

Abstract
The Salmonella type III secretory system secretes virulence proteins, called effectors. Effectors are responsible for the alteration of tight junctions (TJ) and epithelial functions in intestinal infection and inflammation. In a previous study, we have demonstrated that a bacterial effector AvrA plays a role in stabilizing TJs and balancing the opposing action of other bacterial effectors. However, the molecular mechanisms by which AvrA-modulates TJ protein expression remain unknown. AvrA possesses acetyltransferase activity toward specific mitogen-activated protein kinase kinases (MAPKKs) and potently inhibits the c-Jun N-terminal kinase (JNK) pathway in inflammation. Inhibition of the JNK pathway is known to inhibit the TJ protein disassemble. Therefore, we hypothesize that AvrA stabilizes intestinal epithelial TJs via c-Jun and JNK pathway blockage. Using both in vitro and in vivo models, we showed that AvrA targets the c-Jun and JNK pathway that in turn stabilizes TJ protein ZO-1. Inhibition of JNK abolished the effect of AvrA on ZO-1. We further determined that AvrA suppressed the transcription factor activator protein-1, which was regulated by activated JNK. Moreover, we identified the functional domain of AvrA that directly regulated TJs using a series of AvrA mutants. The role of AvrA represents a highly refined bacterial strategy that helps the bacteria survive in the host and dampens the inflammatory response of the host. Our findings have uncovered a novel role of the bacterial protein AvrA in suppressing the inflammatory response of the host through JNK-regulated blockage of epithelial cell barrier function.
Keywords: AvrA, c-Jun, epithelial cell, inflammation, intestine, JNK, permeability, Salmonella, tight junction, ZO-1
Abbreviations: JNK, c-jun N-terminal kinase; TTSS III, type III secretory system; TJ, tight junctions; MAPKKs, mitogen-activated protein kinase kinases; ZO, zonula occludens; IBD, inflammatory bowel diseases; CD, crohn's disease; TER, transepithelial resistance; CFU, colony-forming unit; FBS, fetal bovine serum; HBSS, Hank's balanced salt solutions; DMEM, Dulbecco's modified eagle's medium; PBS, phosphate buffered saline; AP-1, activator protein-1
Introduction
The intestinal epithelium cells play barrier, structural, and host defense roles.1-3 Epithelial cells are consistently exposed to bacteria, which play a key role in normal intestinal development and innate immunity.4-11 The tight junctions (TJs) seal the space between adjacent epithelial cells. TJs can be altered by various pathogens, as well as by their toxins.12-14 These effects may result from direct modification of TJ proteins, such as occludin, claudin, and ZO-1, or by alteration of the perijunctional actomyosin ring.14-16 Defective epithelial barrier function has been implicated in infectious and inflammatory diseases, such as inflammatory bowel diseases (IBD).17-23 Increased permeability is also present in a subset of unaffected first-degree relatives of patients with Crohn's disease.24
The type-3-secretion system (TTSS) is a protein transport device used by Gram-negative pathogenic bacteria, such as Salmonella. TTSS allows bacteria to inject virulent proteins through a molecular needle system into the eukaryotic host cells.25 These virulent factors will paralyze or reprogram the eukaryotic cell to the benefit of the pathogen. Bacterial proteins display a large repertoire of biochemical activities and modulate the function of crucial host regulatory molecules, including TJ proteins and their up-stream regulators.26-28
AvrA is a bacterial effector that stabilizes cell permeability and TJs in intestinal epithelial cells.29 Unlike Salmonella effector SopB and SopD, AvrA does not increase physiologic fluid secretion into infected calf ileal loops.30,31 In a previous study, we demonstrated that AvrA plays a role in stabilizing TJs and balancing the opposing action of other bacterial effectors that decreased TJs expression.29 However, the molecular mechanism of AvrA in increasing TJ protein expression remains unknown. AvrA possesses enzyme activity toward specific mitogen-activated protein kinase kinases (MAPKKs) and potently inhibits the c-Jun N-terminal kinase (JNK) pathway.32-34 Inhibition of the JNK pathway suppresses the disassembly of TJ proteins.35 Therefore, we hypothesize that AvrA stabilizes intestinal epithelial TJs via JNK pathway blockage.
To determine the molecular mechanism by which AvrA modulates the TJ protein expression and distribution, we focused on bacterial strains sufficient or deficient in AvrA–parental PhoPc, PhoPc AvrA mutant (AvrA−) or the AvrA complementary strain (PhoPc AvrA−/AvrA+). PhoPc is a PhoP-PhoQ constitutive mutation of a wild-type Salmonella Typhimurium strain 14028s that increases the expression of PhoP-activated genes.36 Reed et al. showed that PhoPc has similar adherence ability as the WT 14028s Salmonella and is less invasive than the WT Salmonella using the MDCK and T84 cell models.37 We also used a wild-type SL1344 strain with AvrA gene expression and its AvrA knockout mutant (SL1344 AvrA−). Our previous study demonstrated that PhoPc is able to inhibit the activation of the proinflammatory NF-κB pathway. Further study showed that AvrA expression in PhoPc plays an important role in attenuating the inflammatory responses.38-40 In the current study, we continue to use this Salmonella-epithelial interaction system. We have tested Salmonella Typhimurium with AvrA sufficient or deficient expression in a cultured polarized human epithelial cell model, an animal model, and in AvrA-transfected cells. We demonstrated that AvrA-deficient Salmonella decreased TJ protein expression in both cultured colonic epithelial cell and bacterial infected mouse models. A JNK inhibitor was able to rescue Salmonella-AvrA− strain induced-TJ dissemble and degradation. We assessed TJ protein expression at the protein level, as well as TJ protein distribution in both AvrA-deficient and -sufficient bacterial strains infected cellular models (in vitro) and animal model (in vivo). Our findings suggest an important role of bacterial AvrA in regulating the structure and function of TJs in intestinal infection and inflammation.
Results
AvrA expression increases TJ ZO-1 but not α-catenin at the protein level in colon using a bacterial infected animal model
First, we determined whether bacteria modulate TJ expression in streptomycin-pretreated C57BL/6 mice colonized with different bacterial strains. Streptomycin treatment is known to diminish the intestinal flora and to render mice susceptible to intestinal colonization by various microorganisms. In previous studies, we have established the streptomycin-pretreated Salmonella-colitis mice in order to understand the host-pathogen interactions.41,42 Post infection 8 hours is correlated with the early stage of in vitro bacterial colonization. As shown in Figure 1A, TJ ZO-1 protein was significantly increased by wild-type S. Typhimurium SL1344 (AvrA+) 8 hours post-infection, the early stage of infection in vivo (Fig. 1A and B), whereas AvrA mutant strain SL1344 (AvrA−) infection significantly reduced ZO-1 expression in the colon (Fig.1A and B). We also examined the adherens junction protein α-catenin in Salmonella infected colon and found that it was not changed. In addition, we found the ZO-1 mRNA level was not changed by Salmonella infection in vivo (Fig. 1C), indicating that the AvrA regulation of ZO-1 was not at the transcriptional level.
Figure 1.
