

Abstract
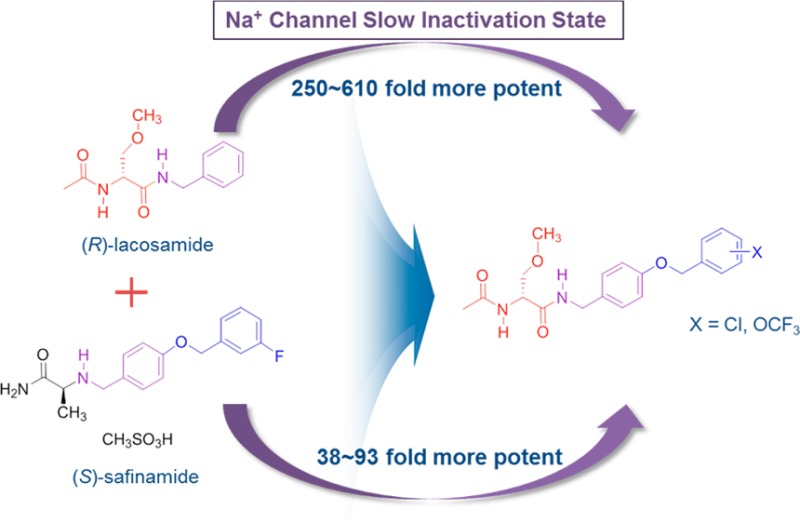
The functionalized amino acid, lacosamide ((R)-2), and the α-aminoamide, safinamide ((S)-3), are neurological agents that have been extensively investigated and have displayed potent anticonvulsant activities in seizure models. Both compounds have been reported to modulate voltage-gated sodium channel activity. We have prepared a series of chimeric compounds, (R)-7–(R)-10, by merging key structural units in these two clinical agents, and then compared their activities with (R)-2 and (S)-3. Compounds were assessed for their ability to alter sodium channel kinetics for inactivation, frequency (use)-dependence, and steady-state activation and fast inactivation. We report that chimeric compounds (R)-7–(R)-10 in catecholamine A-differentiated (CAD) cells and embryonic rat cortical neurons robustly enhanced sodium channel inactivation at concentrations far lower than those required for (R)-2 and (S)-3, and that (R)-9 and (R)-10, unlike (R)-2 and (S)-3, produce sodium channel frequency (use)-dependence at low micromolar concentrations. We further show that (R)-7–(R)-10 displayed excellent anticonvulsant activities and pain-attenuating properties in the animal formalin model. Of these compounds, only (R)-7 reversed mechanical hypersensitivity in the tibial-nerve injury model for neuropathic pain in rats.
Keywords: Chimeric compounds, functionalized amino acids (lacosamide), α-aminoamides (safinamide), sodium channel activity, antiseizure activity, antinociception activity
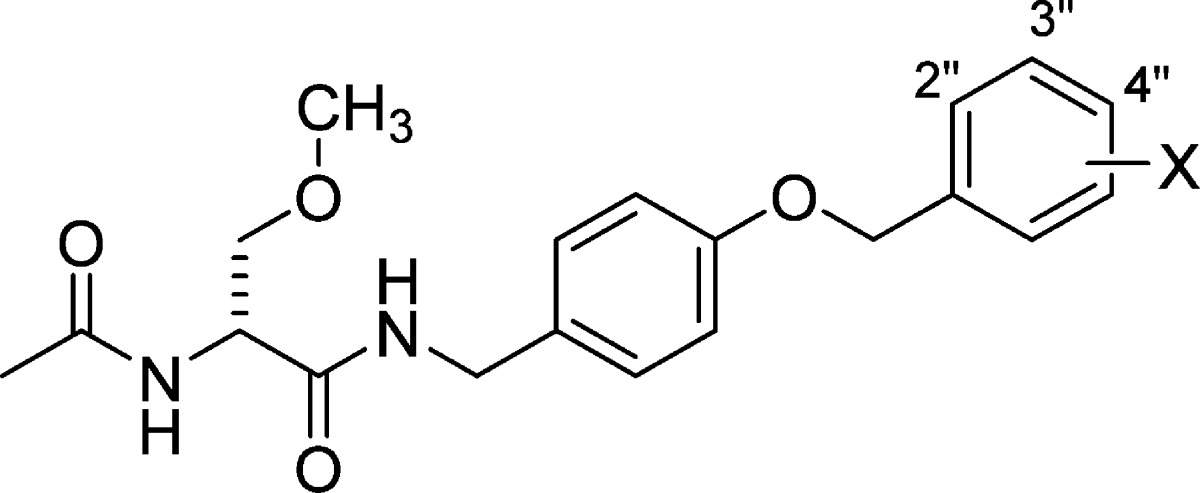
We have reported a novel chimeric compound, (R)-1,1 derived by merging key structural units present in lacosamide2 ((R)-N-benzyl 2-N-acetamido-3-methoxypropionamide, (R)-2) and safinamide3 ((S)-2-(4′-((3″-fluoro)benzyloxy)benzyl)aminopropionamide, (S)-3), two neurological agents that have been extensively investigated (Figure 1). Lacosamide is the archetypal functionalized amino acid (FAA) and is a first-in-class antiseizure drug (ASD) used for the adjunctive and monotherapy treatment of partial-onset seizures in adults.4 Safinamide is a principal example of the class of compounds dubbed α-aminoamide (AAA) and has pharmacological properties relevant to the treatment of Parkinson’s disease.5,6 In (R)-1, we retained the (R)-N-benzyl 2-acetamido-3-methoxypropionamide (A) unit found in (R)-2 and the N-(4′-((3″-fluoro)benzyloxy)benzylamino (B) moiety in (S)-3 (Figure 1). Both (R)-2 and (S)-3 modulated neuronal hyperexcitability by inhibiting voltage-gated sodium channel (VGSC) activity.7−10 In merging these structural units from (R)-2 and (S)-3, we sought compounds with improved and broader functions.
Figure 1.

Structures of (R)-1, lacosamide ((R)-2), and safinamide ((S)-3). Box A represents lacosamide ((R)-2) derived component of (R)-1, and box B represents safinamide ((S)-3) derived component of (R)-1.
Utilizing patch-clamp electrophysiology in the whole-cell configuration, we observed that (R)-1 enhanced the transition of sodium channels in a mouse neuron model cell line, catecholamine A-differentiated (CAD) cells, into inactivated states and also produced frequency (use)-dependent inhibition.11 Compound (R)-1 exhibited effective anticonvulsant activity in the maximal electroshock seizure12 (MES) test in mice (intraperitoneal, ip), but its activity was not better than that of either (R)-22,13 or (S)-35 (MES ED50 (mice, ip, mg/kg): (R)-1, 13, (R)-2, 4.5, (S)-3, 4.1).1 Cellular and anticonvulsant activities similar to (R)-1 were observed for the nonfluorinated (R)-1 analogue, (R)-4, and the two (R)-1 fluorine-regioisomers, (R)-5 and (R)-6.1,11 When (R)-1 was tested in the rat tibial-nerve injury (TNI) model of neuropathic pain,14 a single ip administration completely reversed mechanical hypersensitivity.11

The cellular and animal pharmacological profile for (R)-1 steered us to prepare (R)-7–(R)-10, four chimeric compounds that differed from (R)-1 in the substitution pattern of the terminal benzyloxy (OCH2C6H4X) moiety. We evaluated the compounds by voltage-clamp electrophysiology in CAD cells15 and rat embryonic cortical neurons and compared their cellular properties with the parent compounds, (R)-2 and (S)-3, to determine if the structural units (A and B, Figure 1) in the parent compounds endow sodium channel-mediating properties. We show that (R)-7–(R)-10 enhanced transition of sodium channels to inactivated conformational states at concentrations far lower than those required by (R)-2 and (S)-3 and that several chimeric compounds produced frequency (use)-dependent inhibition at low micromolar concentrations. We also report excellent activity for (R)-7–(R)-10 in the MES and 6 Hz psychomotor16 animal seizure models and that many of the chimeric compounds displayed activity in the formalin pain model17 in mice. However, only (R)-7 reversed mechanical hypersensitivity in the TNI model in rats.
Results
Chimeric Compound Selection
Our earlier studies showed that chimeric compounds (R)-1 and (R)-5, each of which contains an electron-withdrawing fluorine group in the terminal aromatic ring, modulated Na+ channel properties11 and exhibited excellent anticonvulsant activities.1 Thus, we prepared the four benzyloxy analogues in which either a chlorine ((R)-7, (R)-8) or a trifluoromethoxy-group ((R)-9, (R)-10) was positioned at either the 3″- or 4″-aryl site.
Chemistry
Compounds (R)-7–(R)-10 were all prepared by the general procedure described in Scheme 1. Using the mixed anhydride coupling (MAC) method,18 we coupled (R)-N-tert-butoxycarbonyl-d-serine ((R)-24) with the substituted (4-(benzyloxy)phenyl)methanamines 20–23 to give amides (R)-25–(R)-28, respectively, without racemization of the C(2) chiral center. The substituted ((benzyloxy)phenyl)methanamines were prepared by treating 4-cyanophenol (11) with the substituted benzyl bromides 12–15 and K2CO3 in acetone to provide nitriles 16–19,19,20 respectively, which were then reduced (LiAlH4) to give amines 20–23. Methylation (CH3I, Ag2O) of the serine hydroxyl group in (R)-25–(R)-28 yielded ethers (R)-29–(R)-32, respectively. Removal of the tert-butoxycarbonyl group in (R)-29–(R)-32 with acid (HCl/dioxane) and then acetylation (AcCl, Et3N) gave the desired products (R)-7–(R)-10, respectively. The enantiomeric purities of (R)-7–(R)-10 were assessed by a 1H NMR method using the chiral resolving agent, (R)-(−)-mandelic acid.21 Parent compound (S)-3 was prepared as previously reported.3 The Methods section provides the synthetic procedures and physical, spectral, and analytical properties for the final compounds evaluated in the pharmacological studies. The Supporting Information gives similar data for all the synthetic intermediates and final products.
Scheme 1. Synthesis of (R)-7–(R)-10.

