

Summary
Heterotopic ossification (HO) is a debilitating condition in which cartilage and bone forms in soft tissues such as muscle, tendon, and ligament causing immobility. This process is induced by inflammation associated with traumatic injury. In an extremely rare genetic disorder called fibrodysplasia ossificans progessiva (FOP), a combination of inflammation associated with minor soft tissue injuries and a hereditary genetic mutation causes massive HO that progressively worsens throughout the patients’ lifetime leading to the formation of an ectopic skeleton. An activating mutation in the BMP type I receptor ALK2 has been shown to contribute to the heterotopic lesions in FOP patients, yet recent studies have shown that other events are required to stimulate HO including activation of sensory neurons, mast cell degranulation, lymphocyte infiltration, skeletal myocyte cell death, and endothelial-mesenchymal transition (EndMT). In this review, we discuss the recent evidence and mechanistic data that describe the cellular and molecular mechanisms that give rise to heterotopic bone.
Keywords: Heterotopic ossification, HO, Fibrodysplasia ossificans progressiva, FOP, Bone
Introduction
Heterotopic ossification (HO) occurs in cases of traumatic injury, severe burns, and is commonly observed after invasive surgeries. At the site of soft tissue injury, a mesenchymal condensation occurs followed by chondrogenesis and endochondral ossification (Shore and Kaplan, 2010). The inflammatory response in the injured area is thought to induce HO, however, the precise sensory and immune signals involved in this disorder are not completely understood.
Fibrodysplasia ossificans progressiva (FOP) is a devastating genetic disorder that causes progressive HO in the muscle, tendon, and ligament tissue over time until the patients are rendered completely immobile. Patients with FOP have congenital malformations of the great toes (hallux valgus) and develop “flare ups” of painful inflammatory nodules within the fascia, striated muscles, tendons, and ligaments. These nodules form in response to viral infection, minor injury, immunizations and injections, and over-exertion (Pignolo et al., 2011). Histologically, these nodules have increased vascularity and perivascular lymphocytic infiltrates consisting of both B and T lymphocytes that overexpress bone morphogenetic protein 4 (Gannon et al., 1997). Bone morphogenetic proteins (BMP) such as BMP2, BMP4, and BMP9 have been shown to be the primary inducers of HO when injected into the muscle tissue of mice (Lounev et al., 2009; Medici et al., 2010; Leblanc et al., 2011) and are highly expressed in human HO lesions (Gannon et al., 1997; Grenier et al., 2013).
Clinical aspects of HO
The first flare-up that leads to HO usually occurs before the age of 10 years. A child with FOP will typically develop bone at the neck, shoulders, arms, chest, and finally the feet. Later the disease progresses in the ventral, appendicular, caudal and distal regions of the body. However, it may not occur in this order in all patients due to injury-related flare-ups. This condition causes loss of mobility to affected joints, including inability to fully open the mouth, limiting speech and eating. HO around the rib cage restricts the expansion of the lungs and diaphragm causing respiratory complications. FOP patients have moderate immobility by the age of 20 years and decreased life expectancy to approximately 40 years of age (Connor and Evans, 1982). Death is typically the result of respiratory complications due to restricted motion of the chest wall and diaphragm (Kussmaul et al., 1998). As of now, there is no significant treatment for FOP. Surgical removal of HO is not recommended, as trauma can increase the size of the lesion and induce more ectopic bone formation. NSAIDs (indomethacin), cyclo-oxygenase-2 (COX-2) inhibitors, leukotriene inhibitors, mast cell stabilizers and bisphosphonates have been used to manage the inflammation and chronic discomfort. Irradiation for postoperative prophylaxis has also been used in management of HO (Kitterman et al., 2005).
Genetic basis of HO
FOP is caused by an autosomal dominant mutation in a BMP type I receptor called activin-like kinase 2 (ALK2) and acute inflammation (Shore et al., 2006). Most FOP patients carry a heterozygous germ-line mutation in the ACVR1 gene that causes an argenine-to-histidine change in amino acid 206 (R206H) located in the glycine-serine-rich (GS) domain of the ALK2 receptor (Shore et al., 2006). Studies have shown that this change represents a gain-of-function mutation that causes constitutive phosphorylation of the receptor with continuous signal transduction (Shen et al., 2009). In a transgenic mouse model of FOP, adenoviral Cre recombinase-dependent expression of the mutant ALK2 gene forms heterotopic bone in muscle tissue (Yu et al., 2008; Lounev et al., 2009; Medici et al., 2010; Kaplan et al., 2012). More recently a heterozygous R206H knock-in mouse model of FOP was generated and showed similar clinical features of patients with FOP including spontaneous HO in areas of mechanical stress like the joints, as well as the toe malformation. Injection of cardiotoxin to induce inflammation into the muscle tissue of these mice showed dramatic induction of HO at the injection site (Chakkalakal et al., 2012). Other mutations (Table 1) in the GS or kinase domains have been described in patients who exhibit FOP-like phenotypes with additional clinical manifestations (Furuya et al., 2008; Kaplan et al., 2009; Petrie et al., 2009; Bocciardi et al., 2009; Gregson et al., 2011; Whyte et al., 2012; Nakahara et al., 2014).