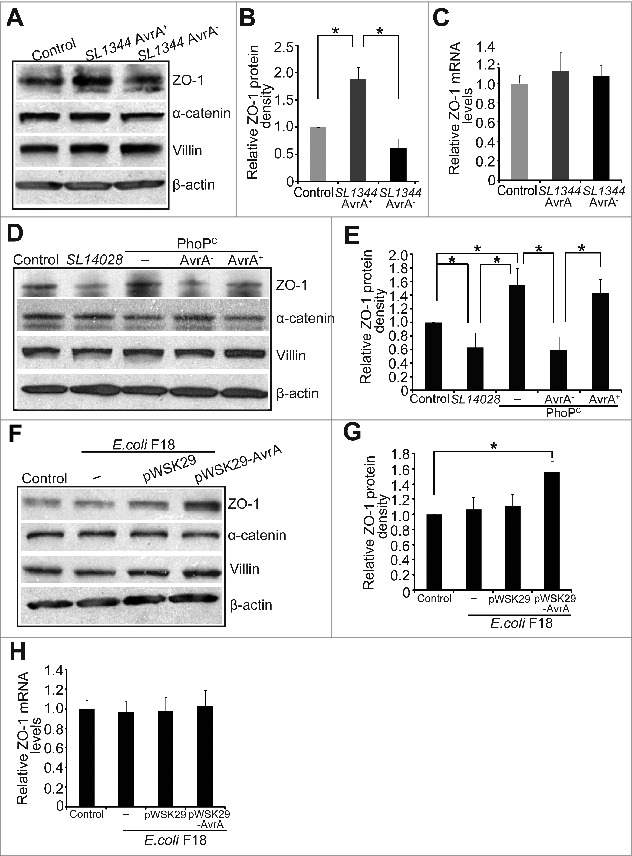
AvrA expression increases ZO-1 but not α-catenin in colon using a bacterial infected animal model (A) Western blot of ZO-1 and α-catenin in colon after mouse infected with Salmonella SL1344 AvrA+ or SL1344 AvrA−. (B) Densitometry of ZO-1. Relative band intensity was determined using NIH Image 1.63 software. ZO-1 expression significantly increased in the SL1344 AvrA+ group compared to the control and SL1344 AvrA− groups in absence of AvrA protein. * P< 0.05. Data are reported as mean ± SD of 3 independent experiments. (C) ZO-1 mRNA level was not changed by Salmonella infection in vivo. (D) Western blot of ZO-1 and α-catenin in colon after mouse infected with Salmonella SL14028/PhoPc, PhoPc AvrA−, PhoPc AvrA−/AvrA+. (E) Densitometry of ZO-1 protein band. * P< 0.05. Data are reported as mean ± SD of 3 independent experiments. (F) Western blot of ZO-1 and α-catenin in colon after germ-free mice were mono-associated with E. coli F18 or E. coli F18 AvrA+. (G) Densitometry of ZO-1. E. coli F18 AvrA+ increased ZO-1 expression in vivo. *P< 0.05. Data are reported as mean ± SD of 3 independent experiments. (H) The mRNA level of ZO-1 in colon by real-time PCR. Mice were infected with Salmonella for 8 hours and intestinal epithelial cells were harvested for immunoblot and Real-time PCR.
We then determined whether bacterial strain complemented with AvrA could increase the level of ZO-1 in the colon. We chose bacterial strains sufficient or deficient in AvrA—parental PhoPc, PhoPc AvrA mutant (AvrA−) or the AvrA complementary strain (PhoPc AvrA−/AvrA+). PhoPc is a PhoP-PhoQ constitutive mutation of a WT Salmonella Typhimurium strain 14028s that increases the expression of PhoP-activated genes.36 WT Salmonella Typhimurium strain 14028s has low AvrA expression.32,40 It is not surprising that 14028s infection did not increase ZO-1 expression in the colon. PhoPc with AvrA expression and the AvrA complementary strain (PhoPc AvrA−/AvrA+) both significantly enhanced ZO-1. In contrast, PhoPc AvrA mutant (AvrA−) significantly reduced ZO-1 expression (Fig. 1D and E).
E. coli F18 is a human commensal bacterial strain.43 We further tested the effect of AvrA expression in the E. coli F18 on ZO-1 using previously germ-free mice mono-associated with E. coli F18 or E. coli F18 AvrA.44 We found that E. coli F18 expressed AvrA was able to increase ZO-1, but not α-catenin at the protein level in these colons (Fig. 1F and G). Moreover, we found that this regulation was not through the mRNA level of ZO-1, evidenced by the real-time PCR data in Figure 1H. Thus, our in vivo data strongly suggest that bacterial effector protein AvrA up-regulates TJ protein ZO-1.
Distribution of ZO-1 protein in Salmonella infected intestine in vivo
ZO-1 is a TJ protein on the surface of intestinal epithelial cells. The immunostaining data showed ZO-1 located on a cytoplasmic membrane surface of intercellular TJs on the top of the intestinal crypts in AvrA sufficient strain-infected WT intestine, but ZO-1 became disorganized on the AvrA deficient strain-infected intestinal epithelial surface (Fig 2A). In Figure 2B, high magnitude images further show that green staining of ZO-1 protein was disorganized in the colonic epithelial cells infected with AvrA− bacterial strain (yellow arrow). These results indicate that the level of bacterial expressed AvrA regulates the distribution of ZO-1 in the intestine.
Figure 2.
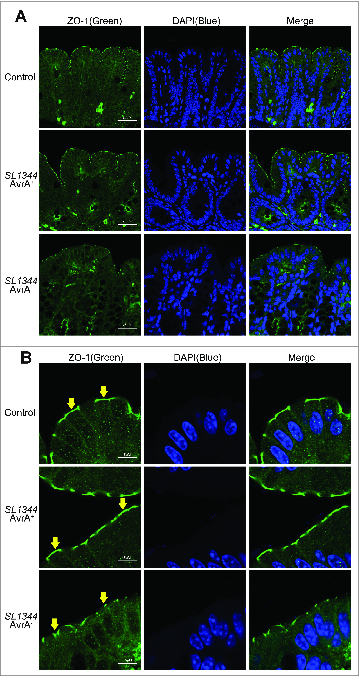
Immunostaining of ZO-1 in vivo. Immunostaining on colonic epithelial cells was performed 8 hours after mouse infection with Salmonella SL1344 AvrA+ or SL1344 AvrA−. Tissues were fixed, permeabilized, and stained with ZO-1 antibodies, followed by Alexa Fluor 488 goat-anti-mouse secondary antibody. (A) AvrA− infected mice display disruption of the TJ structure. (B) Arrows in Panel show the green staining of ZO-1 protein on the top of the intestinal crypts. Please note disorganized structure of ZO-1 in the colonic epithelial cells infected with AvrA− bacterial strain (yellow arrow). Images shown are from a single experiment and are representative of 3 separate experiments. n = 3 animals in each experimental group.
AvrA expression regulates p-JNK (including p-JNK1 and p-JNK2) in vivo and in vitro
ZO-1 is known to be regulated by the JNK signaling pathway. We further hypothesized that the AvrA enhanced ZO-1 through the JNK signaling pathway. We assessed whether the ZO-1 level was correlated with the amount of phosphorylated JNK (p-JNK) and total JNK that in the colon infected by the AvrA sufficient or deficient Salmonella. As shown in Fig. 3A, infection of AvrA sufficient Salmonella did not change ZO-1 and reduced p-JNK (Lane 2), whereas AvrA deficient Salmonella reduced ZO-1 and elevated p-JNK (Lane 3), in the mouse intestinal epithelial cells in vivo. Using the human intestinal epithelial Caco2 BBE cell line, we also detected the correlation between increased p-JNK and reduced ZO-1 regulated by AvrA (Fig. 3B): PhoPc with AvrA expression (Lane 3), the AvrA complementary strain (PhoPc AvrA−/AvrA+) (Lane 5), and SL1344 AvrA+ (Lane 6) all significantly enhanced ZO-1 and reduced p-JNK. In contrast, 14028s with low AvrA expression (Lane 2), PhoPc AvrA mutant (AvrA−) (Lane 4) and SL1344 AvrA− (Lane 7) significantly reduced ZO-1 expression and increased p-JNK. There was no altered total JNK 1 and 2 in cells with or without Salmonella treatment. Moreover, the HT29C19A cells showed a trend similar to the Caco2 BBE cells (Fig. 3C). Both in vivo and in vitro data indicate that AvrA deficiency leads to down-regulation of ZO-1 and this effect is associated with significantly increased JNK phosphorylation in intestinal epithelial cells.