Whole-Cell, Patch-Clamp Electrophysiology
We have previously reported that (R)-1 altered sodium channel kinetics in CAD cells by enhancing the transition to inactivated states in response to protocols that estimate both the fast- and the slow-inactivated states. Compound (R)-1 also limited sodium channel current density in frequency (use)-dependent fashion.11 All three proceses are well studied kinetic properties of sodium channels that collectively can exert control over neuronal hyperexcitability.7−9,22 Accordingly, we examined if the same sodium channel properties were affected by (R)-7–(R)-10 and compared these activities with our previous findings with compounds (R)-1, (R)-4, and (R)-511 and with the parent compounds (R)-29,11,23 and (S)-3. In the earlier studies, we used CAD cells because they express endogenous tetrodotoxin-sensitive sodium channels well suited for the study of activation and inactivation kinetics15 and are likely composed of a majority of NaV1.7 channels with very minor contributions by NaV1.1, NaV1.3, and NaV1.9 channels.9,24,25 We established that (R)-2-mediated effects on CAD cell sodium channels9 are similar to those reported for cultured neurons and for mouse N1E-115 neuroblastoma cells.7 Importantly, the NaV1.7 channel is involved in conducting pain impulses in peripheral nerves, and loss of NaV1.7 function restricts pain in rodent models of inflammatory and neuropathic pain.26 We also evaluated (R)-7–(R)-10 on VGSCs in rat embryonic cortical neurons. These central nervous system (CNS) neurons express a subset of VGSC isoforms, NaV1.1, NaV1.2, NaV1.3, and NaV1.6;27 dysregulation as well as mutations in these isoforms have been reported in human epilepsies.28
A. CAD Cells
In Figures 2–4, we illustrate the effects of (R)-7–(R)-10 in CAD cells on sodium channel biophysical properties of inactivation, steady-state activation and fast inactivation, and frequency (use)-dependence. We have previously reported the effects of (R)-2 on these sodium channel properties in CAD cells9,11,23 and provide herein the corresponding results for (S)-3 (Figure 5). To test the ability of (R)-7–(R)-10 and (S)-3 to modulate transition to an inactivated state, CAD cells were subjected to conditioning prepulses ranging from −120 to +20 mV in 10 mV increments for 5 s.9,11,23 During this conditioning pulse, channels enter both slow- and fast-inactivated states. To estimate slow-inactivated current, a pulse to −120 mV was given for 150 ms to allow fast-inactivated channels to recover. A 15 ms depolazrization to 0 mV is then applied to test the available fraction of current (Figure 2A). Differences in available current between control and compound treated cells represent altered sodium channel inactivation via either altered transition into slow-inactivated state or block of fast-inactivated channels with slow off kinetics.29,30 Unseparated in this study, these two possibilities have the same physiological outcome on current generation by affected channels. Representative traces (at −120 and −50 mV) from CAD cells treated with 0.1% DMSO (control) or 10 μM (R)-7–(R)-10 are plotted in Figure 2B. The −50 mV potential is strongly affected by compounds that mediate slow inactivation kinetics and is within a range of voltages that affect several channel and cellular properties including inactivation,30−32 resting membrane potential and action potential firing,33,34 as well as activation and inactivation kinetics.31,35 To determine the relative extent to which each compound facilitated this inactivation kinetics, we graphed the −50 mV data points for several concentrations and calculated the IC50 inactivation value (Figure 2C–F). Compared with our recently reported IC50 value of 85 μM for (R)-2,9,11 the inactivation protocol IC50 values for the newly prepared chimeric compounds were 250–610-fold lower (IC50 (μM): (R)-7, 0.34; (R)-8, 0.31; (R)-9, 0.24; (R)-10, 0.14). The IC50 values for the unsubstituted ((R)-4) and the fluorine-substituted chimeric compounds ((R)-1 and (R)-5) were between 1.6 and 1.7 μM (IC50 (μM): (R)-1, 1.7; (R)-4, 1.6; (R)-5, 1.6).11 When we tested (S)-3, we determined a 13 μM IC50 value for the inactivation protocol (Figure 5C), a value between that of chimeric compounds (R)-7–(R)-10 and (R)-2. In addition, (R)-7–(R)-10 displayed enhanced efficacy over (R)-2 and (S)-3 (i.e., the maximal transition to inactivated channel state was increased). Together, these findings indicated that appendage of the substituted benzyloxy moiety (OCH2C6H4X), similar to that found in (S)-3 to (R)-2, provided compounds that powerfully promoted sodium channel inactivation transition and that the terminal aryl substituent X affected the degree of inactivation facilitation.
Figure 2.

Effects of the chimeric compounds (R)-7–(R)-10 on inactivation state of Na+ currents in CAD cells. (A) Inactivation voltage protocol. Currents were elicited by 5 s prepulses between −120 and −20 mV and then fast-inactivated channels were allowed to recover for 150 ms at a hyperpolarized pulse to −120 mV. The fraction of channels available at 0 mV was analyzed. (B) Representative current traces from CAD cells without (control, 0.1% DMSO) or in the presence of 10 μM of the compounds as indicated. The red and blue traces represent the currents evoked at −120 and −50 mV (arrows), respectively (also highlighted in the voltage protocol as a dashed line). (C–F) Summary of inactivation curves for CAD cells treated with 0.1% DMSO (control) or various concentrations of the compounds as indicated. The concentrations of half maximal effect for −50 mV conditioning pulse (see text for detailed explanations), the IC50, are indicated in boxes within each panel. Data are from 4 to 8 cells per condition. Some error bars are smaller than the symbols.
Figure 4.
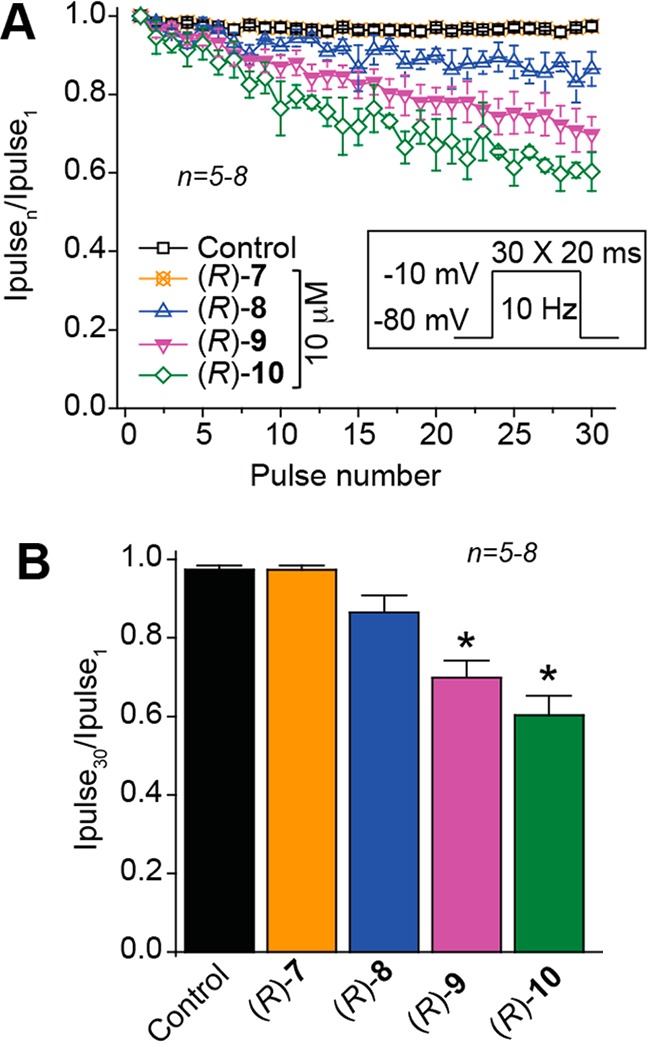
Effect on frequency (use)-dependent block by the chimeric compounds (R)-7–(R)-10 on Na+ currents in CAD cells. (A) Frequency dependence of block was examined by holding cells at the hyperpolarized potential of −80 mV and evoking currents at 10 Hz by 20 ms test pulses to −10 mV (inset middle). Summary of average frequency (use)-dependent decrease in current amplitude over time (±SEM) produced by control (0.1% DMSO) or by the presence of 10 μM of the indicated compounds are shown (p > 0.05, one-way ANOVA with Dunnett’s post hoc test). (B) Summary of the maximal decrement in current amplitude observed at the 30th pulse for control (0.1% DMSO) or the indicated compounds. (R)-9 and (R)-10 caused a significant decrease in current amplitude compared with control (*p < 0.05, one-way ANOVA with Dunnett’s post hoc test; n = 5–8 cells).
Figure 5.

Effect of (S)-3 on sodium channel properties in CAD cells. (A, D, G) Voltage protocols for examining inactivation, fast inactivation, steady-state activation, and frequency (use)-dependent block. (B, E, H) Representative current traces from CAD cells in the absence (control, 0.1% DMSO) or presence of 100 μM (S)-3. (B) Solid and black dashed traces represent the currents induced at −120 and −50 mV, respectively (−50 mV step highlighted in the voltage protocol). (C) Summary of the inactivation curves for CAD cells treated with 0.1% DMSO (control) or 0.5−200 μM (S)-3. The concentrations of half maximal effect for −50 mV conditioning pulse (see text for detailed explanations), the IC50, is indicated. (E) Representative current traces (top, fast inactivation; bottom, steady-state activation). (F) Representative Boltzmann fits for steady-state fast inactivation and steady-state activation for CAD cells treated with 0.1% DMSO (control) or 10–200 μM of (S)-3. Values for V1/2, the voltage of half-maximal inactivation and activation, and the slope factors (k) were derived from Boltzmann distribution fits to the individual recordings and were averaged to determine the mean (±SEM) voltage dependence of steady-state inactivation and activation, respectively. Statistically significant differences between fits of fast inactivation from control cells (0.1% DMSO) compared to 10–200 μM of (S)-3 are indicated by the asterisks (*, p < 0.05, one-way ANOVA). (H) Representative overlaid traces are illustrated by pulses 1 and 30 for control (predrug) and in the presence of (S)-3 (100 μM). (I) Summary of average frequency (use)-dependent decrease in current amplitude over time (±SEM) produced by control (0.1% DMSO) or 100 μM (S)-3. Data are from 4–13 cells per condition.
