursors. Oncogene 1992; 7:1471-80; PMID: [PubMed] [Google Scholar]
- 3.Dumont DJ, Gradwohl G, Fong GH, Puri MC, Gertsenstein M, Auerbach A, Breitman ML. Dominant-negative and targeted null mutations in the endothelial receptor tyrosine kinase, tek, reveal a critical role in vasculogenesis of the embryo. Genes Dev 1994; 8:1897-909; PMID:; http://dx.doi.org/ 10.1101/gad.8.16.1897 [DOI] [PubMed] [Google Scholar]
- 4.Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, Compton D, McClain J, Aldrich TH, Papadopoulos N, et al. Angiopoietin-2, a natural antagonist for tie2 that disrupts in vivo angiogenesis. Science 1997; 277:55-60; PMID:; http://dx.doi.org/ 10.1126/science.277.5322.55 [DOI] [PubMed] [Google Scholar]
- 5.Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD. Requisite role of angiopoietin-1, a ligand for the tie2 receptor, during embryonic angiogenesis. Cell 1996; 87:1171-80; PMID:; http://dx.doi.org/ 10.1016/S0092-8674(00)81813-9 [DOI] [PubMed] [Google Scholar]
- 6.Yuan HT, Venkatesha S, Chan B, Deutsch U, Mammoto T, Sukhatme VP, Woolf AS, Karumanchi SA. Activation of the orphan endothelial receptor tie1 modifies tie2-mediated intracellular signaling and cell survival. FASEB J 2007; 21:3171-83; PMID:; http://dx.doi.org/ 10.1096/fj.07-8487com [DOI] [PubMed] [Google Scholar]
- 7.Seegar TC, Eller B, Tzvetkova-Robev D, Kolev MV, Henderson SC, Nikolov DB, Barton WA. Tie1-tie2 interactions mediate functional differences between angiopoietin ligands. Mol Cell 2010; 37:643-55; PMID:; http://dx.doi.org/ 10.1016/j.molcel.2010.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jeltsch M, Leppanen VM, Saharinen P, Alitalo K. Receptor tyrosine kinase-mediated angiogenesis. Cold Spring Harb Perspect Biol 2013; 5(9); PMID:; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones N, Dumont DJ. The tek/tie2 receptor signals through a novel dok-related docking protein, dok-r. Oncogene 1998; 17:1097-108; PMID:; http://dx.doi.org/ 10.1038/sj.onc.1202115 [DOI] [PubMed] [Google Scholar]
- 10.Harfouche R, Hassessian HM, Guo Y, Faivre V, Srikant CB, Yancopoulos GD, Hussain SN. Mechanisms which mediate the antiapoptotic effects of angiopoietin-1 on endothelial cells. Microvasc Res. 2002; 64:135-147; PMID:; http://dx.doi.org/ 10.1006/mvre.2002.2421 [DOI] [PubMed] [Google Scholar]
- 11.Li X, Stankovic M, Bonder CS, Hahn CN, Parsons M, Pitson SM, Xia P, Proia RL, Vadas MA, Gamble JR. Basal and angiopoietin-1-mediated endothelial permeability is regulated by sphingosine kinase-1. Blood. 2008; 111:3489-97; PMID:; http://dx.doi.org/ 10.1182/blood-2007-05-092148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mammoto T, Parikh SM, Mammoto A, Gallagher D, Chan B, Mostoslavsky G, Ingber DE, Sukhatme VP. Angiopoietin-1 requires p190 rhogap to protect against vascular leakage in vivo. J Biol Chem. 2007; 282:23910-8; PMID:; http://dx.doi.org/ 10.1074/jbc.M702169200 [DOI] [PubMed] [Google Scholar]
- 13.Ghosh CC, Mukherjee A, David S, Knaus UG, Stearns-Kurosawa DJ, Kurosawa S, Parikh SM. Impaired function of the tie-2 receptor contributes to vascular leakage and lethality in anthrax. Proc Natl Acad Sci U S A. 2012; 109:10024-9; PMID:; http://dx.doi.org/ 10.1073/pnas.1120755109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yuan HT, Khankin EV, Karumanchi SA, Parikh SM. Angiopoietin 2 is a partial agonist/antagonist of tie2 signaling in the endothelium. Mol Cell Biol 2009; 29:2011-22; PMID:; http://dx.doi.org/ 10.1128/MCB.01472-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bogdanovic E, Nguyen VP, Dumont DJ. Activation of tie2 by angiopoietin-1 and angiopoietin-2 results in their release and receptor internalization. J Cell Sci 2006; 119:3551-60; PMID:; http://dx.doi.org/ 10.1242/jcs.03077 [DOI] [PubMed] [Google Scholar]