Figure 3.
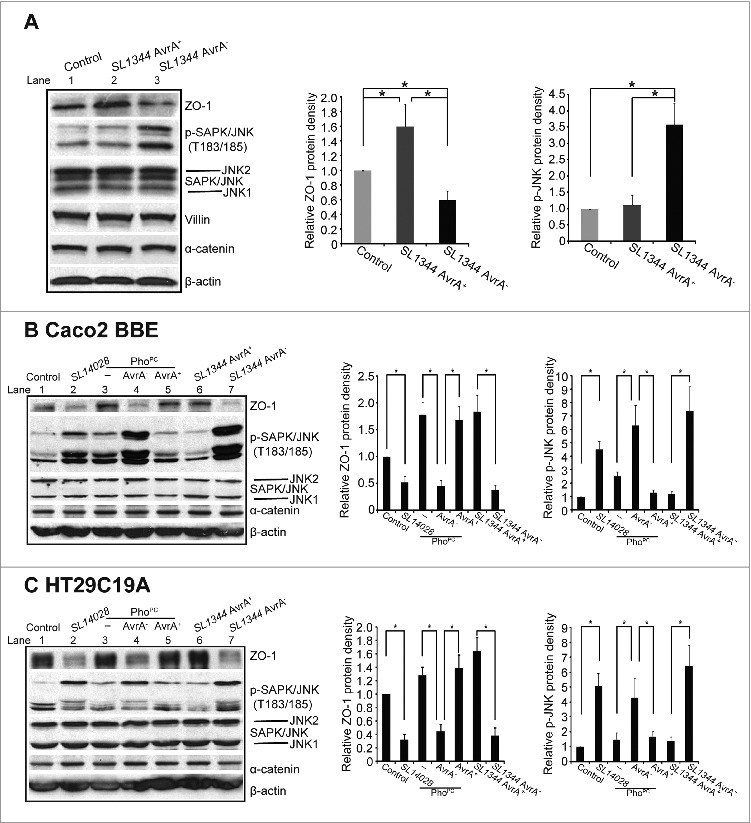
AvrA regulated the protein expressions of ZO-1 through JNK pathway. (A) Western blot of ZO-1, p-JNK, JNK, and α-catenin in vivo after mouse infected with Salmonella SL1344 AvrA+/SL1344 AvrA−. *P< 0.05. n = 3 separate experiments. (B) Western blot of ZO-1, p-JNK, JNK, and α-catenin in vitro. Polarized human colonic epithelial Caco2 BBE cells were colonized with AvrA deficient or sufficient bacterial strains for 30 minutes, washed with HBSS and incubated in DMEM for 30 minutes. Equal volumes of total cell lysate were processed for immunoblotting. (C) Western blot of ZO-1, p-JNK, JNK, and α-catenin in HT29C19A cells colonized with AvrA deficient or sufficient bacterial strains. Results are representative of 3 independent experiments. *P < 0.05. n = 3 separate experiments.
AvrA preserves intestinal barrier function in cultured epithelial cells and a salmonella-colitis animal model
We measured the transepithelial resistance (TER) of cultured epithelial cells to determine whether AvrA expression was associated with preserved permeability. In our study, alteration of TER occurred within the initial 6 hours. As shown in Figure 4A, the TER values of cultured epithelial Caco2 BBE cells from the control group remained relatively stable over the 30 to 300 minute incubation period. There was a decrease in TER after Salmonella colonization. Cells colonized with the AvrA− deficient bacterial strain had the lowest TER compared to the control and AvrA+ groups. Multiple comparisons of mean TER were performed using Two-Way ANOVA. There was significant difference between the AvrA− and AvrA+ groups.
Figure 4.
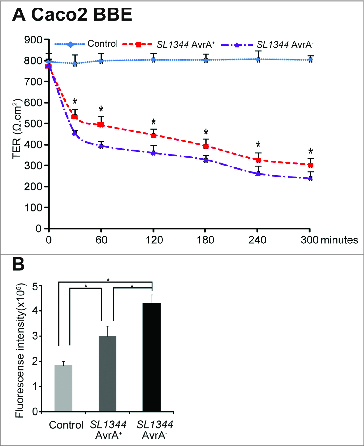
AvrA regulated permeability in the colonic epithelial cells. (A) Salmonella with AvrA preserved permeability in human colonic epithelial cells. Caco2 BBE cells were colonized with Salmonella SL1344 AvrA+ or SL1344 AvrA− for 30 minutes and then incubated for indicated times. Multiple comparisons of mean TER were performed using Two-Way ANOVA. The interested comparison was mean differences between SL1344 AvrA− and AvrA+, the tested p-values were provided for all follow-up measurements at: 30, 60, 120, 160, 240, and 300 minutes with * indicating all time points. *P< 0.05. for SL1344 AvrA+ vs SL1344 AvrA−. n = 3 separate experiments. (B) Permeability of the intestine in vivo. n = 5 mice per group. *P < 0.05 for control vs SL1344 AvrA+, control vs SL1344 AvrA−, and SL1344 AvrA+ vs SL1344 AvrA− after infection for 8 hours.
To determine whether AvrA expression was associated with a preserved intestinal barrier function, paracellular intestinal permeability was assessed using oral administration of the fluorescent tracer FITC-dextran (3 kDa) to all the mice 8 hours post Salmonella infection. The concentrations of fluorescent FITC-dextran in the blood were then measured as a reflection of intestinal permeability in vivo. As we expected, the mice infected with AvrA sufficient Salmonella had lower levels of FITC-dextran compared to the mice infected with AvrA deficient Salmonella (Fig. 4B).
Inhibition of JNK abolished the effect of AvrA on ZO-1
We then treated the cells with a JNK inhibitor to examine whether Inhibition of JNK abolished the effect of AvrA on ZO-1. As shown in Figure 5A, AvrA sufficient Salmonella infection (Lane 2) did not change ZO-1 and p-JNK compared with control without any treatment (Lane1), whereas AvrA deficient Salmonella (Lane 3) significantly reduced ZO-1 and elevated p-JNK in the Salmonella infected Caco2 BBE cells. SP600125 treatment in Lane 5 abolished the effect of AvrA on p-JNK and ZO-1. Thus, no difference was shown between AvrA sufficient Salmonella (Lane 5) and AvrA deficient Salmonella infection (Lane 6). Using the AvrA plasmid transfected in the Caco2 BBE cell line, we also detected the correlation between increased p-JNK and reduced ZO-1 regulated by AvrA (Fig. 5B): TNF-α treatment (Lane 5–7) induced p-JNK. It was found that AvrA suppressed the TNF-α induced p-JNK, thus enhancing the ZO-1 expression (Fig. 5B Lane 8). The transcription factor AP-1 (activator protein-1) was regulated by JNK.45,46 Using a luciferase assay, we further determined that the AP-1 transcriptional activity was significantly suppressed by AvrA sufficient Salmonella, compared to that in cells treated with the AvrA deficient Salmonella (Fig. 5C), In contrast, the AP-1 activity was significantly increased by the AvrA deficient Salmonella, compared to the control without treatment (Fig. 5C).
Figure 5.