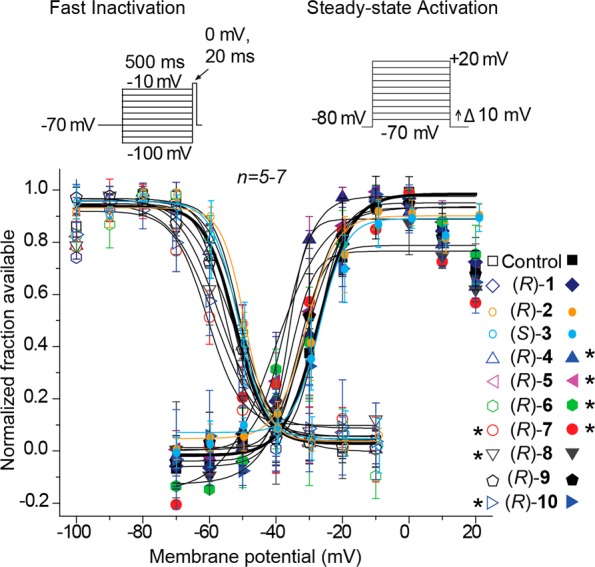
We next asked if (R)-7–(R)-10 and (S)-3 compounds mediate the steady-state fast inactivation property of sodium channels. A fast-inactivated state was induced by the previously described elecrophysiological voltage protocol (Figure 3A, left) and fitted to Boltzmann equation which was then used to derive the V1/2 value of fast inactivation and k, a slope value of the curve (Figure 3B–E).9,11,23 The V1/2 value for inactivation for 0.1% DMSO (control)-treated cells was −68.8 ± 0.4 mV (n = 4), which differed from that of (R)-9 (10 μM)-treated cells (−76.5 ± 0.6 mV; n = 5; p < 0.05; Student’s t test; Figure 3D). The ∼7.7 mV hyperpolarizing shift observed in the presence of (R)-9 was the only V1/2 value significantly different from control (Figure 3B, C, F) (p < 0.05 vs 0.1% DMSO control; Student’s t test). By comparison, we previously observed that (R)-1 affected sodium channel fast inactivation11 but that (R)-2 did not.9,11,23 For (S)-3, compared with control, we observed a pronounced concentration-dependent hyperpolarizing change in the V1/2 value of fast inactivation reaching a maximum at ∼21.6 mV in the presence of 200 μM (S)-3 (Figure 5F). Similarly, we observed that (R)-4 induced a shift of ∼4.2 mV (with 10 μM) and ∼19.7 mV (with 100 μM), and that (R)-5 induced a shift of ∼8.0 mV (with 10 μM) and ∼14.1 mV (with 100 μM), compared with control (∼0.1% DMSO). We found with the chimeric compounds (R)-7–(R)-10, VGSC steady-state activation properties (Figure 3A, right) were unchanged from 0.1% DMSO (control) (Figure 3B–E).
Figure 3.
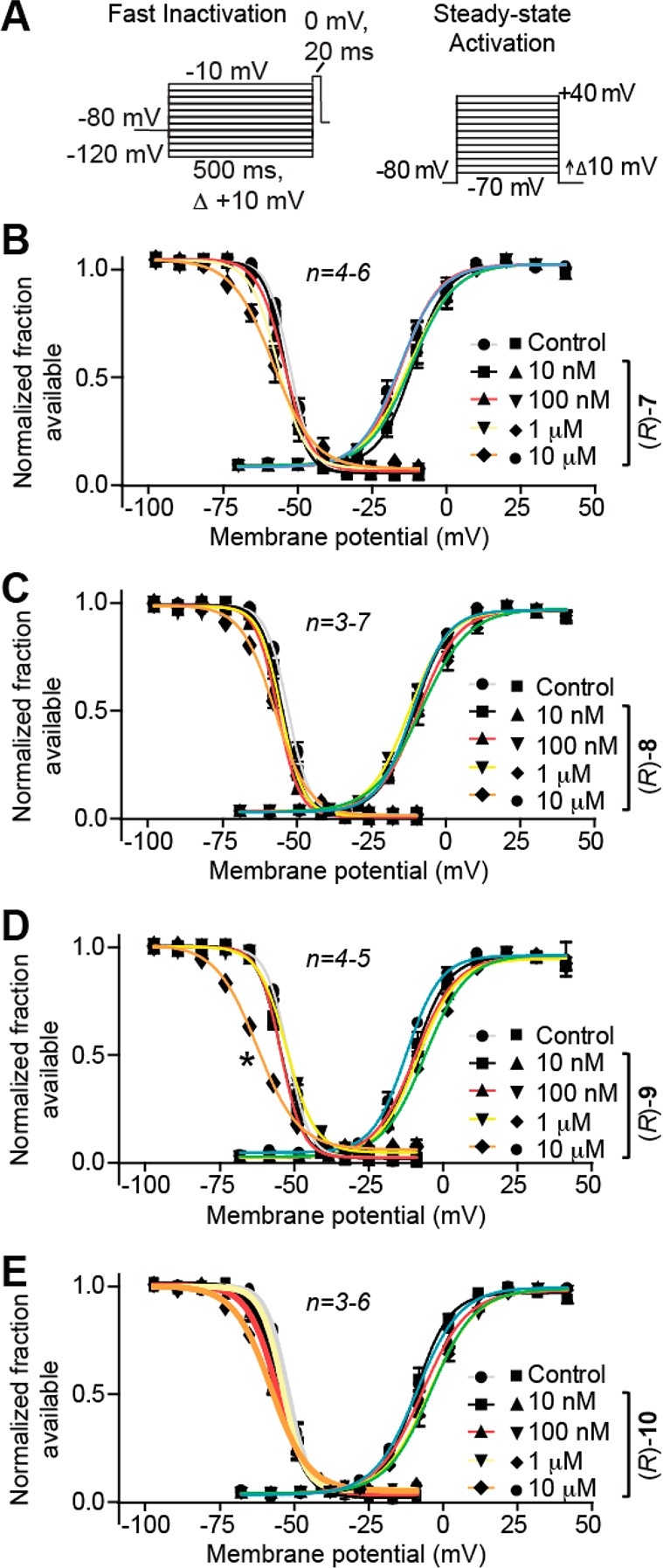
Effects of the chimeric compounds (R)-7–(R)-10 on fast inactivation and steady-state activation states of Na+ currents in CAD cells. (A) Fast inactivation (left) and steady-state activation (right) voltage protocols. (B–E) Representative Boltzmann fits for steady-state fast inactivation and steady-state activation for currents recorded from CAD cells treated with 0.1% DMSO (control) and various concentrations of the indicated compounds are graphed. Values for V1/2, the voltage of half-maximal inactivation and activation, and the slope factors (k) were derived from Boltzmann distribution fits to the individual recordings and averaged to determine the mean (±SEM) voltage dependence of steady-state inactivation and activation, respectively. The V1/2 value of cells treated with 10 μM (R)-9 of −76.5 ± 0.6 (n = 5) was signigifantly greater than that of control (0.1% DMSO) cells (−63.8 ± 0.4 (n = 4); p < 0.05, Student’s t test). The V1/2 and k of steady-state fast inactivation or steady-state fast activation were not different among any of the other compounds tested (p > 0.05, one-way ANOVA). Data from n = 3–7 cells per condition.
Lastly, we tested if (R)-7–(R)-10 and (S)-3 could cause frequency (use)-dependent Na+ current block. The specific property of frequency- or use-dependent block is beneficial for ASDs because it allows for potent sodium channel block during periods of high-firing frequency (i.e., during seizure episodes), but not during periods of low-firing frequency.22 We previously showed for (R)-2, and several structurally related compounds, that concentrations from 5–8-fold greater than the determined inactivation IC50 value were needed to observe frequency (use)-inhibition in CAD cells.23 Here, compounds were tested at 10 μM, a concentration ≥29-fold higher than their inactivation IC50 value, to guarantee observation of use-dependency if the property is present. To monitor this property, 30 repeated test pulses were applied at 10 Hz (Figure 4A inset).7,9,23 The difference in available current was measured by comparing peak current recorded during each pulse as a fraction of the initial pulse current ((pulsen)/(pulse1)). Compounds (R)-9 and (R)-10 revealed frequency (use)-dependent inhibition of Na+ currents (Figure 4B). Currents at the final pulse in cells treated with (R)-9 measured ∼28% lower than 0.1% DMSO control, while the current at the last pulse in cells treated with (R)-10 was ∼39% lower. As a comparsion, our previous study showed 40% reduction of Na+ currents in cells treated with (R)-1 (8.5 μM) and ∼8.8% reduction with (R)-2 (100 μM).23 When, CAD cells were treated with 100 μM (S)-3, we observed a modest level of inhibition (∼13%) of Na+ current (Figure 5I).
B. Cortical Neurons
The effects of (R)-7–(R)-10 in rat embryonic cortical neurons on inactivation, steady-state activation and fast inactivation, and frequency (use)-dependence are reported in Figures 6–8. We add to these figures, newly measured values for the chimeric compounds (R)-1 and (R)-4 – (R)-6 and the two parent compounds (R)-2 and (S)-3. The cortical embryonic neurons were grown for 7–10 days in vitro and then examined using previously described protocols.36 In contrast to CAD cells that express predominantly one isoform of sodium channels (NaV1.7),9,24,25 cortical neurons express varying amounts of NaV1.1–1.3 and NaV1.6.27 Determining the strict contribution of each current type in the additional presence of (R)-7–(R)-10 was not practical, as any results may be confounded by interactions between blockers of various NaV channels and the compounds themselves. We therefore presume the kinetics and measured peaks represent the totality of each CNS NaV isoform, and it is this total recorded current that will be compared between experimental conditions. The compounds were tested only at 10 μM due to experimental constraints largely governed by cortical neuron viability in patch-clamp conditions and limited obtainability of culture preparations.
Figure 6.

Effects of the chimeric compounds ((R)-1, (R)-4–(R)-10) and the parent compounds ((R)-2, (S)-3) on inactivation state of Na+ currents in rat embryonic cortical neurons. (A) Inactivation voltage protocol. Currents were elicited by 5 s prepulses between −100 and +20 mV (in 10 mV increments), and then fast-inactivated channels were allowed to recover for 1000 ms at a hyperpolarized pulse to −70 mV before testing for the fraction of available channels for 20 ms at −10 mV. Finally, the fraction of channels available at −10 mV was analyzed. Representative current traces from cortical neurons in the absence (control, 0.1% DMSO) or presence of 10 μM (R)-4 or (R)-10 are illustrated. The black and pink traces represent the peak current evoked (between −100 to −80 mV and −50 mV, respectively (also highlighted in the voltage protocol as a dashed pink line). (B) Summary of steady-state activation curves for neurons treated with 0.1% DMSO (control) or 10 μM (R)-4. For compounds that mediate inactivation, (R)-4 shown, significant enhancement of inactivation is evident by separation of the curves starting at −80 mV. (C) Summary of the fraction of current available at −50 mV for neurons treated with 0.1% DMSO (control) or 10 μM of the indicated compounds. Asterisks (*) indicate statistically significant differences in fraction of current available between control (0.1% DMSO) and the indicated compounds (p < 0. 05, Student’s t test; n = 5–6 cells per condition).