- 16.Cascone I, Napione L, Maniero F, Serini G, Bussolino F. Stable interaction between alpha5beta1 integrin and tie2 tyrosine kinase receptor regulates endothelial cell response to ang-1. J Cell Biol 2005; 170:993-1004; PMID:; http://dx.doi.org/ 10.1083/jcb.200507082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Felcht M, Luck R, Schering A, Seidel P, Srivastava K, Hu J, Bartol A, Kienast Y, Vettel C, Loos EK, et al. Angiopoietin-2 differentially regulates angiogenesis through tie2 and integrin signaling. J Clin Invest 2012; 122:1991-2005; PMID:; http://dx.doi.org/ 10.1172/JCI58832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gale NW, Thurston G, Hackett SF, Renard R, Wang Q, McClain J, Martin C, Witte C, Witte MH, Jackson D, et al. Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter role is rescued by angiopoietin-1. Dev Cell 2002; 3:411-23; PMID:; http://dx.doi.org/ 10.1016/S1534-5807(02)00217-4 [DOI] [PubMed] [Google Scholar]
- 19.Wong AL, Haroon ZA, Werner S, Dewhirst MW, Greenberg CS, Peters KG. Tie2 expression and phosphorylation in angiogenic and quiescent adult tissues. Circ Res 1997; 81:567-74; PMID:; http://dx.doi.org/ 10.1161/01.RES.81.4.567 [DOI] [PubMed] [Google Scholar]
- 20.Thurston G, Rudge JS, Ioffe E, Zhou H, Ross L, Croll SD, Glazer N, Holash J, McDonald DM, Yancopoulos GD. Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat Med 2000; 6:460-3; PMID:; http://dx.doi.org/ 10.1038/74725 [DOI] [PubMed] [Google Scholar]
- 21.Thurston G, Suri C, Smith K, McClain J, Sato TN, Yancopoulos GD, McDonald DM. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science 1999; 286:2511-4; PMID:; http://dx.doi.org/ 10.1126/science.286.5449.2511 [DOI] [PubMed] [Google Scholar]
- 22.Parikh SM, Mammoto T, Schultz A, Yuan HT, Christiani D, Karumanchi SA, Sukhatme VP. Excess circulating angiopoietin-2 may contribute to pulmonary vascular leak in sepsis in humans. PLoS Med 2006; 3:e46; PMID:; http://dx.doi.org/ 10.1371/journal.pmed.0030046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhandari V, Choo-Wing R, Lee CG, Zhu Z, Nedrelow JH, Chupp GL, Zhang X, Matthay MA, Ware LB, Homer RJ, et al. Hyperoxia causes angiopoietin 2-mediated acute lung injury and necrotic cell death. Nat Med 2006; 12:1286-93; PMID:; http://dx.doi.org/ 10.1038/nm1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fiedler U, Reiss Y, Scharpfenecker M, Grunow V, Koidl S, Thurston G, Gale NW, Witzenrath M, Rosseau S, Suttorp N, et al. Angiopoietin-2 sensitizes endothelial cells to tnf-alpha and has a crucial role in the induction of inflammation. Nat Med 2006; 12:235-9; PMID:; http://dx.doi.org/ 10.1038/nm1351 [DOI] [PubMed] [Google Scholar]
- 25.Parikh SM. Dysregulation of the angiopoietin-tie-2 axis in sepsis and ards. Virulence 2013; 4:517-24; PMID:; http://dx.doi.org/ 10.4161/viru.24906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trejo J, Altschuler Y, Fu HW, Mostov KE, Coughlin SR. Protease-activated receptor-1 down-regulation: A mutant hela cell line suggests novel requirements for par1 phosphorylation and recruitment to clathrin-coated pits. J Biol Chem 2000; 275:31255-65; PMID:; http://dx.doi.org/ 10.1074/jbc.M003770200 [DOI] [PubMed] [Google Scholar]