Table 1.
Genetic mutations involved in heterotopic ossification.
| ACVR1 Mutation | ALK2 Domain | Amino Acid Change | Clinical Features |
|---|---|---|---|
| c.617 G>A | GS | R206H | Classical FOP; HO of tendons, ligaments, skeletal muscle; congenital hallux valgus of the great toes; short broad femoral necks; cervical spine fusion; osteochondromas |
| c.605 G>T | GS | R202I | One short great toe; fixed right shoulder and elbow; fusion of neck; relatively late age onset (14 years) |
| c.619 C>G | GS | Q207E | Classical FOP phenotype plus growth retardation with height and weight below the 5th percentile for age |
| c.983 G>A | Kinase | G328E | Severe reduction deformities in all digits; spinal fixation; shoulders fixed in adduction; elbows fixed in flexion; jaw gape; mild cognitive impairment; diffuse scalp hair thinning |
| c.982 G>A | Kinase | G328R | FOP variant; normal toes; short thumbs; no HO during childhood; stooped posture at 30 years of age; hypoplastic cerebellum |
| c.982 G>T | Kinase | G328W | Severe reduction deficits of great toes with lack of toe nails in affected digits; severe malformation of thumb; thin scalp hair and slowed growth rate in second decade of life; sparse eyebrows; mild cognitive impairment with difficulty in abstract thinking; no difficulty in attention span; anatomic abnormalities of the cerebellum |
| c.1067 G>A | Kinase | G356D | Persistence of primary teeth into adulthood; primary amenorrhea; malformation of thumb and great toes |
| c.774 G>C | Kinase | R258S | Absence of great toe malformation; disease onset at age 4 with painful swelling in cervical vertebral region; long remission period; flare-up at age 18 with progressive HO |
| c.1124 G>C | Kinase | R375P | Normal toes; slow progression of disease; flare-ups at age 14; limited motion of spine and shoulders at age 40 |
| c.587 T>C | GS | L196P | No great toe malformation; no heterotopic ossification in skeletal muscle until motorcycle accident at age 21 |
| c.974 G>C | Kinase | G325A | Hallux valgus; late-onset HO provoked by viral illness |
| c.1067 G>A | Kinase | R356D | Slowly progressive respiratory dysfunction and abnormal heterotopic ossification; rigid spine; baldness; sensorineural hearing loss; severe hypodactyly |
Cellular origins of HO
Although the genetic basis of FOP has been known for several years, only recently has the cellular origin of heterotopic skeletal cells been elucidated. Lineage tracing studies have been performed using transgenic mice that use Cre recombinase driven by cell type-specific gene promoters crossed with GFP or LacZ reporter mice in order to identify which cells in the muscle tissue give rise to ectopic chondrocytes and osteocytes. Since inflammation triggers HO, immune cells were investigated using CD19-Cre to label B-cell lineage, LCK-Cre to mark T-cell lineage, and Lyz-Cre to label the monocyte/macrophage lineage, but no ectopic skeletal cells were found to be of immune cell origin (Kan et al., 2009). Similar negative results were found using Nestin-Cre reporter mice that label somite-derived cells, as well as Myf5-Cre (Kan et al., 2009) and MyoD-Cre (Lounev et al., 2009), which labeled cells of skeletal myocyte origin. The lack of involvement of skeletal myocytes in generating ectopic bone was expected, since it has been shown that the immune response in the early lesions causes myocyte cell death (Shore and Kaplan, 2010). Therefore, the muscle itself does not become bone, but rather dies and is replaced by bone. This is consistent with observed HO in mouse models of Duchenne muscular dystrophy (Mu et al., 2013), suggesting that skeletal myocyte cell death may be an essential initiator of HO.