Figure 8.
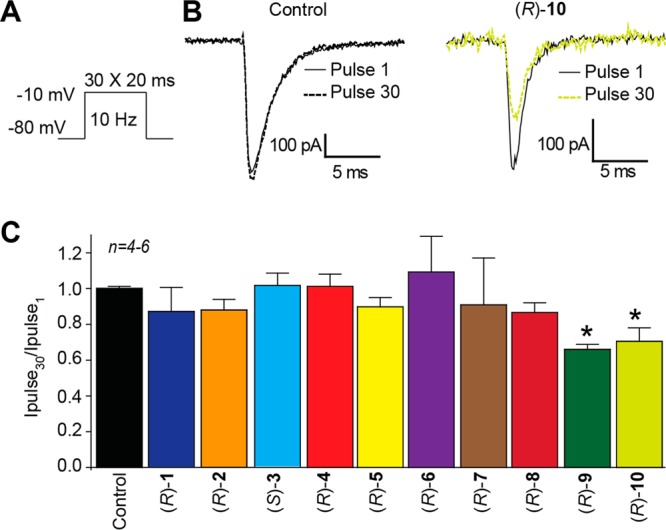
Effects of the chiral compounds ((R)-1, (R)-4–(R)-10) and the parent compounds ((R)-2, (S)-3) on frequency (use)-dependent block of Na+ currents in rat embryonic cortical neurons. (A) Frequency (use)-dependence of block was examined by holding cells at the hyperpolarized potential of −80 mV and evoking currents at 10 Hz by 20 ms test pulses to −10 mV. (B) Representative overlaid traces are illustrated by pulses 1 (black) and 30 (dashed) for control (0.1% DMSO) and in the presence of (R)-10 (10 μM). (C) Summary of the maximal decrement in current amplitude observed at the end of the 30th pulse train for control or 10 μM of the indicated compounds. (R)-9 and (R)-10 caused a significant decrease in current amplitude compared with control (0.1% DMSO) (*p < 0.05, one-way ANOVA with Dunnett’s post hoc test; n = 4–6 cells per condition).
First, we evaluated the ability of the chimeric compounds (R)-1 and (R)-4–(R)-10 to promote an inactivated conformation in response to an inactivation voltage protocol. Voltage-gated sodium channel availability was conditioned by 5 s prepulses between −100 and +20 mV in 10 mV increments.23,34 Channels that undergo fast inactivation but not slow inactivation are allowed to recover during a 1 s pulse to −70 mV before available current is measured at −10 mV for 20 ms. (Figure 6A, left). Hyperpolarization allowed the channels to recover from fast inactivation while limiting recovery from slow inactivation. Differences in available current between control and compound treated cells represent altered sodium channel inactivation via either altered transition into slow-inactivated state or block of fast-inactivated channels with slow off kinetics.29,30 As before, here we did not distinguish between these possibilities; however, both have the same physiological outcome on generation of Na+ currents. Representative traces from cells in the absence and presence of 10 μM (R)-4 and (R)-10 are shown in Figure 6A (right). Full inactivation curves for control (0.1% DMSO) and (R)-4 are shown in Figure 6B. At −50 mV for the control (0.1% DMSO)-treated cells, 0.79 ± 0.06 fractional units (n = 5) of the Na+ current was available and suggested a small fraction (i.e., 0.21 ± 0.06; calculated as 1 minus the normalized INa) of the channels entered inactivated conformational states. This can be compared to (R)-4 (Figure 6B,C), where a large fraction (0.63 ± 0.13) underwent inactivation. In addition to (R)-4, chimeric compounds (R)-1 and (R)-5–(R)-10 significantly enhanced sodium channel inactivation with maximal effects in response to (R)-9 (0.92 ± 0.09, n = 5) and (R)-10 (0.91 ± 0.05, n = 5) (p < 0.01, Mann–Whitney U test). The rank order of chimeric compound effectiveness for promoting inactivation in the cortical neurons mirrored the IC50 values obtained in CAD cells, with the chlorine and trifloromethoxy-group substituted derivatives ((R)-7–(R)-10) being more potent than the unsubstituted compound ((R)-4) or the three fluorine substituted analogues ((R)-1, (R)-5, (R)-6). When (R)-2 and (S)-3 were tested at 10 μM with cortical neurons, neither parent compound affected inactivation, indicating that this concentration was insufficient to facilitate sodium channel inactivation in these cells (Figure 6C).
We then asked if the eight chimeric compounds could enhance steady-state fast inactivation. Fast-inactivated state was induced by the previously described elecrophysiological voltage protocol, and data were fitted to Boltzmann equations to derive V1/2 value of fast inactivation and slope, k (Figure 7, top left).34,36 The inactivation V1/2 for 0.1% DMSO-treated cells was −53.2 ± 1.1 mV (n = 4), which differed significantly from (R)-7- (−60.0 ± 2.1 mV; n = 5), (R)-8- (−57.9 ± 0.7 mV; n = 7), and (R)-10- (−58.4 ± 1.0 mV; n = 8) treated cells (p < 0.05 vs control; Student’s t-test; Figure 7). By comparison, we did not see evidence that the remaining chimeric compounds affected V1/2 or k values of steady-state fast inactivation (Figure 7). At 10 μM, neither (R)-2 nor (S)-3 affected fast inactivation (Figure 7).
Figure 7.
Effects of the chimeric compounds ((R)-1, (R)-4–(R)-10) and the parent compounds ((R)-2, (S)-3) on fast inactivation and steady-state activation states of Na+ currents in rat embryonic cortical neurons. Voltage protocol for fast inactivation (top left) and steady-state activation (top right). Representative Boltzmann fits for steady-state fast inactivation and steady-state activation for cortical neurons treated with 0.1% DMSO (control) and various concentrations of the indicated compounds are shown. Values for V1/2, the voltage of half-maximal inactivation and activation, and the slope factors (k) were derived from Boltzmann distribution fits to the individual recordings and averaged to determine the mean (±SEM) voltage dependence of steady-state inactivation and activation, respectively. Statistically significant differences between control and fast inactivation or steady-state activation are indicated by the asterisks in symbol key (*p < 0.05, one-way ANOVA; n = 5–7 cells per condition).
Next, we tested whether the chimeric compounds could alter the voltage-dependence of sodium channel activation in cortical neurons. Changes in activation for the cortical neurons treated with the compounds were analyzed by comparison of Boltzmann curve derived values V1/2 and slope k (Figure 7, top right).23,34 The steady-state activation V1/2 for 0.1% DMSO-treated (control) neurons was −27.4 ± 1.6 mV (n = 5). Four compounds, (R)-4 (−36.3 ± 4.9 mV; n = 5), (R)-5 (−37 ± 4.4 mV; n = 5), (R)-6 (−32.2 ± 1.9 mV; n = 4), and (R)-7 (−40.4 ± 6.8 mV; n = 5) (p < 0.05 vs control; Student’s t test; Figure 7), displayed significant hyperpolarized shifts. Correspondingly, neither 10 μM (R)-2 nor 10 μM (S)-3 affected the steady-state activation of VGSCs (Figure 7).
Finally, we tested if the chimeric compounds could affect frequency (use)-dependent block of Na+ currents in cortical neurons. Thirty repeated test pulses were applied in rapid succession at 10 Hz (Figure 8A).23 The difference in available current was measured by comparing peak current recorded during each pulse and plotted as a fraction compared to the current recorded at the initial pulse ((pulsen)/(pulse1)). Representative currents are illustrated for control (0.1% DMSO) and 10 μM (R)-10-treated cells (Figure 8B). Of the eight chimeric compounds tested, (R)-9 and (R)-10 displayed significant frequency (use)-dependent inhibition (Figure 8C); peak current recorded at the final pulse was reduced by ∼35% in the presence of 10 μM (R)-9 and ∼31% in the presence of 10 μM (R)-10 compared with control. A comparable finding was observed in CAD cells (Figure 4). At 10 μM, neither of the parent compounds ((R)-2, (S)-3) displayed frequency (use)-inhibition of voltage-gated Na+ currents (Figure 8C).
Whole Animal Pharmacological Activity
The anticonvulsant activity37 for compounds (R)-7–(R)-10 in the MES model12 (mice, ip; rats, po) and the psychomotor 6 Hz (32 mA) seizure test for therapy-resistant limbic seizures16 (mice, ip) were determined at the Anticonvulsant Screening Program (ASP) of the NIH’s National Institute of Neurological Disorders and Stroke (NINDS). The data are summarized in Table 1 along with similar results obtained for chimeric compounds (R)-11 and (R)-4–(R)-6,1 parent compounds (R)-22,13 and (S)-3,5 and the ASDs phenytoin,38 phenobarbital,38 and valproate.38 We report, where possible, the compounds’ 50% effective dose (ED50) values obtained in quantitative screening evaluations, the median doses for 50% neurological impairment (TD50) in either the rotorod test in mice39 or the behavioral toxicity assessment in rats,40 and the corresponding protective index value (PI = TD50/ED50). Compounds (R)-7–(R)-10 were evaluated in the subcutaneous Metrazol (scMet) seizure model.41 No activity was observed at 300 mg/kg (data not shown). Similar findings were found for (R)-1,1 (R)-2,2,13 and structurally related compounds,42 but (S)-3 did display activity in this model (scMet ED50 = 27 mg/kg).5 With the promising activity observed for (R)-1 in the TNI pain model,1 we evaluated (R)-1, (R)-4, (R)-5, and (R)-7–(R)-10 in the formalin pain model17 at the NINDS ASP, and (R)-7–(R)-10 in the TNI model14 at the Indiana University School of Medicine.