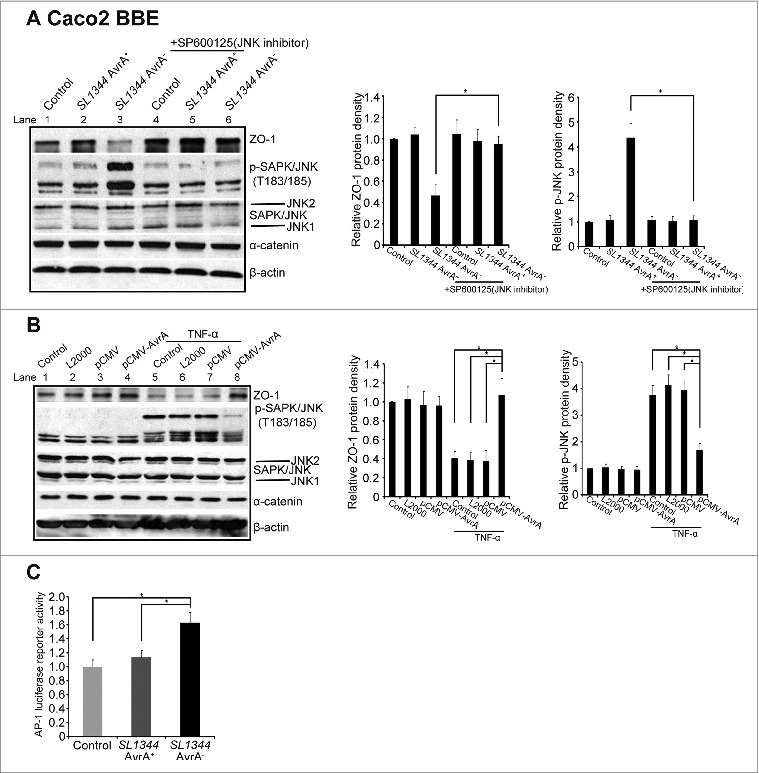
AvrA regulated ZO-1 expression is dependent on the SAPK/JNK pathway. (A) The effect of AvrA on ZO-1 expression was abolished by the expression induced by the SAPK/JNK inhibitor (SP600125) in Caco2 BBE cells. Cells were treated with SP600125 (50μM) for 1 hour, followed by colonization of Salmonella (SP600125) for 30 minutes and incubation for 30 minutes (SP600125). *P< 0.05. Data are reported as mean ± SD of 3 independent experiments. (B) Transfected Salmonella AvrA protein was able to modulate ZO-1 expression and block the TNF-α induced p-JNK. Caco2 BBE cells were transfected with pCMV-AvrA plasmids for 24 hours and then treated with TNF-α (10ng/ml) for 30 minutes. *P< 0.05. Data are reported as mean ± SD of 3 independent experiments. (C). AP-1 transcriptional activity by luciferase assay in Caco2 BBE cells colonized with Salmonella SL1344 AvrA+ or SL1344 AvrA−. Data are expressed as mean ± SD. *P< 0.05. n = 3 separate experiments.
AvrA mutations and their effects on ZO-1 and JNK activity in vitro
AvrA is injected into the intestinal epithelial cells by bacteria. We then explored the mechanism by which AvrA interacts with the host. We have generated a series of AvrA mutations in the current study (Fig. 6A) and transfected the Caco2 BBE cells with pCMV plasmids containing either WT AvrA or AvrA truncated mutants. Whereas WT AvrA expression (Lane 4) increased the expression of the TJ protein ZO-1 and suppressed p-JNK and the truncated mutation A3 and A4 (Lane 5/6) showed a trend similar to the WT AvrA, the AvrA truncated mutation A3 and A4 (Lane 7/8) abolished the effect of WT AvrA on TJ proteins and JNK activity in vitro (Fig. 6B). The ZO-1 level was significantly reduced and the p-JNK was significantly increased in the cells transfected with A3 and A4 (Fig. 6C/D). Thus, we identified the functional domain C-terminal of AvrA in regulating TJs.
Figure 6.
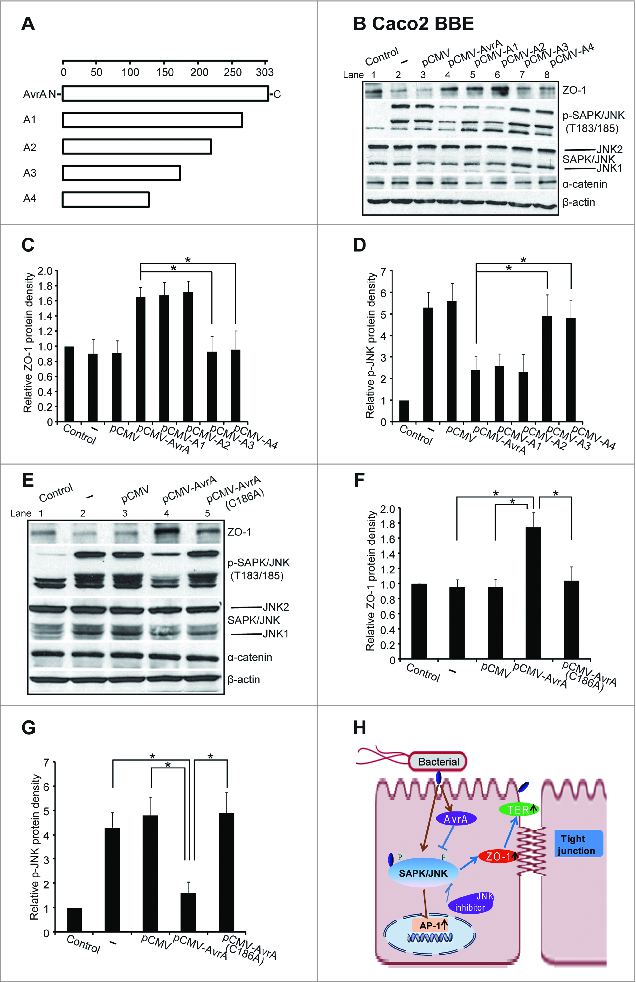
The functional domain of AvrA in regulated ZO-1 in vitro. (A) Diagrams of AvrA constructs used in this study. A series of the C-terminal of AvrA truncated mutation plasmids were constructed. (B–D) Caco2 BBE cells were transfected with pCMV-myc-AvrA wild-type construct, a series deletion of the C-terminal of AvrA gene constructs, or control empty pCMV-myc plasmid. Deletion of the C-terminal AvrA pCMV-A3/pCMV-A4 construct did not increase ZO-1 expression. It indicates that deletion C-terminal AvrA (>88 amino acid) abolished the effects of wild-type AvrA on TJ protein expression. *P< 0.05. Data are reported as mean ± SD of 2–3 independent experiments. (E–G) Caco2 BBE cells were transfected with a pCMV-myc-AvrA wild-type gene construct, a pCMV-myc-AvrAC186A AvrA mutant construct, or control empty pCMV-myc plasmid. The AvrA mutant C186A is a single amino acid residue transition which is mutated at the key cysteine required for AvrA's activity. AvrA mutant C186A expression did not increase ZO-1 expression. It indicates that cysteine mutation abolished the effects of wild-type AvrA on TJ protein expression. *P< 0.05. Data are reported as mean ± SD of 2–3 independent experiments. (H) A diagram indicates that AvrA stabilizes ZO-1 expression dependent on the SAPK/JNK pathway.
Based on the sequence alignment of representative AvrA members including the adenovirus-like proteases (human adenovirus type 2, fowl adenovirus 8, Hemorrhagic enteritis virus), as well as Yersinia virulence factor YopJ, and Xanthomonas campestris pv. vesicatoria (AvrBsT), the catalytic triad for the cysteine protease presents in all AvrA family members. These residues include Cys186. We transfected a pCMV plasmid with wild-type (WT) AvrA or AvrA Cys186 point mutant (C186A) in the human epithelial cells. We found that the single mutation of Cys186 was able to abolish AvrA-induced upregulation of ZO-1 and blockage of the JNK activity (Fig. 6E, Lane 5), whereas WT AvrA (Lane 4) significantly enhanced ZO-1 and suppressed p-JNK (Fig. 6F, 6G). The adherens junction protein α-catenin was not changed by WT AvrA or AvrA C186A mutant.