Histological analyses of heterotopic lesions from FOP patients have demonstrated positive staining for endothelial-specific biomarkers in the ectopic chondrocytes and osteoblasts, whereas normal cartilage and bone cells do not (Medici et al., 2010). Similar results have been found when analyzing heterotopic bone in BMP ligand-induced and transgenic mouse models of HO (Lounev et al., 2009; Medici et al., 2010). These endothelial biomarkers include Tie1, Tie2, vWF, and VE-cadherin. Lineage tracing studies using Tie2-Cre reporter mice have shown positive GFP or LacZ reporter expression in approximately 50% of the mesenchymal cells, chondrocytes, and osteoblasts found in the lesions, strongly suggesting that these cells are of endothelial origin (Lounev et al., 2009; Medici et al., 2010; Chakkalakal et al., 2012). Expression of the mutant (R206H) ALK2 receptor in cultured vascular endothelial cells caused them to undergo an endothelial-mesenchymal transition (EndMT). These cells acquired most of the anatomical, biochemical, and physiological properties of mesenchymal stem cells and could be readily differentiated into osteoblasts, chondrocytes or adipocytes when exposed to the appropriate differentiation medium. Similar results were observed upon treating cultured endothelial cells with ALK2-activating ligands TGF-β2 or BMP4 (Medici et al., 2010). These findings suggest that the vascular endothelium in muscle tissue may be transforming into cartilage and bone through a mesenchymal stem cell intermediate. A recent study demonstrated that the same mechanism of EndMT gives rise to osteoprogenitor cells that induce vascular calcifications (Yao et al., 2013).
Other studies have suggested that resident skeletal muscle stem cells may also play a role in generating some of the ectopic bone tissue (Wosczyna et al., 2012). A novel lineage tracing study has shown that Glast-Cre mice have positive expression of the reporter in heterotopic skeletal cells. Glast is most commonly expressed in neural cells, but can also be found in many other cell types. Although the study does not specify the exact cellular origin, it is clear that approximately 35% of the ectopic skeletal cells in the lesions are of the Glast-expressing lineage (Kan et al., 2013).
Therapeutic targets for HO
Two recent studies have investigated the role of sensory neurons in HO. TRPV1 knockout mice have dysfunctional sensory neurons and show dramatically impaired BMP-induced HO compared to wild-type mice (Salisbury et al., 2011). Substance P, a chemical released by sensory neurons, is highly expressed in early HO lesions prior to the mesenchymal condensation, chondrogenesis, and endochondral ossification (Kan et al., 2011). Injection of an adenoviral construct expressing BMP2 into muscle tissue of mice shows elevated expression of Substance P in the nerves and increased recruitment of mast cells (Salisbury et al., 2011). RP-6758, a chemical inhibitor of Substance P, was shown to dramatically reduce BMP4-induced HO in mice, suggesting it has strong therapeutic potential (Kan et al., 2011). In response to injury, neurons release Substance P, which causes degranulation of mast cells and triggers an inflammatory response. The antihistimine Cromolyn, which blocks mast cell degranulation, significantly reduces HO in mice, demonstrating the importance of mast cells in the disease progression (Salisbury et al., 2011).
Perhaps the most obvious therapeutic target to prevent HO is ALK2, the receptor that is activated by BMPs and mutated in FOP patients. Small molecule drugs called dorsomorphin and LDN-193189 have been shown to bind and inhibit the activity of ALK2. These inhibitors successfully reduced the incidence of HO in mutant ALK2 transgenic mice (Yu et al., 2008). Use of dorsomorphin or siRNA-mediated knockdown of ALK2 expression has been shown to prevent EndMT (Medici et al., 2010). A more recent study has demonstrated that the BMP type II receptor is essential for HO in mutant ALK2 transgenic mice (Bagarova et al., 2013), identifying a role for both type I and type II receptors and suggesting that the type II receptor may be a novel therapeutic target.
Despite the significance of these inhibitory studies, none of them were able to completely prevent HO. However, recent evidence has shown that retinoic acid receptor-γ (RAR-γ) agonists can eradicate HO formation in mice. These synthetic drugs, along with retinoic acid itself, prevent mesenchymal stem cell differentiation into chondrocytes (Shimono et al., 2011). Therefore, RAR-γ agonists presumably prevent HO by inhibiting the chondrogenic stage, without which endochondral ossification cannot occur. Currently, these drugs represent the most effective pharmacological inhibitors for potential clinical application to prevent HO (Table 2).
Table 2.