- 27.Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014; 510:92-101; PMID:; http://dx.doi.org/ 10.1038/nature13479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Springer TA, Thompson WS, Miller LJ, Schmalstieg FC, Anderson DC. Inherited deficiency of the mac-1, lfa-1, p150,95 glycoprotein family and its molecular basis. J Exp Med 1984; 160:1901-18; PMID:; http://dx.doi.org/ 10.1084/jem.160.6.1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malinin NL, Zhang L, Choi J, Ciocea A, Razorenova O, Ma YQ, Podrez EA, Tosi M, Lennon DP, Caplan AI, et al. A point mutation in kindlin3 ablates activation of three integrin subfamilies in humans. Nat Med 2009; 15:313-8; PMID:; http://dx.doi.org/ 10.1038/nm.1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Majno G, Palade GE, Schoefl GI. Studies on inflammation. Ii. The site of action of histamine and serotonin along the vascular tree: A topographic study. J Biophys Biochem Cytol 1961; 11:607-26; PMID:; http://dx.doi.org/ 10.1083/jcb.11.3.607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Majno G, Palade GE. Studies on inflammation. 1. The effect of histamine and serotonin on vascular permeability: An electron microscopic study. J Biophys Biochem Cytol 1961; 11:571-605; PMID:; http://dx.doi.org/ 10.1083/jcb.11.3.571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vandenbroucke E, Mehta D, Minshall R, Malik AB. Regulation of endothelial junctional permeability. Ann N Y Acad Sci 2008; 1123:134-45; PMID:; http://dx.doi.org/ 10.1196/annals.1420.016 [DOI] [PubMed] [Google Scholar]
- 33.Dejana E, Orsenigo F, Molendini C, Baluk P, McDonald DM. Organization and signaling of endothelial cell-to-cell junctions in various regions of the blood and lymphatic vascular trees. Cell Tissue Res 2009; 335:17-25; PMID:; http://dx.doi.org/ 10.1007/s00441-008-0694-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corada M, Mariotti M, Thurston G, Smith K, Kunkel R, Brockhaus M, Lampugnani MG, Martin-Padura I, Stoppacciaro A, Ruco L, et al. Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc Natl Acad Sci U S A 1999; 96:9815-20; PMID:; http://dx.doi.org/ 10.1073/pnas.96.17.9815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yano K, Liaw PC, Mullington JM, Shih SC, Okada H, Bodyak N, Kang PM, Toltl L, Belikoff B, Buras J, et al. Vascular endothelial growth factor is an important determinant of sepsis morbidity and mortality. J Exp Med 2006; 203:1447-58; PMID:; http://dx.doi.org/ 10.1084/jem.20060375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.London NR, Zhu W, Bozza FA, Smith MC, Greif DM, Sorensen LK, Chen L, Kaminoh Y, Chan AC, Passi SF, et al. Targeting robo4-dependent slit signaling to survive the cytokine storm in sepsis and influenza. Sci Transl Med 2010; 2:23ra19; PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Puneet P, Yap CT, Wong L, Lam Y, Koh DR, Moochhala S, Pfeilschifter J, Huwiler A, Melendez AJ. Sphk1 regulates proinflammatory responses associated with endotoxin and polymicrobial sepsis. Science 2010; 328:1290-4; PMID:; http://dx.doi.org/ 10.1126/science.1188635 [DOI] [PubMed] [Google Scholar]
- 38.Sanchez T, Estrada-Hernandez T, Paik JH, Wu MT, Venkataraman K, Brinkmann V, Claffey K, Hla T. Phosphorylation and action of the immunomodulator fty720 inhibits vascular endothelial cell growth factor-induced vascular permeability. J Biol Chem 2003; 278:47281-90; PMID:; http://dx.doi.org/ 10.1074/jbc.M306896200 [DOI] [PubMed] [Google Scholar]