Table 1. Structure–Activity Relationship for Substituted (R)-N-4-(Benzyloxy)benzyl 2-Acetamido-3-methoxypropionamide Derivativesa.
| mice (ip)b |
rat (po)g |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| compd no. | X | MES, ED50 (mg/kg)c | 6 Hz ED50 (mg/kg)d | Tox, TD50 (mg/kg)e | PIf | MES, ED50 (mg/kg)c | Tox, TD50 (mg/kg)h | PIf | IC value (μM)i |
| (R)-4j | H | 5.8 [0.25] (4.4–7.2) | <15 [0.25–5.0] | 22 [0.25] (19–25) | 3.8 | 5.6 [0.25] (4.2–6.4) | >250 [1.0] | >45 | 1.6k |
| (R)-5j | 2″-F | 6.7 [0.25] (4.8–9.1) | NDl | 37 [0.5] (29–48) | 5.5 | 11 [0.5] (7.9–13) | >500 | >45 | 1.6k |
| (R)-1j | 3″-F | 13 [0.25] (11–16) | ∼10 [0.25] | 26 [0.5] (21–34) | 2.0 | 14 [0.5] (6.1–27) | >500 [0.25–6.0] | >36 | 1.7k |
| (R)-6j | 4″-F | >10, <30 [0.5] | NDl | >30, <100 [0.5] | 5.8 [0.5] (4.3–7.3) | >500 [0.25–6.0] | >86 | NDl | |
| (R)-7 | 3″-Cl | 16 [0.5] (10–26) | 13 [0.5] (7.7–23) | 190 [2.0] (140–260) | 12 | 39 [6.0] (25–63) | >500 | >13 | 0.34 |
| (R)-8 | 4″-Cl | 7.2 [0.5] (4.3–13) | 7.6 [0.25] (4.5–11) | 49 [0.5] (29–66) | 6.8 | 17 [1.0] (12–25) | >500 | >29 | 0.31 |
| (R)-9 | 3″-OCF3 | 12 [0.5] (6.6–21) | 12 [0.5] (6.8–24) | 38 [0.5] (31–47) | 3.2 | 9.8 [2.0] (4.8–17) | >500 | >51 | 0.24 |
| (R)-10 | 4″-OCF3 | 8.3 [1.0] (7.4–9.8) | 23 [1.0] (14–31) | 39 [0.5] (33–47) | 4.7 | 20 [2.0] (8.9–52) | 250–500 [1.0–6.0] | >13 | 0.14 |
| (R)-2mn | 4.5 [0.5] (3.7–5.5) | 10 [0.5] (7.8–13) | 27 [0.25] (26–28) | 6.0 | 3.9 [2.0] (2.6–6.2) | >500 [0.5] | >130 | 85 | |
| (S)-3o | 4.1 (3.0–5.5) | NRp | NRp | – | 12 (10–14) | NRp | – | 13 | |
| phenytoinq | 9.5 [2.0] (8.1–10) | 66 [2.0] (53–72) | 6.9 | 30 [4.0] (22–39) | >100 | ||||
| phenobarbitalq | 22 [1.0] (15–23) | 69 [0.5] (63–73) | 3.2 | 9.1 [5.0] (7.6–12) | 61 [0.5] (44–96) | 6.7 | |||
| valproateq | 270 [0.25] (250–340) | 430 [0.25] (370–450) | 1.6 | 490 [0.5] (350–730) | 280 [0.5] (190–350) | 0.6 | |||
The compounds were tested through the NINDS ASP.
The compounds were administered intraperitoneally. ED50 and TD50 values are in milligrams per kilogram. Numbers in parentheses are 95% confidence intervals. A dose–response curve was generated for all compounds that displayed sufficient activity. The dose–effect for these compounds was obtained at the “time of peak effect” (indicated in hours in the brackets).
MES = maximal electroshock seizure test.
6 Hz = 6 Hz psychomotor seizure test.
TD50 value determined from the rotorod test.
PI = protective index (TD50/ED50) in the MES test.
The compounds were administered orally. ED50 and TD50 values are in milligrams per kilogram. Numbers in parentheses are 95% confidence intervals. A dose–response curve was generated for all compounds that displayed sufficient activity. The dose–effect for these compounds was obtained at the “time of peak effect” (indicated in hours in the brackets).
Tox = behavioral toxicity.
IC50, concentration at which half of the Na+ channels have transitioned to an inactivated state.
Reference (1).
Reference (11).
ND = not determined.
Reference (2).
Reference (13).
Reference (5).
NR = not reported.
Reference (38).
A. Anticonvulsant Activity
Compounds (R)-7–(R)-10 exhibited excellent activity in the MES seizure model. The ED50 values for (R)-7–(R)-10 in mice (ip) were 7.2–16 mg/kg, and in rats (po) 9.8–39 mg/kg, which were similar to the values observed for phenytoin,38 but higher than those for (R)-22,13 and (S)-35 (ED50 (mice, ip, mg/kg): (R)-7, 16; (R)-8, 7.2; (R)-9, 12; (R)-10, 8.3; (R)-2, 4.5; (S)-3, 4.1; phenytoin, 9.5. ED50 (rat, po, mg/kg): (R)-7, 39; (R)-8, 17; (R)-9, 9.8; (R)-10, 20; (R)-2, 3.9; (S)-3, 12; phenytoin, 30). While no clear structural patterns emerged for the chimeric compounds, we found that replacing the terminal 3″-fluorine substituent in (R)-1 by either a 3″- or 4″-chlorine group provided compounds with substantially higher PI values in mice (PI: (R)-1, 2.0; (R)-7, 12; (R)-8, 6.8). Finally, (R)-7–(R)-10 exhibited potent activity in the 6 Hz (32 mA) psychomotor seizure assay in mice (ip) that compared favorably with (R)-11 and (R)-213 (ED50 mice, ip, mg/kg: (R)-7, 13; (R)-8, 7.6; (R)-9, 12; (R)-10, 23; (R)-1, ∼10; (R)-2, 10).
B. Formalin Pain Model
The formalin model has been advanced as an effective method to prescreen agents for persistent clinical pain17 because of its ease of administration and standardization. Administering formalin to mice leads to biphasic changes of nociceptive behavior,43−45 with many clinically used drugs for neuropathic pain being active in the second (inflammation) phase of this model. In brief, mice are given the test compound ip prior to the formalin (5%) injection into a posterior paw. The formalin injection leads to paw licking in control animals, and possibly the test animals, and then the licking time is counted in 5 min bins.
Compounds (R)-1, (R)-4, (R)-5, and (R)-7–(R)-10 were evaluated at a single dose (5–12 mg/kg) based, in part, on the MES ED50 (mice, ip) values. Compounds (R)-1, (R)-4, (R)-5, (R)-7, and (R)-10 administration led to significant decreases in the percent licking time (49–72% of control) in the second (inflammation) phase of the model (Table 2), suggesting that these compounds may be effective in controlling neuropathic pain. The compounds also showed activity in the first (acute) phase of the test (39–72% of control). Of the chimeric compounds tested, only (R)-8 and (R)-9 did not show activity in this model; we did not examine (R)-2 and (S)-3, but previous studies reported that (R)-2 was effective in the second (inflammation) phase at 8 mg/kg.46
Table 2. Pharmacological Activity of the Substituted (R)-N-4-(Benzyloxy)benzyl 2-Acetamido-3-methoxypropionamide Derivatives in the Formalin Pain Model.
| compd | dose (mg/kg)a | phase I (acute)b | phase II (inflammatory)b |
|---|---|---|---|
| (R)-7 | 5.0 | 41 | 72 |
| (R)-8 | 7.0 | 85 | 103 |
| (R)-9 | 12 | 114 | 98 |
| (R)-10 | 8.0 | 66 | 55 |
| (R)-1 | 9.0 | 39 | 63 |
| (R)-4 | 5.8 | 77 | 52 |
| (R)-5 | 7.0 | 69 | 49 |
| (R)-6 | NDc |
Compounds administered to mice by ip.
Percent time spent licking (s) of control.
ND = not determined.
C. Tibial-Nerve Injury (TNI) Model
Next, we examined the effects of (R)-7–(R)-10 on chronic nociceptive behavior in an animal model of TNI-mediated neuropathic pain (Figure 9A).14 Presurgical response to tactile stimulus evoked hindpaw withdrawal at 70.7 ± 3.1 mN force (n = 6). Drug candidates administered systemically to uninjured animals failed to produce a change in the paw withdrawal threshold (data not shown). Two weeks after TNI, animals exhibited pronounced mechanical allodynia (31.1 ± 2.4 mN; n = 6) in response to von Frey hair stimulation of the injured hindpaw. Compared with postinjury baseline behavioral measurements, we observed pronounced reversal of tactile hypersensitivity 1 h after systemic administration of (R)-7 (10 mg/kg, ip) (∼89%) (Figure 9B). Significant reversal of hypersensitivity was also observed 4 h after systemic injection of (R)-7 (43% of preinjury values). In contrast, systemic administration of (R)-8–(R)-10 was ineffective at reducing hypersensitivity (n = 5–6). By comparison, we reported that after the systemic administration of (R)-1 (10 mg/kg, ip) the near complete reversal of tactile hypersensitivity lasted ∼4 h.11 Correspondingly, (R)-2 significantly reduced tactile hypersensitivity (∼72% of preinjury values 1 h after administration), and (S)-3 showed no effect (Figure 9). The ASD carbamazepine (CBZ; 65 mg/kg, ip) was ineffective (Figure 9B).
Figure 9.