Established pharmacological inhibitors of heterotopic ossification.
| Inhibitor | Target | Function |
|---|---|---|
| Dorsomorphin | ALK2 | Binds and inhibits phosphorylation and activity of the ALK2 receptor |
| LDN-193189 | ALK2 | Binds and inhibits phosphorylation and activity of the ALK2 receptor |
| RP-6758 | Substance P | Non-peptide Substance P antagonist; inhibits mast cell degranulation |
| Cromolyn | Mast cells | Anti-histamine; prevents mast cell degranulation |
| Retenoic acid | Retinoic acid receptors | Inhibits chondrogenesis; blocks BMP signaling |
| NRX204647 | Retinoic acid receptor-γ | RAR-γ agonist; inhibits chondrogenesis; blocks BMP signaling |
| CD1530 | Retinoic acid receptor-γ | RAR-γ agonist; inhibits chondrogenesis; blocks BMP signaling |
Taken together, these findings suggest a complex mechanism involving injury-induced activation of neurons and various immune cells promote EndMT and subsequent differentiation of progenitor cells into cartilage and bone cells to form the ectopic lesions (Fig. 1). This knowledge may provide a foundation for therapeutic intervention through targeted inhibition of the various phases of HO progression.
Fig. 1.
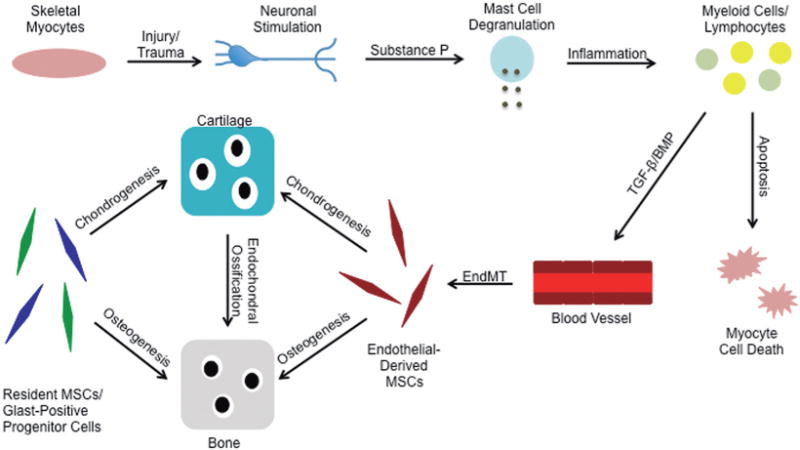
An overview of the mechanisms of heterotopic ossification. Trauma to muscle tissue stimulates sensory neurons to release Substance P, which will promote mast cell degranulation and subsequent inflammation in the area of injury. Myeloid cells and lymphocytes invade the injured tissue and promote skeletal myocyte cell death. These immune cells also release cytokines such as TGF-β2 and BMP4, which cause the vascular endothelial cells to undergo endothelial-mesenchymal transition (EndMT) to form multipotent mesenchymal stem-like cells (MSCs). Inflammatory signals and the muscle microenvironment foster these MSCs to differentiate into chondrocytes and osteoblasts to form heterotopic lesions of cartilage and bone. Resident skeletal muscle MSCs and Glast-positive progenitor cells may also contribute to the formation of the ectopic skeletal tissue.
Acknowledgments
This work was supported by grants R01HL112860 (to D.M.) and P20GM104937 (to D.M. and A.M.R.) from the National Institutes of Health and a grant from the John Butler Mulliken Foundation (to D.M.).