- 39.Witzenbichler B, Westermann D, Knueppel S, Schultheiss HP, Tschope C. Protective role of angiopoietin-1 in endotoxic shock. Circulation 2005; 111:97-105; PMID:; http://dx.doi.org/ 10.1161/01.CIR.0000151287.08202.8E [DOI] [PubMed] [Google Scholar]
- 40.David S, Ghosh CC, Mukherjee A, Parikh SM. Angiopoietin-1 requires iq domain gtpase-activating protein 1 to activate rac1 and promote endothelial barrier defense. Arterioscler Thromb Vasc Biol 2011; 31:2643-52; PMID:; http://dx.doi.org/ 10.1161/ATVBAHA.111.233189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dallabrida SM, Ismail N, Oberle JR, Himes BE, Rupnick MA. Angiopoietin-1 promotes cardiac and skeletal myocyte survival through integrins. Circ Res 2005; 96:e8-24; PMID:; http://dx.doi.org/ 10.1161/01.RES.0000158285.57191.60 [DOI] [PubMed] [Google Scholar]
- 42.Xu Y, Yu Q. Angiopoietin-1, unlike angiopoietin-2, is incorporated into the extracellular matrix via its linker peptide region. J Biol Chem 2001; 276:34990-8; PMID:; http://dx.doi.org/ 10.1074/jbc.M103661200 [DOI] [PubMed] [Google Scholar]
- 43.Lampugnani MG, Resnati M, Dejana E, Marchisio PC. The role of integrins in the maintenance of endothelial monolayer integrity. J Cell Biol 1991; 112:479-90; PMID:; http://dx.doi.org/ 10.1083/jcb.112.3.479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tournaire R, Simon MP, le Noble F, Eichmann A, England P, Pouyssegur J. A short synthetic peptide inhibits signal transduction, migration and angiogenesis mediated by tie2 receptor. EMBO reports 2004; 5:262-7; PMID:; http://dx.doi.org/ 10.1038/sj.embor.7400100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.David S, Ghosh CC, Kumpers P, Shushakova N, Van Slyke P, Khankin EV, Karumanchi SA, Dumont D, Parikh SM. Effects of a synthetic peg-ylated tie-2 agonist peptide on endotoxemic lung injury and mortality. Am J Physiol Lung Cell Mol Physiol 2011; 300:L851-62; PMID:; http://dx.doi.org/ 10.1152/ajplung.00459.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kumpers P, Gueler F, David S, Van Slyke P, Dumont DJ, Park JK, Bockmeyer CL, Parikh SM, Pavenstadt H, Haller H, et al. The synthetic tie2 agonist peptide vasculotide protects against vascular leakage and reduces mortality in murine abdominal sepsis. Crit Care 2011; 15:R261; PMID:; http://dx.doi.org/ 10.1186/cc10523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jeansson M, Gawlik A, Anderson G, Li C, Kerjaschki D, Henkelman M, Quaggin SE. Angiopoietin-1 is essential in mouse vasculature during development and in response to injury. J Clin Invest 2011; 121:2278-89; PMID:; http://dx.doi.org/ 10.1172/JCI46322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saharinen P, Eklund L, Miettinen J, Wirkkala R, Anisimov A, Winderlich M, Nottebaum A, Vestweber D, Deutsch U, Koh GY, et al. Angiopoietins assemble distinct tie2 signalling complexes in endothelial cell-cell and cell-matrix contacts. Nat Cell Biol 2008; 10:527-37; PMID:; http://dx.doi.org/ 10.1038/ncb1715 [DOI] [PubMed] [Google Scholar]
- 49.Fukuhara S, Sako K, Minami T, Noda K, Kim HZ, Kodama T, Shibuya M, Takakura N, Koh GY, Mochizuki N. Differential function of tie2 at cell-cell contacts and cell-substratum contacts regulated by angiopoietin-1. Nat Cell Biol 2008; 10:513-26; PMID:; http://dx.doi.org/ 10.1038/ncb1714 [DOI] [PubMed] [Google Scholar]