Discussion
In the current study, we have demonstrated that the bacterial effector protein AvrA stabilizes the expression and distribution of TJ protein ZO-1 through inhibition of the JNK pathway in vitro and in vivo. Inhibition of JNK abolished the effect of AvrA on ZO-1. We further determined that AvrA expression suppressed the transcription factor activator protein-1, which was regulated by JNK activity. Moreover, we identified the functional domain of AvrA in regulating TJs using a series of AvrA mutants. Our data on AvrA stabilization of TJs further suggest a distinguishing role of a bacterial effector in regulating host responses in the intestine.
AvrA attenuates the proinflammatory activities of JNK and NF-κB34,41,47, activates the β-catenin transcription factor38,39,42, inhibits cell apoptosis in mouse epithelial cells41 and activates p53 signaling.40 In contrast, other bacterial effectors do the opposite. Salmonella effectors, such as SopB, SopE, SopE2, are know to activate the proinflammatory response by directly stimulating proinflammatory signaling events in host cells.48-51 Many proteins coordinate distinct signaling pathways in the cells by having multiple functions. Bacterial effectors may have multiple protease activities to modify different eukaryotic proteins and maximize the bacterial effector's ability to modulate cellular functions. As a multifunctional protease, AvrA targets multiple key proteins in the host cells and uses different strategies to manipulate the signaling transduction pathways that utilize ubiquitin38,39,42 and acetylation40, including JNK, NF-κB46, β-catenin38,39,42, and p53.40 Therefore, AvrA may function as an anti-inflammatory protein to stabilize TJs, prevent cell death, and help the bacteria survive in the host. Characterization of the synergistic functions of AvrA in regulating multiple pathways will give new clues concerning microbial–host interaction in inflammation.
Our data on AvrA point and truncation mutants indicate that the key 186 amino acid cysteine is required for AvrA regulation of TJ expression. AvrA acts as a deubiquitinase to inhibit the degradation of inflammatory regulators IκBα and β-catenin.41 However, it is not clear whether AvrA regulates these TJ proteins through phosphorylation or through ubiquitination. The half-life of the TJ proteins is very long, such as 9-12 hours for claudin.52 Therefore, protein synthesis inhibitor is not a good choice to study the role of AvrA in regulating the stabilization of TJ proteins.
The intestinal barrier is a complex environment exposed to dietary components and commensal bacteria. Intestinal TJs seal the paracellular space between epithelial cells and play a critical role in regulating barrier function and preventing diffusion of microorganisms and other antigens across the epithelium. TJs are highly dynamic structures that are constantly being remodeled due to interactions with external stimuli, such as pathogenic and commensal bacteria. Commensal bacteria and probiotics have been shown to promote intestinal barrier integrity both in vitro and in vivo, through JNK, Ark or Rho GTPase signaling pathways.27,53,54 Based on our data, AvrA likely targets the expression of ZO-1 through blocking the JNK pathway. In theory, p-JNK should be activated first, then AP-1 is activated.45 Our transcriptional data of AP-1 indicates that blockage of JNK leads to suppressed AP-1 activity. Our previous study indicates that AvrA also targets the expression of occludin. Occludin is a functional target of the E3 ligase Itch.55 It is unclear whether Rho GTPase is influenced by the AvrA expression. The mechanisms through which occludin expression and distribution are altered have yet to be elucidated.
In summary, our current study on AvrA and JNK provides a mechanism by which bacterial protein blocks the JNK activation, thus preventing TJ protein dis-assemble and preserving intestinal epithelial permeability (Fig. 6H). Inhibition of JNK by an inhibitor could abolish the effect of bacterial AvrA on ZO-1. Our findings indicate an important role of bacterial protein in microbial-epithelial interactions in regulating the structure and function of TJs during infection and inflammation. Manipulation of JNK activity could be a novel therapeutic approach to treat infectious diseases.
Materials and Methods
Ethics statement
C57BL/6 mice (Female, 6-7 weeks) were obtained from the Jackson Laboratory (Jackson Laboratory, Bar Harbor, ME, USA). GF 129/SvEv mice were obtained from the Center for Gastrointestinal Biology and Disease Gnotobiotic Core Facility and the National Gnotobiotic Rodent Resource Center (University of North Carolina, Chapel Hill, NC, USA).44 All animal work was approved by the Rush University Committee on Animal Resources. Euthanasia method was sodium pentobarbital (100 mg per kg body weight) I.P. followed by cervical dislocation.
Bacterial strains and growth condition
Bacteria strains included wild-type (WT) S. Typhimurium ATCC 14028; S. Typhimurium PhoPC, a derivative of wild-type Salmonella SL14028 with AvrA gene and protein expression; Salmonella PhoPC mutant strain lacking the AvrA gene (PhoPC AvrA−); PhoPC AvrA- transcomplemented with a plasmid encoding WT AvrA (PhoPC AvrA−/AvrA+); wild-type S. Typhimurium SL1344 (AvrA+) and its AvrA mutant strain SL1344 (AvrA−); Escherichia coli F18 (a flagellated nonpathogenic strain), F18 AvrA+ was generated by transforming with the pWSK29-AvrA plasmid (Table 1). Bacterial growth conditions were as follows: non-agitated microaerophilic bacterial cultures were prepared by inoculating 10 ml of Luria-Bertani broth with 0.01 ml of a stationary phase culture followed by overnight incubation (∼18 h) at 37°C, as previously described.41
Table 1.
PCR Primers for construct truncation AvrA gene plasmids
| Primers name | Sequence |
|---|---|
| ACΔF | 5’ -CGGAATTCAGATGATATTTTCGGTG- 3’ |
| ACΔR1 | 5’ -GGGGTACCTCATATACTGCTTCC- 3’ |
| ACΔR2 | 5’ -GGGGTACCTCAACGTTCACAAAT- 3’ |
| ACΔR3 | 5’ -GGGGTACCTCAATAACAATCAGG- 3’ |
| ACΔR4 | 5’ -GGGGTACCTCAGACGACTGAAAT- 3’ |
Cell culture
Human epithelial Caco2 BBE and HT29C19A cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS), streptomycin-penicillin and L-glutamine. Monolayers of Caco2 BBE and HT29C19A cells were grown on permeable supports (0.33 or 4.67 cm2, 0.4 mm pore. Costar, Cambridge, MA, USA).
Streptomycin pre-treated mouse model
Water and food were withdrawn 4 h before oral gavage with 7.5 mg/mouse of streptomycin (75 μl of sterile solution or 75 μl of sterile water in control). Afterward, animals were supplied with water and food ad libitum. Twenty hours after streptomycin treatment, water and food were withdrawn again for 4 hours before the mice were infected with 1×107 CFU of S. typhimurium (100 μl suspension in HBSS) or treated with sterile HBSS (control) by oral gavage as previously described. At the indicated times post-infection, mice were sacrificed, and tissue samples from the intestinal tracts were removed for analysis.15,29
Bacterial colonization in the polarized epithelial cells in vitro
Polarized human epithelial cells were colonized with equal numbers of the indicated bacteria for 30 min, washed with Hank's balanced salt solution (HBSS), and incubated in Dulbecco's modified eagle's medium (DMEM) containing gentamicin (500 mg/ml) for the times indicated in our previous study.40 The first 30-minute incubation allowed bacteria to contact the surface of the epithelial cells and inject the effectors in the host cells. After extensive HBSS washing, the extracellular bacteria were washed away. Incubation with gentamicin inhibited the growth of bacteria.
AvrA WT and mutant plasmids transfection
Caco2 BBE cells were grown in 12-well plates. At 70–80% confluence, cells were transfected with a pCMV-myc-AvrA wild-type gene construct, a pCMV-myc-AvrA mutant construct, or control empty pCMV-myc plasmid using LipofectAMINE (Invitrogen, Grand Island, NY, USA)).