References
- Bagarova J, Vonner AJ, Armstrong KA, Borgermann J, Lai CS, Deng DY, Beppu H, Alfano I, Filippakopoulos P, Morrell NW, Bullock AN, Knaus P, Mishina Y, Yu PB. Constitutively active ALK2 receptor mutants require type II receptor cooperation. Mol Cell Biol. 2013;33:2413–2424. doi: 10.1128/MCB.01595-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocciardi R, Bordo D, Di Duca M, Di Rocco M, Ravazzolo R. Mutational Analysis of the ACVR1 gene in Italian patients affected with fibrodysplasia ossificans progressiva: confirmations and advancements. Eur J Hum Genet. 2009;17:311–318. doi: 10.1038/ejhg.2008.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakkalakal SA, Zhang D, Culbert AL, Convente MR, Caron RJ, Wright AC, Maidment AD, Kaplan FS, Shore EM. An Acvr1 R206H knock-in mouse has fibrodysplasia ossificans progressiva. J Bone Miner Res. 2012;27:1746–1756. doi: 10.1002/jbmr.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor JM, Evans DA. Fibrodysplasia ossificans progressiva. The clinical features and natural history of 34 patients. J Bone Joint Surg Br. 1982;64:76–83. doi: 10.1302/0301-620X.64B1.7068725. [DOI] [PubMed] [Google Scholar]
- Furuya H, Ikezoe K, Wang L, Ohyagi Y, Motomura K, Fujii N, Kira J, Fukumaki Y. A unique case of fibrodysplasia ossificans progressiva with an ACVR1 mutation, G356D, other than the common mutation (R206H) Am J Med Genet A. 2008;146A:459–463. doi: 10.1002/ajmg.a.32151. [DOI] [PubMed] [Google Scholar]
- Gannon FH, Kaplan FS, Olmsted E, Finkel GC, Zasloff MA, Shore E. Bone morphogenetic protein 2/4 in early fibromatous lesions of fibrodysplasia ossificans progressiva. Hum Pathol. 1997;28:339–343. doi: 10.1016/s0046-8177(97)90133-7. [DOI] [PubMed] [Google Scholar]
- Gregson CL, Holingworth P, Williams M, Petrie KA, Bullock AN, Brown MA, Tobias JH, Triffitt JT. A novel ACVR1 mutation in the glycine/serine rich domain found in the most benign case of fibrodysplasia ossificans progressiva variant reported to date. Bone. 2011;48:654–658. doi: 10.1016/j.bone.2010.10.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenier G, Leblanc E, Faucheux N, Lauzier D, Kloen P, Hamdy RC. BMP-9 expression in human traumatic heterotopic ossification: a case report. Skeletal Muscle. 2013;3:29. doi: 10.1186/2044-5040-3-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan L, Liu Y, McGuire TL, Berger DM, Awatramani RB, Dymecki SM, Kessler JA. Dysregulation of local stem/progenitor cells as a common cellular mechanism of heterotopic ossification. Stem Cells. 2009;27:150–156. doi: 10.1634/stemcells.2008-0576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan L, Lounev VY, Pignolo RJ, Duan L, Liu Y, Stock SR, McGuire TL, Lu B, Gerard NP, Shore EM, Kaplan FS, Kessler JA. Substance P signaling mediates BMP-dependent heterotopic ossification. J Cell Biochem. 2011;112:2795–2772. doi: 10.1002/jcb.23259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan L, Peng CY, McGuire TL, Kessler JA. Glast-expressing progenitor cells contribute to heterotopic ossification. Bone. 2013;53:194–203. doi: 10.1016/j.bone.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan FS, Xu M, Seemann P, Connor JM, Glaser DL, Carroll L, Delai P, Fastnaucht-Urban E, Forman SJ, Gillessen-Kaesbach G, Hoover-Fong J, Koster B, Pauli RM, Reardon W, Zaidi SA, Zasloff M, Morhart R, Mundlos S, Groppe J, Shore EM. Classic and atypical fibrodysplasia ossificans progressive (FOP) phenotypes are caused by mutations in the bone morphogenic protein (BMP) type I receptor ACVR1. Hum Mutat. 2009;30:379–390. doi: 10.1002/humu.20868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan FS, Chakkalakal SA, Shore EM. Fibrodysplasia ossificans progressiva: mechanisms and models of skeletal metamorphosis. Dis Model Mech. 2012;5:756–762. doi: 10.1242/dmm.010280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitterman JA, Kantanie S, Rocke DM, Kaplan FS. Latrogenic harm caused by diagnostic errors in fibrodysplasia ossificans progressiva. Pediatrics. 2005;116:e654–e661. doi: 10.1542/peds.2005-0469. [DOI] [PubMed] [Google Scholar]
- Kussmaul WG, Esmail AN, Sagar Y, Ross J, Gregory S, Kaplan FS. Pulmonary and cardiac function in advanced fibrodysplasia ossificans progressiva. Clin Orthop Relat Res. 1998;346:104–109. [PubMed] [Google Scholar]