- 50.Aslan A, Jongman RM, Moser J, Stegeman CA, van Goor H, Diepstra A, van den Heuvel MC, Heeringa P, Molema G, Zijlstra JG, et al. The renal angiopoietin/tie2 system in lethal human sepsis. Crit Care 2014; 18:423; PMID:; http://dx.doi.org/ 10.1186/cc13806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.David S, Mukherjee A, Ghosh CC, Yano M, Khankin EV, Wenger JB, Karumanchi SA, Shapiro NI, Parikh SM. Angiopoietin-2 may contribute to multiple organ dysfunction and death in sepsis. Crit Care Med 2012; 40:3034-41; PMID:; http://dx.doi.org/ 10.1097/CCM.0b013e31825fdc31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fiedler U, Scharpfenecker M, Koidl S, Hegen A, Grunow V, Schmidt JM, Kriz W, Thurston G, Augustin HG. The tie-2 ligand angiopoietin-2 is stored in and rapidly released upon stimulation from endothelial cell weibel-palade bodies. Blood 2004; 103:4150-6; PMID:; http://dx.doi.org/ 10.1182/blood-2003-10-3685 [DOI] [PubMed] [Google Scholar]
- 53.Stiehl T, Thamm K, Kaufmann J, Schaeper U, Kirsch T, Haller H, Santel A, Ghosh CC, Parikh SM, David S. Lung-targeted rna-interference against angiopoietin-2 ameliorates multi-organ dusfunction and death in sepsis. Crit Care Med 2014; (accepted); PMID: [DOI] [PubMed] [Google Scholar]
- 54.Daly C, Wong V, Burova E, Wei Y, Zabski S, Griffiths J, Lai KM, Lin HC, Ioffe E, Yancopoulos GD, et al. Angiopoietin-1 modulates endothelial cell function and gene expression via the transcription factor fkhr (foxo1). Genes Dev 2004; 18:1060-71; PMID:; http://dx.doi.org/ 10.1101/gad.1189704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.David S, Kumpers P, van Slyke P, Parikh SM. Mending leaky blood vessels: The angiopoietin-tie2 pathway in sepsis. J Pharmacol Exp Ther 2013; 345:2-6; PMID:; http://dx.doi.org/ 10.1124/jpet.112.201061 [DOI] [PubMed] [Google Scholar]
- 56.van der Heijden M, Pickkers P, van Nieuw Amerongen GP, van Hinsbergh VW, Bouw MP, van der Hoeven JG, Groeneveld AB. Circulating angiopoietin-2 levels in the course of septic shock: Relation with fluid balance, pulmonary dysfunction and mortality. Intensive Care Med 2009; 35:1567-74; PMID:; http://dx.doi.org/ 10.1007/s00134-009-1560-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van der Heijden M, van Nieuw Amerongen GP, Koolwijk P, van Hinsbergh VW, Groeneveld AB. Angiopoietin-2, permeability oedema, occurrence and severity of ali/ards in septic and non-septic critically ill patients. Thorax 2008; 63:903-9; PMID:; http://dx.doi.org/ 10.1136/thx.2007.087387 [DOI] [PubMed] [Google Scholar]
- 58.Meyer NJ, Li M, Feng R, Bradfield J, Gallop R, Bellamy S, Fuchs BD, Lanken PN, Albelda SM, Rushefski M, et al. Angpt2 genetic variant is associated with trauma-associated acute lung injury and altered plasma angiopoietin-2 isoform ratio. Am J Respir Crit Care Med 2011; 183:1344-53; PMID:; http://dx.doi.org/ 10.1164/rccm.201005-0701OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim KT, Choi HH, Steinmetz MO, Maco B, Kammerer RA, Ahn SY, Kim HZ, Lee GM, Koh GY. Oligomerization and multimerization are critical for angiopoietin-1 to bind and phosphorylate tie2. J Biol Chem 2005; 280:20126-31; PMID:; http://dx.doi.org/ 10.1074/jbc.M500292200 [DOI] [PubMed] [Google Scholar]
- 60.Davis S, Papadopoulos N, Aldrich TH, Maisonpierre PC, Huang T, Kovac L, Xu A, Leidich R, Radziejewska E, Rafique A, et al. Angiopoietins have distinct modular domains essential for receptor binding, dimerization and superclustering. Nat Struct Biol 2003; 10:38-44; PMID:; http://dx.doi.org/ 10.1038/nsb880 [DOI] [PubMed] [Google Scholar]
- 61.Barton WA, Tzvetkova-Robev D, Miranda EP, Kolev MV, Rajashankar KR, Himanen JP, Nikolov DB. Crystal structures of the tie2 receptor ectodomain and the angiopoietin-2-tie2 complex. Nat Struc Mol Biol 2006; 13:524-32; PMID:; http://dx.doi.org/ 10.1038/nsmb1101 [DOI] [PubMed] [Google Scholar]