A series of plasmids with C-terminal truncation mutants of AvrA gene were constructed. The C-terminal truncation mutants of AvrA were generated by PCR amplification. A single forward primer was used, which includes the AvrA native ribosome binding site as well as an EcoR I site at the 5′ end. The reverse primers were designed to be complementary to the sequences encoding different regions of the AvrA C-terminus and also included a stop codon as well as a Kpn I site at the 5′ end. All PCR primers were designed using Lasergene software (Table 2). The PCR products were cleaved with EcoR I/Kpn I and subcloned into pCMV plasmids.
Table 2.
Salmonella strains used in this study
| Name | Description |
|---|---|
| SL14028 | Wild-type Salmonella |
| PhoPc | Derivative of SL14028 with AvrA expression |
| PhoPc AvrA− | AvrA− mutation derived from PhoPc |
| PhoPc AvrA+ | PhoPc AvrA+ with complemented plasmid encoding AvrA |
| SL1344 AvrA+ | Salmonella 1344 strain with AvrA expression |
| SL1344 AvrA− | Salmonella 1344 strain with AvrA deletion |
| E. coli F18 | Wild-type E. coli |
| E. coli F18 AvrA+ | E. coli F18 strain with transformed plasmid encoding AvrA |
The AvrA mutant C186A is a single amino acid residue transition which is mutated at the key cysteine required for AvrA's activity as previously described41,47 24 h after transfection, cells were lysed in protein loading buffer (50 mM Tris, pH 6.8, 100 mM dithiothreitol, 2% SDS, 0.1% bromophenol blue, 10% glycerol). Equal volumes of total cell lysate were separated by SDS-PAGE, transferred to nitrocellulose, and processed for immunoblotting.
Immunoblotting
Mouse epithelial cells were scraped and lysed in lysis buffer (1% Triton X-100, 150 mM NaCl, 10 mM Tris pH 7.4, 1 mM EDTA, 1 mM EGTA pH 8.0, 0.2 mM sodium orthovanadate, protease inhibitor cocktail), and then the protein concentration was measured.39 Cultured cells were rinsed twice in ice-cold HBSS, lysed in protein loading buffer (50 mM Tris, pH 6.8, 100 mM dithiothreitol, 2% SDS, 0.1% bromophenol blue, 10% glycerol), and then sonicated. Equal amounts of protein were separated by SDS-polyacrylamide gel electrophoresis, transferred to nitrocellulose, and immunoblotted with primary antibodies. The following antibodies were used: anti-ZO-1, anti-α-catenin (Invitrogen, Grand Island, NY, USA); anti-p-SAPK/JNK, anti-SAPK/JNK (Cell Signal, Beverly, MA, USA); anti-Villin (Santa Cruz Biotechnology Inc.., Santa Cruz, CA, USA.); or anti-β-actin (Sigma-Aldrich, Milwaukee, WI, USA.) antibodies and were visualized by ECL (Thermo Scientific, Rockford, IL, USA). Membranes that were probed with more than one antibody were stripped before re-probing.
Immunofluorescence
Colonic tissues from the colon were freshly isolated and embedded in paraffin wax after fixation with 10% neutral buffered formalin. Immunofluorescence was performed on paraffin-embedded sections (4 μm). After preparation of the slides as previously described, slides were incubated for 1 hour in blocking solution (2% bovine serum albumin, 1% goat serum in HBSS) to reduce nonspecific background. The tissue samples were incubated overnight with primary antibodies at 4°C. Samples were then incubated with secondary antibodies for 1 hour at room temperature. Tissues were mounted with SlowFade Antifade Kit (Life technologies, s2828, Grand Island, NY, USA), followed by a coverslip, and the edges were sealed to prevent drying. Specimens were examined with Zeiss laser scanning microscope 710 (Carl Zeiss Inc.., Oberkochen, Germany)
Analysis of ZO-1 distribution
Fluorescence images were analyzed using image analysis software (LSM 710 META, version 4.2; Carl Zeiss Inc.., Oberkochen, Germany). Each analysis was performed in triplicate from each tissue section on a total of 10 images per mouse sample (n = 5).
Treatment with JNK inhibitor SP600125
JNK inhibitor SP600125 (50 μM, EMD Biosciences, San Diego, CA, USA) was added directly to the culture medium one hour before Salmonella treatment. Caco2 BBE cells (with SP600125 pretreatment) were incubated with Salmonella (SP600125 50 μM) 30 minutes, washed 3 times in HBSS and incubated in HBSS (SP600125 50 μM) for 30 minutes, then harvested. Levels of indicated proteins were determined by Western blotting as described above.32
AP-1 transcriptional activity
Caco2 BBE cells were co-transfected with AP-1-luciferase reporter and pRL-TK plasmids using Lipofectamine 2000 (Invitrogen, San Diego, CA, USA) in accordance with the manufacturer's instructions. After a 24-hour transfection period, the cells were lysed and luciferase activity was determined using the Dual Luciferase Reporter Assay System (Promega, Madison, WI, USA) with a TD-20/20 luminometer (Turner Designs, Sunnyvale, CA, USA). Firefly luciferase activity was normalized to Renilla luminescence activity and the activity was presented as relative units.
Real-time quantitative PCR analysis
Total RNA was extracted from mouse epithelial cells or cultured cells using TRIzol reagent (Invitrogen, Grand Island, NY, USA). The RNA integrity was verified by electrophoresis. RNA reverse transcription was performed using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA) according to the manufacturer's protocol. The RT cDNA reaction products were subjected to quantitative real-time PCR using CTFX 96 Real-time system (Bio-Rad, Hercules, CA, USA) and SYBR green supermix (Bio-Rad, Hercules, CA, USA) according to the manufacturer's protocol. All expression levels were normalized to β-actin levels of the same sample. Percent expression was calculated as the ratio of the normalized value of each sample to that of the corresponding untreated control cells. All real-time PCR reactions were performed in triplicate.
Statistical Analysis
All data are expressed as mean ± SD. All statistical tests were 2-sided. P values of less than 0.05 were considered to be statistically significant. Differences between 2 samples were analyzed by Student's t-test. Multiple comparisons of mean TER in Figure 4A were performed using Two-Way ANOVA. Since we are interested in comparison of mean differences between SL1344 AvrA− and AvrA+, the group effects were tested at 0, 30, 60, 120, 160, 240, and 300 minutes. For fluorescence intensity data in Figure 4B, One-Way ANOVA was conducted to determine the mean differences among the control, AvrA− and AvrA+ groups. Statistical analyses were performed using SAS version 9.3 (SAS Institute, Inc.., Cary, NC, USA).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the NIDDK (KO1 DK075386 and 1R03DK089010-01), the American Cancer Society (RSG-09-075-01-MBC), and Swim Across America Cancer Research Award to JS and 1R01HL113640 to AX.