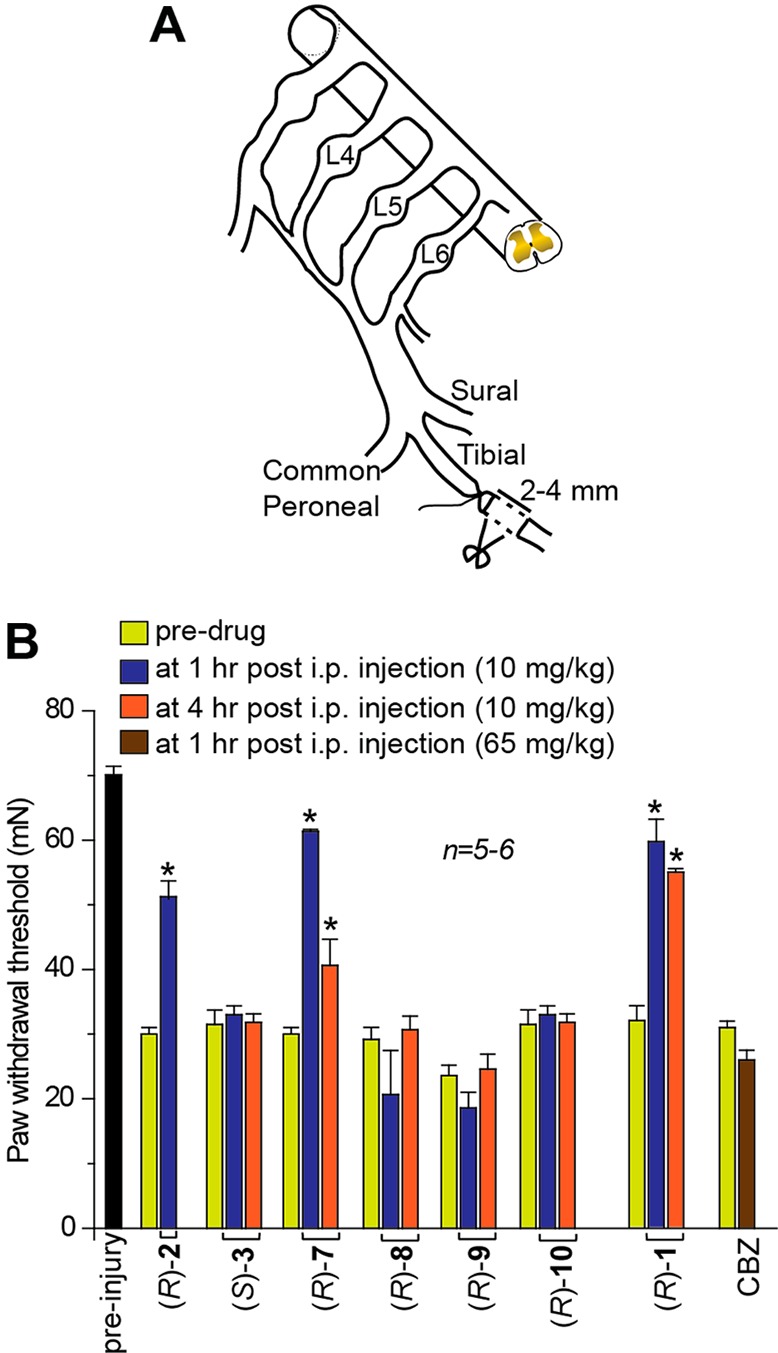
(R)-7 reverses mechanical hypersensitivity in the tibial-nerve injury model of neuropathic pain. (A) Diagram of the sural, tibial, and common peroneal terminal nerve branches of the sciatic nerve and their dorsal root origins. Neuropathic painlike behavior was induced by ligation of the tibial nerve and 2–4 mm of the nerve distal to the ligation was removed. (B) Withdrawal threshold (in millinewtons, mN) in response to von Frey stimulation to the paw ipsilateral to the tibial-nerve injury following a single, intraperitoneal administration of the indicated drugs (10 mg/kg, n = 5–6) on day 14 after injury. A single injection of (R)-7 almost completely reversed tibial-nerve injury induced mechanical hypersensitivity (p < 0.05; Student’s t test) compared with predrug baseline. As a comparison, (R)-2 and (R)-1 also reversed mechanical hypersensitivity.11 Compound (S)-3 and the antiseizure drug carbamazepine (CBZ) were without effect.
Discussion
The chimeric compounds (R)-1 and (R)-4–(R)-10 were designed by merging structural units present in (R)-2 and (S)-3,1 anticonvulsant agents reported to modulate sodium channels (Figure 1).7−10 We and others have shown that (R)-2 enhances the inactivation phase of neuronal VGSCs in response to long (5 s) depolarizations without affecting response to standard fast inactivation protocols using briefer (500 ms) depolarizations.7−9 Compound (R)-2 also inhibited Na+ currents in CAD cells in a frequency (use)-dependent fashion at concentrations above the inactivation IC50 value.23 Correspondingly, Salvati and co-workers showed that (S)-3 reduced whole-cell current density and reduced action potential generation in response to repetative stimuli of cultured hippocampal neurons.10 They did not test whether (S)-3 affected VGSC slow inactivation. Thus, both (R)-2 and (S)-3 affected Na+ currents, but the underlying kinetic modulation of activation or inactivation for each compound may be different. We asked if the chimeric compounds (R)-1 and (R)-4–(R)-10 would display broader and possibly improved cellular and whole animal pharmacological properties compared with either (R)-2 or (S)-3.
The sodium channel properties of the chimeric compounds reported herein were determined in a model cell line (i.e., CAD) as well as in cortical neurons. Direct comparison between these two cell systems was difficult. While the chimeric compounds promoted inactivation, regardless of the cell type, several compounds exhibited differences in modulation of sodium channel fast inactivation (e.g., (R)-1, (R)-7–(R)-10), steady-state activation (e.g., (R)-1, (R)-7), and frequency (use)-inhibition of Na+ currents (e.g., (R)-1) in the two cell systems. These differences may be the result of dissimilar channel isoforms, expression levels of said isoforms, the species, the auxiliary proteins in the two cell types, and the different concentrations of compounds used for each cell type. Accordingly, direct comparisons of sodium channel kinetic properties were restricted to data within the same cell type.
Neuronal hyperexcitability can be contolled by transitioning VGSCs to the fast- and slow-inactivated conformations and by constraining Na+ currents in a frequency (use)-dependent manner.22 The chimeric compounds consistently affected sodium channel activity using voltage protocols designed to affect the slow inactivation pathway. Differences resolved by this protocol, which subjects cells to 5 s depolarizations, can be the result of either enhanced transition into slow-inactivated states, or transition into fast-inactivated state with slow drug off rates.29,30 Enhanced transition to inactivated states in response to this protocol is a favorable characteristic of ASDs. Using this protocol, we found that the chimeric compounds with a terminal substituted aryl group were 50–610-fold more effective than (R)-29,11 and 38–93-fold more effective than (S)-3 in transitioning CAD cells to inactivated states (Figure 2, Table 1). We further showed that the chimeric compound’s terminal X-substituent affected inactivation activity, with the trifluoromethoxy-group ((R)-9, (R)-10) being the most effective, then the chlorine substituent ((R)-7, (R)-8), and last the fluorine substituent ((R)-1, (R)-5). Finally, we found that the 4″-substituted compounds were slightly more potent than the 3″-substituted isomers. Compound (R)-10 was the most potent agent (IC50 (μM): (R)-2, 85; (S)-3, 13; (R)-10, 0.14). We suggest that the enhanced potency for the chimeric compounds compared with (R)-2 and (S)-3 was derived from the synergy gained by judiciously combining, within a single agent, key structural units (Figure 1, A and B) from two compounds that show moderate inactivation properties. Indeed, we have reported that N-(4′-((3′-fluoro)benzyloxy)benzylamine25 (33), a compound that contained the biaryl motif C found in the chimeric agents, transitioned CAD cells to the inactivated state(s) (IC50 μM: (R)-1, 1.7; (R)-2, 85; (S)-3, 13; 33, 31). This result may explain why (S)-3, in part, transitioned CAD cells to the inactivated state more effectively than (R)-2 because (S)-3 alone has an embedded biaryl C motif.

Our determination that (S)-3 promoted inactivation of VGSCs using both fast and slow inactivation protocols and modestly inhibited CAD cells’ Na+ currents in a frequency (use)-dependent manner aligned the electrophysiological properties of this compound with the reported properties of the corresponding 2″-fluorine regioisomer, ralfinamide ((S)-34).47 Stummann and co-workers have described the effects of (S)-34 on dorsal root ganglion Na+ currents. These researchers found that (S)-34 produced a hyperpolarizing move in the steady-state inactivation curve, affected inactivation, and exhibited frequency (use)-dependent inhibition of Na+ currents.

Among the chimeric compounds, we found that, in CAD cells, compounds (R)-1, (R)-4, (R)-5, and (R)-9 affected the VGSC fast inactivation pathway, but (R)-7, (R)-8, and (R)-10 did not (Figure 3). When we tested the two parent compounds, only (S)-3 promoted fast inactivation of VGSCs (Figure 5F). Finally, we observed (R)-1, (R)-9, and (R)-10 repressed Na+ currents in a frequency (use)-dependent manner at concentrations above their inactivation IC50 values similar to (R)-222 and (S)-3 (Figure 5I) but that compounds (R)-4, (R)-5, (R)-7, and (R)-8 did not (Figure 4). Thus, the chimeric compounds all promoted VGSCs to inactivated conformations similar to (R)-2 and (S)-3, but they displayed a range of fast inactivation and frequency (use)-dependent inhibition properties.
We recently described the pharmacological activities of a set of chimeric compounds, (R)-D, similar to (R)-1 and (R)-4–(R)-10, in which a single bond separated the two terminal aromatic rings,23 rather than the oxymethylene (OCH2) linkage in the series described herein.23 Like the present series, (R)-D contained a biaryl C motif (L = single bond).

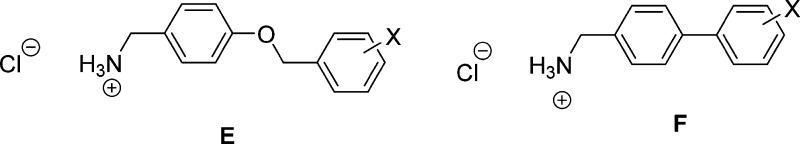
For (R)-D, we prepared the fluorine, chlorine, and trifluoromethoxy-group derivatives where the X-substituent was placed at either the 3″- or the 4″-position. When we compared the CAD cell and rat embryonic cortical neuron data for both series, we found similar results. Both potently supported sodium channel inactivation, with the trifluoromethoxy-group compounds being the most effective, followed by the chlorine and then the fluorine-substituted compounds. In both series, some, but not all, compounds affected fast inactivation protocol results, and some compounds limited Na+ currents in a frequency (use)-dependent manner. These results suggested that the different linkers (L = OCH2, single bond) bridging the two aromatic rings did not markedly affect how these chimeric compounds modulated sodium channel activity. The insensitivity of the composition and length of the linker in the chimeric compound on VGSC inactivation was not surprising. We obtained similar findings for the corresponding biaryl-type amines E and F using rat embryonic cortical neurons.20,36 These results are interesting, since the significant increase in inactivation observed for the chimeric compounds compared with (R)-2 has been attributed, in part, to the chimeric compound’s biaryl C motif and the enhanced interaction of these compounds with the channel (receptor) responsible for inactivation.25 These collective findings reinforce the need for additional information concerning the factors that govern this interaction and the apparent structural latitude for the terminal biaryl C unit in the chimeric compounds.