- 62.Yu X, Seegar TC, Dalton AC, Tzvetkova-Robev D, Goldgur Y, Rajashankar KR, Nikolov DB, Barton WA. Structural basis for angiopoietin-1-mediated signaling initiation. Proc Natl Acad Sci U S A 2013; 110:7205-10; PMID:; http://dx.doi.org/ 10.1073/pnas.1216890110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fuxe J, Tabruyn S, Colton K, Zaid H, Adams A, Baluk P, Lashnits E, Morisada T, Le T, O'Brien S, et al. Pericyte requirement for anti-leak action of angiopoietin-1 and vascular remodeling in sustained inflammation. Am J Pathol 2011; 178:2897-909; PMID:; http://dx.doi.org/ 10.1016/j.ajpath.2011.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ziegler T, Horstkotte J, Schwab C, Pfetsch V, Weinmann K, Dietzel S, Rohwedder I, Hinkel R, Gross L, Lee S, et al. Angiopoietin 2 mediates microvascular and hemodynamic alterations in sepsis. J Clin Invest 2013; epub ahead of print; PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Woo KV, Qu X, Babaev VR, Linton MF, Guzman RJ, Fazio S, Baldwin HS. Tie1 attenuation reduces murine atherosclerosis in a dose-dependent and shear stress-specific manner. J Clin Invest 2011; 121:1624-35; PMID:; http://dx.doi.org/ 10.1172/JCI42040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Korhonen J, Partanen J, Armstrong E, Vaahtokari A, Elenius K, Jalkanen M, Alitalo K. Enhanced expression of the tie receptor tyrosine kinase in endothelial cells during neovascularization. Blood 1992; 80:2548-55; PMID: [PubMed] [Google Scholar]
- 67.Kaipainen A, Vlaykova T, Hatva E, Bohling T, Jekunen A, Pyrhonen S, Alitalo K. Enhanced expression of the tie receptor tyrosine kinase mesenger rna in the vascular endothelium of metastatic melanomas. Cancer Res 1994; 54:6571-7; PMID: [PubMed] [Google Scholar]
- 68.Puri MC, Rossant J, Alitalo K, Bernstein A, Partanen J. The receptor tyrosine kinase tie is required for integrity and survival of vascular endothelial cells. EMBO J 1995; 14:5884-91; PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.D'Amico G, Korhonen EA, Anisimov A, Zarkada G, Holopainen T, Hagerling R, Kiefer F, Eklund L, Sormunen R, Elamaa H, et al. Tie1 deletion inhibits tumor growth and improves angiopoietin antagonist therapy. J Clin Invest 2014; 124:824-34; PMID:; http://dx.doi.org/ 10.1172/JCI68897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van Meurs M, Kurniati NF, Wulfert FM, Asgeirsdottir SA, de Graaf IA, Satchell SC, Mathieson PW, Jongman RM, Kumpers P, Zijlstra JG, et al. Shock-induced stress induces loss of microvascular endothelial tie2 in the kidney which is not associated with reduced glomerular barrier function. Am J Physiol Renal Physiol 2009; 297:F272-81; PMID:; http://dx.doi.org/ 10.1152/ajprenal.00137.2009 [DOI] [PubMed] [Google Scholar]
- 71.Suarez Y, Fernandez-Hernando C, Yu J, Gerber SA, Harrison KD, Pober JS, Iruela-Arispe ML, Merkenschlager M, Sessa WC. Dicer-dependent endothelial micrornas are necessary for postnatal angiogenesis. Proc Natl Acad Sci U S A 2008; 105:14082-7; PMID:; http://dx.doi.org/ 10.1073/pnas.0804597105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li R, Zijlstra JG, Kamps JA, van Meurs M, Molema G. Abrupt reflow enhances cytokine induced pro-inflammatory activation of endothelial cells during simulated shock and resuscitation. Shock 2014; PMID: [DOI] [PubMed] [Google Scholar]
- 73.Wehrle C, Van Slyke P, Dumont DJ. Angiopoietin-1-induced ubiquitylation of tie2 by c-cbl is required for internalization and degradation. Biochem J 2009; 423:375-80; PMID:; http://dx.doi.org/ 10.1042/BJ20091010 [DOI] [PubMed] [Google Scholar]