- Leblanc E, Trensz F, Haroun S, Droulin G, Bergeron E, Penton CM, Montanaro F, Roux S, Faucheux N, Grenier G. BMP-9-induced muscle heterotopic ossification requires changes to the skeletal muscle microenvironment. J Bone Miner Res. 2011;26:1166–1177. doi: 10.1002/jbmr.311. [DOI] [PubMed] [Google Scholar]
- Lounev VY, Ramachandran R, Wosczyna MN, Yamamoto M, Maidment AD, Shore EM, Glaser DL, Goldhamer DJ, Kaplan FS. Identification of progenitor cells that contribute to heterotopic skeletogenesis. J Bone Joint Surg Am. 2009;91:652–663. doi: 10.2106/JBJS.H.01177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med. 2010;16:1400–1406. doi: 10.1038/nm.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu X, Usas A, Tang Y, Lu A, Wang B, Weiss K, Huard J. RhoA mediates defective stem cell function and heterotopic ossification in dystrophic muscle of mice. FASEB J. 2013;27:3619–3631. doi: 10.1096/fj.13-233460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahara Y, Katagiri T, Ogata N, Haga N. ACVR1 (587T>C) mutation in a variant form of fibrodysplasia ossificans progressiva: second report. Am J Med Genet A. 2014;164A:220–224. doi: 10.1002/ajmg.a.36219. [DOI] [PubMed] [Google Scholar]
- Petrie KA, Lee WH, Bullock AN, Pointon JJ, Smith R, Russell RG, Brown MA, Wordsworth BP, Triffitt JT. Novel mutations in ACVR1 result in atypical features in two fibrodysplasia ossificans progressiva patients. PLoS One. 2009;4:e5005. doi: 10.1371/journal.pone.0005005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignolo RJ, Shore EM, Kaplan FS. Fibrodysplasia ossificans progressiva: clinical and genetic aspects. Orphanet J Rare Dis. 2011;6:80. doi: 10.1186/1750-1172-6-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salisbury E, Rodenberg E, Sonnet C, Hipp J, Gannon FH, Vadakkan TJ, Dickinson ME, Olmsted-Davis EA, Davis AR. Sensory nerve induced inflammation contributes to heterotopic ossification. J Cell Biochem. 2011;112:2748–2758. doi: 10.1002/jcb.23225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q, Little SC, Xu M, Haupt J, Ast C, Katagiri T, Mundlos S, Seeman P, Kaplan FS, Mullins MC, Shore EM. The fibrodysplasia ossificans progressive R206H ACVR1 mutation activates BMP-independent chondrogenesis and zebrafish embryo ventralization. J Clin Invest. 2009;119:3462–3472. doi: 10.1172/JCI37412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimono K, Tung W, Macolino C, Chi AH, Didizian JH, Mundy C, Chandraratna RA, Mishina Y, Enomoto-Iwamoto M, Pacifici M, Iwamoto M. Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-γ agonists. Nat Med. 2011;17:454–460. doi: 10.1038/nm.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore EM, Kaplan FS. Inherited human diseases of heterotopic ossification. Nat Rev Rhuematol. 2010;6:518–527. doi: 10.1038/nrrheum.2010.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, Connor JM, Delai P, Glaser DL, LeMerrer M, Morhart R, Rogers JG, Smith R, Triffitt JT, Urtizberea JA, Zasloff M, Brown MA, Kaplan FS. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet. 2006;38:525–527. doi: 10.1038/ng1783. [DOI] [PubMed] [Google Scholar]
- Whyte MP, Wenkert D, Demerzis JL, DiCarlo EF, Westenberg E, Mumm S. Fibrodysplasia ossificans progressiva: middle-age onset of heterotopic ossification from a unique missense mutation (c.974G>C, p G325A) in ACVR1. J Bone Miner Res. 2012;27:729–737. doi: 10.1002/jbmr.1473. [DOI] [PubMed] [Google Scholar]
- Wosczyna MN, Biswas AA, Cogswell CA, Goldhamer DJ. Multipotent progenitors resident in the skeletal muscle interstitium exhibit robust BMP-dependent osteogenic activity and mediate heterotopic ossification. J Bone Miner Res. 2012;27:1004–1017. doi: 10.1002/jbmr.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Jumabay M, Ly A, Radparvar M, Cubberly MR, Bostrom KI. A role for the endothelium in vascular calcification. Circ Res. 2013;113:495–504. doi: 10.1161/CIRCRESAHA.113.301792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu PB, Deng DY, Lai CS, Hong CC, Cuny GD, Bouxsein ML, Hong DW, McManus PM, Katagiri T, Sachidanandan C, Kamiya N, Fukuda T, Mishina Y, Peterson RT, Bloch KD. BMP type I receptor inhibition reduces heterotopic ossification. Nat Med. 2008;14:1363–1369. doi: 10.1038/nm.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]