The enhanced potency of chimeric compounds (R)-1 and (R)-4–(R)-10 to promote sodium channel inactivation compared with (R)-2 and (S)-3 did not translate to an increase in anticonvulsant activity for these agents over the parent compounds. Rather, we observed that the chimeric compounds ((R)-4–(R)-10) were slightly less effective in controlling seizures in the MES model in mice (ip) than either (R)-22,13 or (S)-35 (ED50 mg/kg: (R)-1, 13; (R)-2, 4.5; (S)-3, 4.1; (R)-4, 5.8; (R)-5, 6.7; (R)-16, >10, <30; (R)-7, 16, (R)-8, 7.2; (R)-9, 12; (R)-10, 8.3). A parallel finding was seen in the MES test in rats (po). We have not determined what accounts for this finding, but we recognize that multiple properties beyond the interaction of the agent with the target site(s) determine efficacy in animal models. Similarly, cellular electrophysiological studies do not fully recapitulate the anticonvulsant test events.
The data from the formalin pain model (Table 2) showed that at low doses (5–9 mg/kg) most of the chimeric compounds evaluated ((R)-1, (R)-4, (R)-5, (R)-7, (R)-10) substantially reduced mouse discomfort in phase 2 (49–72% of control) and phase 1 (39–77% of control) of the test. Only (R)-8 and (R)-9 were ineffective in this model. Further support for (R)-7′s pain-attenuating properties was gained by the mechanical hypersensitivity reversal in the TNI model14 (Figure 9). The finding for (R)-7 mirrored that reported for (R)-1.11 Structurally, these compounds are nearly identical with the only difference being the 3″-halogen substituent ((R)-1, 3″-F; (R)-7, 3″-Cl). Unlike the formalin model, the activity profile in the TNI test was sensitive to the terminal aromatic substituent. Repositioning the chlorine moiety in (R)-7 or replacing the chlorine moiety by a trifluoromethoxy group led to complete loss of activity. Earlier, we suggested that (R)-1′s strong effects in promoting fast, and possibly slow, inactivation in CAD cells was responsible for the reversal of tactile hypersensitivity in the TNI model.11 Our finding that (R)-7 is nearly as effective as (R)-1 in this model and that (R)-2 administration also led to hypersensitivity reversal (Figure 9) but that these compounds did not affect fast inactivation in CAD cells (Figure 3) places this hypothesis in doubt.
Conclusions
A select series of substituted (R)-N-(4′-(benzyloxy)benzyl 2-acetamido-3-methoxypropionamides were prepared by merging key units in (R)-2 and (S)-3 and positioning an electron-withdrawing (Cl, OCF3) group at either the 3″- or 4″-site of the terminal aromatic ring. The electrophysiological properties documented that these compounds strongly transitioned sodium channels to the inactivated state. The IC50 values of CAD cells for (R)-7–(R)-10 were 250–610-fold more potent than (R)-2 and 38–93-fold more potent than (S)-3. The chimeric compounds’ high potency has been credited to the strategic merger of units present in the two neurological compounds (R)-2 and (S)-3. The chimeric compounds displayed excellent anticonvulsant activities in animal seizure models and promising activities in pain models.
Methods
General Methods
The general methods employed in this study were the same as those previously reported,2 and are summarized in the Supporting Information. Compounds were checked by TLC, 1H and 13C NMR, MS, and either HR-MS or elemental analysis. For all new compounds evaluated in the pharmacological models, elemental analysis was obtained. The TLC, NMR, and analytical data confirmed the purity of the final products was ≥95%.
General Procedure for the Deprotection and Acetylation of (R)-Substituted N-Benzyl 2-N-(tert-Butoxycarbonyl)amino-3-methoxypropionamide Derivatives (Method 1)
The N-(tert-butoxycarbonyl compound was dissolved in CH2Cl2 (0.1–0.3 M) and then treated with 4 M HCl in dioxane (3–4 equiv) at room temperature (2–12 h). The mixture was concentrated in vacuo, and the residue dissolved in CH2Cl2 (0.1–0.3 M) and then triethylamine (2–3 equiv) and acetyl chloride (1.0–1.2 equiv) were carefully added (0 °C). The resulting solution was stirred at room temperature (2–16 h), and then successively washed with an aqueous 10% citric acid solution and a saturated aqueous NaHCO3 solution. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by column chromatography on SiO2 and/or recrystallized with EtOAc/hexanes.
Preparation of (R)-N-4′-(3″-Chlorobenzyloxy)benzyl 2-Acetamido-3-methoxypropionamide ((R)-7)
Employing Method 1, (R)-29 (8.18 g, 18.3 mmol), 4 M HCl (15.98 mL, 63.9 mmol), Et3N (5.60 mL, 40.2 mmol), and AcCl (1.42 mL, 20.1 mmol) gave (R)-7 as a white solid (5.86 g, 82%): Rf = 0.40 (MeOH/CH2Cl2 1/20); mp 170–172 °C; [α]26D −18.1° (c 1.2, CHCl3). IR (nujol) 3282, 3082, 2923, 2860, 1634, 1551, 1456, 1378, 1246, 1130, 1058, 980, 777, 716 cm–1. 1H NMR (CDCl3) δ 2.00 (s, C(O)CH3), 3.35 (s, OCH3), 3.42–3.46 (m, CHH′OCH3), 3.77 (dd, J = 4.0, 9.0 Hz, CHH′OCH3), 4.32–4.43 (m, CH2N), 4.53–4.58 (m, CH), 5.01 (s, OCH2), 6.55 (d, J = 6.4 Hz, NHCH), 6.83–6.91 (m, 2 ArH, NHCH2), 7.18 (d, J = 8.4 Hz, 2 ArH), 7.26–7.42 (m, 4 ArH); addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-7 gave only one signal for the acetyl methyl and one signal for the ether methyl protons. 13C NMR (CDCl3) δ 23.4 (C(O)CH3), 43.2 (NCH2), 52.6 (OCH2CH), 59.2 (OCH3), 69.4 (OCH2), 72.0 (OCH2CH), 115.2, 125.5, 127.6, 128.3, 129.1, 130.1, 130.8, 134.7, 139.2, 158.0 (ArC), 170.1, 170.5 (2 C(O)). LRMS (ESI+) 413.2 [M + Na]+ (calcd for C20H23ClN2O4Na+ 413.2). Anal. Calcd for C20H23ClN2O4: C, 61.46; H, 5.93; Cl, 9.07; N, 7.17. Found: C, 61.55; H, 5.97; Cl, 8.89; N, 7.17.
Preparation of (R)-N-4′-(4″-Chlorobenzyloxy)benzyl 2-Acetamido-3-methoxypropionamide ((R)-8)
Employing Method 1, (R)-30 (4.80 g, 10.7 mmol), 4 M HCl (9.38 mL, 37.5 mmol), Et3N (3.28 mL, 23.5 mmol), and AcCl (0.83 mL, 11.8 mmol) gave (R)-8 as a white solid (3.65 g, 87%): Rf = 0.40 (MeOH/CH2Cl2 1/20); mp 180–181 °C; [α]26D −17.9° (c 1.1, CHCl3). IR (nujol) 3280, 3103, 2924, 2859, 1635, 1553, 1458, 1375, 1240, 1098, 1047, 815, 727, 608 cm–1. 1H NMR (CDCl3) δ 1.99 (s, C(O)CH3), 3.35 (s, OCH3), 3.42–3.46 (m, CHH’OCH3), 3.76 (dd, J = 4.0, 9.0 Hz, CHH’OCH3), 4.31–4.42 (m, CH2N), 4.54–4.59 (m, CH), 5.00 (s, OCH2), 6.58 (d, J = 6.4 Hz, NHCH), 6.85–6.92 (m, 2 ArH, NHCH2), 7.17 (d, J = 8.0 Hz, 2 ArH), 7.34 (s, 4 ArH); addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-8 gave only one signal for the acetyl methyl and one signal for the ether methyl protons. 13C NMR (CDCl3) δ 23.3 (C(O)CH3), 43.2 (NCH2), 52.6 (OCH2CH), 59.2 (OCH3), 69.4 (OCH2), 72.1 (OCH2CH), 115.2, 128.9, 128.9, 129.0, 130.7, 133.9, 135.6, 158.1 (ArC), 170.1, 170.5 (2 C(O)). LRMS (ESI+) 391.2 [M + H]+ (calcd for C20H23ClN2O4H+ 391.2). Anal. Calcd for C20H23ClN2O4: C, 61.46; H, 5.93; Cl, 9.07; N, 7.17. Found: C, 61.75; H, 5.86; Cl, 9.15; N, 7.11.
Preparation of (R)-N-4′-(3″-Trifluoromethoxybenzyloxy)benzyl 2-Acetamido-3-methoxypropionamide ((R)-9)
Employing Method 1, (R)-31 (4.50 g, 9.0 mmol), 4 M HCl (7.90 mL, 31.6 mmol), Et3N (2.77 mL, 19.9 mmol), and AcCl (0.70 mL, 9.9 mmol) gave (R)-9 as a white solid (3.50 g, 88%): Rf = 0.40 (MeOH/CH2Cl2 1/20); mp 139–140 °C; [α]26D −15.1° (c 1.0, CHCl3); IR (nujol) 3281, 3081, 2924, 2860, 1632, 1550, 1456, 1381, 1281, 1141, 1060, 974, 822, 710, 618 cm–1. 1H NMR (CDCl3) δ 1.98 (s, C(O)CH3), 3.35 (s, OCH3), 3.43–3.47 (m, CHH′OCH3), 3.76 (dd, J = 4.0, 9.2 Hz, CHH′OCH3), 4.32–4.43 (m, CH2N), 4.56–4.61 (m, CH), 5.04 (s, OCH2), 6.63 (d, J = 6.8 Hz, NHCH), 6.89–6.97 (m, 2 ArH, NHCH2), 7.18 (d, J = 8.4 Hz, 2 ArH), 7.29–7.42 (m, 4 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-9 gave only one signal for the acetyl methyl and one signal for the ether methyl protons. 13C NMR (CDCl3) δ 23.3 (C(O)CH3), 43.1 (NCH2), 52.7 (OCH2CH), 59.2 (OCH3), 69.2 (OCH2), 72.1 (OCH2CH), 115.2, 119.3, 120.4 (ArC), 120.6 (q, J = 256.4 Hz, OCF3), 125.6, 129.1, 130.1, 130.9, 139.5, 149.6, 158.0 (ArC), 170.1, 170.5 (2 C(O)). LRMS (ESI+) 441.1 [M + H]+ (calcd for C21H23F3N2O5H+ 441.1). Anal. Calcd for C21H23F3N2O5: C, 57.27; H, 5.26; F, 12.94; N, 6.36. Found: C, 57.08; H, 5.20; F, 12.88; N, 6.30.