- 74.Fachinger G, Deutsch U, Risau W. Functional interaction of vascular endothelial-protein-tyrosine phosphatase with the angiopoietin receptor tie-2. Oncogene 1999; 18:5948-53; PMID:; http://dx.doi.org/ 10.1038/sj.onc.1202992 [DOI] [PubMed] [Google Scholar]
- 75.Baumer S, Keller L, Holtmann A, Funke R, August B, Gamp A, Wolburg H, Wolburg-Buchholz K, Deutsch U, Vestweber D. Vascular endothelial cell-specific phosphotyrosine phosphatase (ve-ptp) activity is required for blood vessel development. Blood 2006; 107:4754-62; PMID:; http://dx.doi.org/ 10.1182/blood-2006-01-0141 [DOI] [PubMed] [Google Scholar]
- 76.Dominguez MG, Hughes VC, Pan L, Simmons M, Daly C, Anderson K, Noguera-Troise I, Murphy AJ, Valenzuela DM, Davis S, et al. Vascular endothelial tyrosine phosphatase (ve-ptp)-null mice undergo vasculogenesis but die embryonically because of defects in angiogenesis. Proc Natl Acad Sci U S A 2007; 104:3243-8; PMID:; http://dx.doi.org/ 10.1073/pnas.0611510104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kuppers V, Vockel M, Nottebaum AF, Vestweber D. Phosphatases and kinases as regulators of the endothelial barrier function. Cell Tissue Res. 2014; 355:577-86; PMID:; http://dx.doi.org/ 10.1007/s00441-014-1812-1 [DOI] [PubMed] [Google Scholar]
- 78.Hayashi M, Majumdar A, Li X, Adler J, Sun Z, Vertuani S, Hellberg C, Mellberg S, Koch S, Dimberg A, et al. Ve-ptp regulates vegfr2 activity in stalk cells to establish endothelial cell polarity and lumen formation. Nat Commun 2013; 4:1672; PMID:; http://dx.doi.org/ 10.1038/ncomms2683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nawroth R, Poell G, Ranft A, Kloep S, Samulowitz U, Fachinger G, Golding M, Shima DT, Deutsch U, Vestweber D. Ve-ptp and ve-cadherin ectodomains interact to facilitate regulation of phosphorylation and cell contacts. EMBO J 2002; 21:4885-95; PMID:; http://dx.doi.org/ 10.1093/emboj/cdf497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Esser S, Lampugnani MG, Corada M, Dejana E, Risau W. Vascular endothelial growth factor induces ve-cadherin tyrosine phosphorylation in endothelial cells. J Cell Sci 1998; 111(Pt 13):1853-65; PMID: [DOI] [PubMed] [Google Scholar]
- 81.Bentley K, Franco CA, Philippides A, Blanco R, Dierkes M, Gebala V, Stanchi F, Jones M, Aspalter IM, Cagna G, et al. The role of differential ve-cadherin dynamics in cell rearrangement during angiogenesis. Nat Cell Biol 2014; 16:309-21; PMID:; http://dx.doi.org/ 10.1038/ncb2926 [DOI] [PubMed] [Google Scholar]
- 82.Broermann A, Winderlich M, Block H, Frye M, Rossaint J, Zarbock A, Cagna G, Linnepe R, Schulte D, Nottebaum AF, et al. Dissociation of ve-ptp from ve-cadherin is required for leukocyte extravasation and for vegf-induced vascular permeability in vivo. J Exp Med 2011; 208:2393-401; PMID:; http://dx.doi.org/ 10.1084/jem.20110525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Winderlich M, Keller L, Cagna G, Broermann A, Kamenyeva O, Kiefer F, Deutsch U, Nottebaum AF, Vestweber D. Ve-ptp controls blood vessel development by balancing tie-2 activity. J Cell Biol 2009; 185:657-71; PMID:; http://dx.doi.org/ 10.1083/jcb.200811159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.De Palma M, Naldini L. Angiopoietin-2 ties up macrophages in tumor angiogenesis. Clin Cancer Res 2011; 17:5226-32; PMID:; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-0171 [DOI] [PubMed] [Google Scholar]