References
- 1.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol 2009; 9:799-809; PMID:; http://dx.doi.org/ 10.1038/nri2653 [DOI] [PubMed] [Google Scholar]
- 2.Abreu MT. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol 2010; 10:131-44; PMID:; http://dx.doi.org/ 10.1038/nri2707 [DOI] [PubMed] [Google Scholar]
- 3.Robinson A, Keely S, Karhausen J, Gerich ME, Furuta GT, Colgan SP. Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology 2008; 134:145-55; PMID:; http://dx.doi.org/ 10.1053/j.gastro.2007.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mshvildadze M, Neu J. The infant intestinal microbiome: Friend or foe? Early Hum Dev 2010; 86(Suppl 1):67-71; PMID:; http://dx.doi.org/ 10.1016/j.earlhumdev.2010.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hooper LV. Do symbiotic bacteria subvert host immunity? Nat Rev Microbiol 2009; 7:367-74; PMID:; http://dx.doi.org/ 10.1038/nrmicro2114 [DOI] [PubMed] [Google Scholar]
- 6.Hooper LV, Midtvedt T, Gordon JI. How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu Rev Nutr 2002; 22:283-307; PMID:; http://dx.doi.org/ 10.1146/annurev.nutr.22.011602.092259 [DOI] [PubMed] [Google Scholar]
- 7.Bates JM, Akerlund J, Mittge E, Guillemin K. Intestinal alkaline phosphatase detoxifies lipopolysaccharide and prevents inflammation in zebrafish in response to the gut microbiota. Cell Host Microbe 2007; 2:371-82; PMID:; http://dx.doi.org/ 10.1016/j.chom.2007.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Preidis GA, Versalovic J. Targeting the human microbiome with antibiotics, probiotics, and prebiotics: gastroenterology enters the metagenomics era. Gastroenterology 2009; 136:2015-31; PMID: ; http://dx.doi.org/ 10.1053/j.gastro.2009.01.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Camp JG, Kanther M, Semova I, Rawls JF. Patterns and scales in gastrointestinal microbial ecology. Gastroenterology 2009; 136:1989-2002; PMID:; http://dx.doi.org/ 10.1053/j.gastro.2009.02.075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neish AS. Microbes in gastrointestinal health and disease. Gastroenterology 2009; 136:65-80; PMID:; http://dx.doi.org/ 10.1053/j.gastro.2008.10.080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Artis D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat Rev Immunol 2008; 8:411-20; PMID:; http://dx.doi.org/ 10.1038/nri2316 [DOI] [PubMed] [Google Scholar]
- 12.Ngendahayo Mukiza C, Dubreuil JD. Escherichia coli heat-stable toxin b impairs intestinal epithelial barrier function by altering tight junction proteins. InfectImmun 2013; 81:2819-27; PMID:; http://dx.doi.org/ 10.1128/IAI.00455-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Popoff MR, Geny B. Multifaceted role of Rho, Rac, Cdc42 and Ras in intercellular junctions, lessons from toxins. Biochim Biophys Acta 2009; 1788:797-812; PMID:; http://dx.doi.org/ 10.1016/j.bbamem.2009.01.011 [DOI] [PubMed] [Google Scholar]
- 14.Berkes J, Viswanathan VK, Savkovic SD, Hecht G. Intestinal epithelial responses to enteric pathogens: effects on the tight junction barrier, ion transport, and inflammation. Gut 2003; 52:439-51; PMID:; http://dx.doi.org/ 10.1136/gut.52.3.439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang YG, Wu S, Xia Y, Sun J. Salmonella infection upregulates the leaky protein claudin-2 in intestinal epithelial cells. PloS one 2013; 8:e58606; PMID:; http://dx.doi.org/ 10.1371/journal.pone.0058606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sousa S, Lecuit M, Cossart P. Microbial strategies to target, cross or disrupt epithelia. Curr Opin Cell Bio 2005; 17:489-98; PMID:; http://dx.doi.org/ 10.1016/j.ceb.2005.08.013 [DOI] [PubMed] [Google Scholar]
- 17.Hollander D. Crohn's disease—a permeability disorder of the tight junction? Gut 1988; 29:1621-4; PMID:; http://dx.doi.org/ 10.1136/gut.29.12.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schulzke JD, Ploeger S, Amasheh M, Fromm A, Zeissig S, Troeger H, Richter J, Bojarski C, Schumann M, Fromm M. Epithelial tight junctions in intestinal inflammation. Ann N Y Acad Sci 2009; 1165:294-300; PMID:; http://dx.doi.org/ 10.1111/j.1749-6632.2009.04062.x [DOI] [PubMed] [Google Scholar]
- 19.Ivanov AI, Nusrat A, Parkos CA. The epithelium in inflammatory bowel disease: potential role of endocytosis of junctional proteins in barrier disruption. Novartis Found Symp 2004; 263:115-24; discussion 24-32, 211-8; PMID:; http://dx.doi.org/ 10.1002/0470090480.ch9 [DOI] [PubMed] [Google Scholar]
- 20.Weber CR, Nalle SC, Tretiakova M, Rubin DT, Turner JR. Claudin-1 and claudin-2 expression is elevated in inflammatory bowel disease and may contribute to early neoplastic transformation. Lab Invest 2008; 88:1110-20; PMID:; http://dx.doi.org/ 10.1038/labinvest.2008.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeissig S, Burgel N, Gunzel D, Richter J, Mankertz J, Wahnschaffe U, Kroesen AJ, Zeitz M, Fromm M, Schulzke JD. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn's disease. Gut 2007; 56:61-72; PMID:; http://dx.doi.org/ 10.1136/gut.2006.094375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arnott ID, Kingstone K, Ghosh S. Abnormal intestinal permeability predicts relapse in inactive Crohn disease. Scand J Gastroenterol 2000; 35:1163-9; PMID:; http://dx.doi.org/ 10.1080/003655200750056637 [DOI] [PubMed] [Google Scholar]
- 23.Wyatt J, Vogelsang H, Hubl W, Waldhoer T, Lochs H. Intestinal permeability and the prediction of relapse in Crohn's disease. Lancet 1993; 341:1437-9; PMID:; http://dx.doi.org/ 10.1016/0140-6736(93)90882-H [DOI] [PubMed] [Google Scholar]
- 24.Katz KD, Hollander D, Vadheim CM, McElree C, Delahunty T, Dadufalza VD, Krugliak P, Rotter JI. Intestinal permeability in patients with Crohn's disease and their healthy relatives. Gastroenterology 1989; 97:927-31; PMID: [DOI] [PubMed] [Google Scholar]
- 25.Mota LJ, Journet L, Sorg I, Agrain C, Cornelis GR. Bacterial injectisomes: needle length does matter. Science 2005; 307:1278; PMID:; http://dx.doi.org/ 10.1126/science.1107679 [DOI] [PubMed] [Google Scholar]
- 26.Sun J. Pathogenic Bacterial Proteins and their Anti-Inflammatory Effects in the Eukaryotic Host. Antiinflamm Allergy Agents Med Chem 2009; 8:214-27; PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ulluwishewa D, Anderson RC, McNabb WC, Moughan PJ, Wells JM, Roy NC. Regulation of tight junction permeability by intestinal bacteria and dietary components. J Nutr 2011; 141:769-76; PMID:; http://dx.doi.org/ 10.3945/jn.110.135657 [DOI] [PubMed] [Google Scholar]
- 28.Bansal T, Alaniz RC, Wood TK, Jayaraman A. The bacterial signal indole increases epithelial-cell tight-junction resistance and attenuates indicators of inflammation. Proc Natl Acad Sci U S A 2010; 107:228-33; PMID:; http://dx.doi.org/ 10.1073/pnas.0906112107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liao AP, Petrof EO, Kuppireddi S, Zhao Y, Xia Y, Claud EC, Sun J. Salmonella type III effector AvrA stabilizes cell tight junctions to inhibit inflammation in intestinal epithelial cells. PloS one 2008; 3:e2369; PMID:; http://dx.doi.org/ 10.1371/journal.pone.0002369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang S, Santos RL, Tsolis RM, Stender S, Hardt WD, Baumler AJ, et al. The Salmonella enterica serotype typhimurium effector proteins SipA, SopA, SopB, SopD, and SopE2 act in concert to induce diarrhea in calves. Infec Immun 2002; 70:3843-55; PMID:; http://dx.doi.org/ 10.1128/IAI.70.7.3843-3855.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schesser K, Dukuzumuremyi JM, Cilio C, Borg S, Wallis TS, Pettersson S, Galyov EE. The Salmonella YopJ-homologue AvrA does not possess YopJ-like activity. Microb Pathog 2000; 28:59-70; PMID:; http://dx.doi.org/ 10.1006/mpat.1999.0324 [DOI] [PubMed] [Google Scholar]
- 32.Ma J, Zhang YG, Xia Y, Sun J. The inflammatory cytokine tumor necrosis factor modulates the expression of Salmonella typhimurium effector proteins. J Inflamm (Lond) 2010; 7:42; PMID:; http://dx.doi.org/ 10.1186/1476-9255-7-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Du F, Galan JE. Selective inhibition of type III secretion activated signaling by the Salmonella effector AvrA. PLoS Pathog 2009; 5:e1000595; PMID:; http://dx.doi.org/ 10.1371/journal.ppat.1000595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones RM, Wu H, Wentworth C, Luo L, Collier-Hyams L, Neish AS. Salmonella AvrA Coordinates Suppression of Host Immune and Apoptotic Defenses via JNK Pathway Blockade. Cell Host Microbe 2008; 3:233-44; PMID:; http://dx.doi.org/ 10.1016/j.chom.2008.02.016 [DOI] [PubMed] [Google Scholar]
- 35.Naydenov NG, Hopkins AM, Ivanov AI. c-Jun N-terminal kinase mediates disassembly of apical junctions in model intestinal epithelia. Cell Cycle 2009; 8:2110-21; PMID:; http://dx.doi.org/ 10.4161/cc.8.13.8928 [DOI] [PubMed] [Google Scholar]
- 36.Miller SI, Mekalanos JJ. Constitutive expression of the phoP regulon attenuates Salmonella virulence and survival within macrophages. Journal of bacteriology 1990; 172:2485-90; PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reed KA, Hobert ME, Kolenda CE, Sands KA, Rathman M, O'Connor M, Lyons S, Gewirtz AT, Sansonetti PJ, Madara JL. The Salmonella typhimurium flagellar basal body protein FliE is required for flagellin production and to induce a proinflammatory response in epithelial cells. J Biol Chem 2002; 277:13346-53; PMID:; http://dx.doi.org/ 10.1074/jbc.M200149200 [DOI] [PubMed] [Google Scholar]
- 38.Sun J, Hobert ME, Rao AS, Neish AS, Madara JL. Bacterial activation of β-catenin signaling in human epithelia. Am J Physiol Gastrointest Liver Physiol 2004; 287:G220-7; PMID:; http://dx.doi.org/ 10.1152/ajpgi.00498.2003 [DOI] [PubMed] [Google Scholar]
- 39.Liu X, Lu R, Wu S, Sun J. Salmonella regulation of intestinal stem cells through the Wnt/β-catenin pathway. FEBS Lett 2010; 584:911-6; PMID:; http://dx.doi.org/ 10.1016/j.febslet.2010.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu S, Ye Z, Liu X, Zhao Y, Xia Y, Steiner A, Petrof EO, Claud EC, Sun J. Salmonella typhimurium infection increases p53 acetylation in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol 2010; 298:G784-94; PMID:; http://dx.doi.org/ 10.1152/ajpgi.00526.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ye Z, Petrof EO, Boone D, Claud EC, Sun J. Salmonella effector AvrA regulation of colonic epithelial cell inflammation by deubiquitination. Am J Pathol 2007; 171:882-92; PMID:; http://dx.doi.org/ 10.2353/ajpath.2007.070220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duan Y, Liao AP, Kuppireddi S, Ye Z, Ciancio MJ, Sun J. β-Catenin activity negatively regulates bacteria-induced inflammation. Lab Invest 2007; 87:613-24; PMID:; http://dx.doi.org/ 10.1038/labinvest.3700497 [DOI] [PubMed] [Google Scholar]
- 43.Sweeney NJ, Laux DC, Cohen PS. Escherichia coli F-18 and E. coli K-12 eda mutants do not colonize the streptomycin-treated mouse large intestine. Infect Immun 1996; 64:3504-11; PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu X, Lu R, Wu S, Zhang YG, Xia Y, Sartor RB, et al. Wnt2 inhibits enteric bacterial-induced inflammation in intestinal epithelial cells. Inflamm Bowel Dis 2012; 18:418-29; PMID:; http://dx.doi.org/ 10.1002/ibd.21788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rui HL, Wang YY, Cheng H, Chen YP. JNK-dependent AP-1 activation is required for aristolochic acid-induced TGF-beta1 synthesis in human renal proximal epithelial cells. Am J Renal Physiol 2012; 302:F1569-75; PMID:; http://dx.doi.org/ 10.1152/ajprenal.00560.2011 [DOI] [PubMed] [Google Scholar]
- 46.Das KC, Muniyappa H. c-Jun-NH2 terminal kinase (JNK)-mediates AP-1 activation by thioredoxin: phosphorylation of cJun, JunB, and Fra-1. Mol Cell Biochem 2010; 337:53-63; PMID:; http://dx.doi.org/ 10.1007/s11010-009-0285-0 [DOI] [PubMed] [Google Scholar]
- 47.Collier-Hyams LS, Zeng H, Sun J, Tomlinson AD, Bao ZQ, Chen H, Madara JL, Orth K, Neish AS. Cutting edge: Salmonella AvrA effector inhibits the key proinflammatory, anti-apoptotic NF-kappa B pathway. J Immunol 2002; 169:2846-50; PMID:; http://dx.doi.org/ 10.4049/jimmunol.169.6.2846 [DOI] [PubMed] [Google Scholar]
- 48.Steele-Mortimer O, Knodler LA, Marcus SL, Scheid MP, Goh B, Pfeifer CG, et al. Activation of Akt/protein kinase B in epithelial cells by the Salmonella typhimurium effector sigD. J Biol Chem 2000; 275:37718-24; PMID:; http://dx.doi.org/ 10.1074/jbc.M008187200 [DOI] [PubMed] [Google Scholar]
- 49.Friebel A, Ilchmann H, Aepfelbacher M, Ehrbar K, Machleidt W, Hardt WD. SopE and SopE2 from Salmonella typhimurium activate different sets of RhoGTPases of the host cell. J Biol Chem 2001; 276:34035-40; PMID:; http://dx.doi.org/ 10.1074/jbc.M100609200 [DOI] [PubMed] [Google Scholar]
- 50.Zhang S, Kingsley RA, Santos RL, Andrews-Polymenis H, Raffatellu M, Figueiredo J,Nunes J, Tsolis RM, Adams LG, Bäumler AJ. Molecular pathogenesis of salmonella enterica serotype typhimurium-induced diarrhea. Infect Immun 2003; 71:1-12; PMID:; http://dx.doi.org/ 10.1128/IAI.71.1.1-12.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang FC, Werne A, Li Q, Galyov EE, Walker WA, Cherayil BJ. Cooperative interactions between flagellin and SopE2 in the epithelial interleukin-8 response to salmonella enterica serovar typhimurium infection. Infec Immun 2004; 72:5052-62; PMID:; http://dx.doi.org/ 10.1128/IAI.72.9.5052-5062.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Van Itallie CM, Colegio OR, Anderson JM. The cytoplasmic tails of claudins can influence tight junction barrier properties through effects on protein stability. J Membr Biol 2004; 199:29-38; PMID:; http://dx.doi.org/ 10.1007/s00232-004-0673-z [DOI] [PubMed] [Google Scholar]
- 53.Wilson BA, Ho M. Recent insights into Pasteurella multocida toxin and other G-protein-modulating bacterial toxins. Future Microbiol 2010; 5:1185-201; PMID:; http://dx.doi.org/ 10.2217/fmb.10.91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoon SS, Sun J. Probiotics, nuclear receptor signaling, and anti-inflammatory pathways. Gastroenterol Res Pract 2011; 2011:971938; PMID:; http://dx.doi.org/ 10.1155/2011/971938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Traweger A, Fang D, Liu YC, Stelzhammer W, Krizbai IA, Fresser F, Bauer HC, Bauer H. The tight junction-specific protein occludin is a functional target of the E3 ubiquitin-protein ligase itch. J Biol Chem 2002; 277:10201-8; PMID:; http://dx.doi.org/ 10.1074/jbc.M111384200 [DOI] [PubMed] [Google Scholar]