Preparation of (R)-N-4′-(4″-Trifluoromethoxybenzyloxy)benzyl 2-Acetamido-3-methoxypropionamide ((R)-10)
Employing Method 1, (R)-32 (3.68 g, 7.4 mmol), 4 M HCl (6.47 mL, 25.9 mmol), Et3N (2.27 mL, 16.3 mmol), and AcCl (0.58 mL, 8.1 mmol) gave (R)-10 as a white solid (3.10 g, 95%): Rf = 0.40 (MeOH/CH2Cl2 1/20); mp 172–173 °C; [α]26D −16.0° (c 1.1, CHCl3); IR (nujol) 3281, 3102, 2923, 2860, 1635, 1552, 1457, 1378, 1275, 1233, 1148, 1021, 835, 730, 609 cm–1. 1H NMR (CDCl3) δ 1.99 (s, C(O)CH3), 3.35 (s, OCH3), 3.43–3.47 (m, CHH′OCH3), 3.76 (dd, J = 4.0, 9.2 Hz, CHH′OCH3), 4.32–4.43 (m, CH2N), 4.56–4.60 (m, CH), 5.03 (s, OCH2), 6.61 (d, J = 6.4 Hz, NHCH), 6.89–6.94 (m, 2 ArH, NHCH2), 7.18 (d, J = 8.4 Hz, 2 ArH), 7.22 (d, J = 8.4 Hz, 2 ArH), 7.44 (d, J = 8.4 Hz, 2 ArH); addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-10 gave only one signal for the acetyl methyl and one signal for the ether methyl protons. 13C NMR (CDCl3) δ 23.3 (C(O)CH3), 43.1 (NCH2), 52.7 (OCH2CH), 59.2 (OCH3), 69.3 (OCH2), 72.0 (OCH2CH), 115.1 (ArC), 120.6 (q, J = 255.7 Hz, OCF3), 121.3, 129.0, 129.1, 130.8, 135.8, 149.0, 158.1 (ArC), 170.1, 170.5 (2 C(O)). LRMS (ESI+) 441.1 [M + H]+ (calcd for C21H23F3N2O5H+ 441.1). Anal. Calcd for C21H23F3N2O5: C, 57.27; H, 5.26; F, 12.94; N, 6.36. Found: C, 57.35; H, 5.28; F, 12.78; N, 6.38.
Pharmacology
Compounds were screened under the auspices of the NINDS’ ASP. Experiments were performed in male rodents (albino Carworth Farms No. 1 mice (ip), albino Sprague–Dawley rats (ip, po)). Housing, handling, and feeding complied with recommendations contained in the Guide for the Care and Use of Laboratory Animals. Anticonvulsant activity was determined using the MES test,12 6 Hz,16 and the scMet test,41 and pain-attenuating activity using the formalin test17 according to previously reported methods.1,2
Tibial-Nerve Injury
Pathogen-free, adult female Sprague–Dawley (S/D) rats (150–200 g; Harlan Laboratories, Madison, WI) were housed in temperature (23 ± 3 °C) and light (12 h light/12 h dark cycle; lights on at 07:00 h) controlled rooms with standard rodent chow and autoclaved tap water available. Experiments were performed during the light cycle. Animals were randomly assigned to the treatment groups. All animal experiments were approved by the Institutional Animal Care and Use Committees of Indiana University School of Medicine. All procedures were conducted in accordance with the Guide for Care and Use of Laboratory Animals published by the NIH and the ethical guidelines established by the International Association for the Study of Pain.
To model neuropathic pain, we performed a tibial-nerve injury (TNI) as previously described.11,14,48,49 Under anesthesia, rodents were subjected to a tibial nerve ligation with 5–0 silk and the nerve transected distal to the ligation. In addition, removal of 2–4 mm of distal nerve stump prevented reinnervation by the proximal nerve. The overlying tissue layers of muscle and skin were sutured and animals left to recover.
Mechanical threshold was tested before, during and following administration of candidate coumpounds by an experimenter blinded to treatment groups. Behavioral baseline measurements were collected from at least 2 separate days prior to the surgery. The rats were then tested once weekly after the surgery. The incidence of foot withdrawal was measured as a function in response to mechanical indentation of the plantar surface of each hind paw with custom Von Frey-type filaments as described previously.50
Catecholamine A-Differentiated (CAD) Cells
CAD cells were grown at 37 °C and in 5% CO2 (Sarstedt, Newton, NC) in Ham’s F12/DMEM (GIBCO, Grand Island, NY), supplemented with 10% fetal bovine serum (Sigma, St. Louis, MO) and 1% penicillin/streptomycin (100% stocks, 10 000 U/mL penicillin G sodium and 10 000 μg/mL streptomycin sulfate).9,15 Cells were passaged every 3–5 days at a 1:5 dilution.
Cortical Neurons
Rat cortical neuron cultures were prepared from cortices dissected from embryonic day 19 brains exactly as described.51,52
Electrophysiology
Whole-cell voltage clamp recordings were performed at room temperature on CAD cells and cortical neurons using an EPC 10 amplifier (HEKA Electronics, Lambrecht/Pfalz Germany). Electrodes were pulled from thin-walled borosilicate glass capillaries (Warner Instruments, Hamden, CT) with a P-97 electrode puller (Sutter Instrument, Novato, CA) such that final electrode resistances were 1–2 MΩ when filled with internal solutions. The internal solution for recording Na+ currents contained (in mM): 110 CsCl, 5 MgSO4, 10 EGTA, 4 ATP Na2-ATP, 25 HEPES (pH 7.2, 290–310 mOsm/L). The external solution contained (in mM): 100 NaCl, 10 tetraethylammonium chloride (TEA-Cl), 1 CaCl2, 1 CdCl2, 1 MgCl2, 10 d-glucose, 4 4-AP, 0.1 NiCl2, 10 HEPES (pH 7.3, 310–315 mOsm/L). Whole-cell capacitance and series resistance were compensated with the amplifier. Series resistance error was always compensated to be less than ±3 mV. Cells were considered only when the access resistance was less than 3 MΩ. Linear leak currents were digitally subtracted by −P/4 leak subtraction. Unless otherwise specified, protocols were preformed with 3 s between the ending of a voltage pulse and beginning of the next. Maximum current tested at the end of each protocol verified that 3 s was sufficient to re-equilibrate channel availability.
Data Acquisition and Analysis
Signals were filtered at 10 kHz and digitized at 10–20 kHz. Analysis was performed using Fitmaster and origin8.1 (OriginLab Corporation, Northampton, MA). For activation curves, conductance (G) through sodium channels was calculated using the equation G = I/(Vm – Vrev), where Vrev is the reversal potential, Vm is the membrane potential at which the current was recorded, and I is the peak current. Activation and inactivation curves were fitted to a single-phase Boltzmann function G/Gmax = 1/{1 + exp[(V – V50)/k]}, where G is the peak conductance, Gmax is the fitted maximal G, V50 is the half activation voltage, and k is the slope factor. Additional details of specific pulse protocols are described in the Results text or figure legends.
Statistical Analyses
Differences between means were compared by either paired or unpaired, two-tailed Student’s t tests or an analysis of variance (ANOVA), when comparing multiple groups (repeated measures whenever possible). If a significant difference was determined by ANOVA, then a Dunnett’s or Tukey’s post hoc test was performed. Data are expressed as mean ± SEM, with p < 0.05 considered as the level of significance.
Acknowledgments
We are grateful to the NINDS and the ASP at the National Institutes of Health with Drs. Tracy Chen, Jeffrey Jiang, and John Kehne for kindly conducting the pharmacological studies through the ASP’s contract site at the University of Utah with Drs. H. S. White, H. Wolfe, and K. Wilcox.
Glossary
Abbreviations
- AAA
α-aminoamide
- ASD
antiseizure drug
- ASP
Anticonvulsant Screening Program
- CAD
catecholamine A-differentiated
- CF3O
trifluoromethoxy
- CNS
central nervous system
- ED50
effective dose (50%)
- FAA
functionalized amino acid
- IBCF
isobutyl chloroformate
- IC50
concentration at which half of the channels have transitioned to a inactivated state
- ip
intraperitoneally
- MAC
mixed anhydride coupling
- MES
maximal electroshock seizure
- NINDS
National Institutes of Neurological Disorders and Stroke
- NMM
N-methylmorpholine
- PI
protective index
- po
orally
- scMet
scMetrazol
- TD50
neurological impairment (toxicity, 50%)
- TEA-Cl
tetraethylammonium chloride
- TNI
tibial-nerve injury
- V1/2
voltage of half-maximal (in)activation
- VGSC
voltage-gated sodium channel
Supporting Information Available
Synthetic procedures, experimental and spectral data for the intermediates and final products evaluated in this study including elemental analyses, high-resolution MS data; 1H NMR and 13C NMR spectra for (R)-7–(R)-10 and (S)-3. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
∇ K.D.P.: Center for Neuro-Medicine, Brain Science Institute, Korea Institute of Science and Technology, Seoul, Korea.
Author Contributions
K.D.P. synthesized the compounds. X.-F.Y., E.T.D., and Y.W. preformed whole-cell electrophysiology experiments. M.S.R. performed animal behavior experiments. F.A.W. analyzed the behavior data. K.D.P., R.K., and H.K. designed this study, and R.K. and H.K. wrote the manuscript and F.A.W. contributed to writing the behavioral section.
This work is supported by grants, in part, from the NINDS (1 R41 NS080278) and a National Scientist Development Award from the American Heart Association (SDG5280023 to R.K.) and NIDA (DA026040 to F.A.W.), and NIDDK (DK091694 to F.A.W.).
The content of this paper is solely the responsibility of the authors and does not constitute the official views of the National Center for Research Resources, NIH.
The authors declare the following competing financial interest(s): Harold Kohn has a royalty-stake position in (R)-2 and is the founder of NeuroGate Therapeutics, Inc.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Salome C.; Salome-Grosjean E.; Stables J. P.; Kohn H. (2010) Merging the structural motifs of functionalized amino acids and α-aminoamides: compounds with significant anticonvulsant activities. J. Med. Chem. 53, 3756–3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi D.; Stables J. P.; Kohn H. (1996) Synthesis and anticonvulsant activities of N-benzyl-2-acetamidopropionamide derivatives. J. Med. Chem. 39, 1907–1916. [DOI] [PubMed] [Google Scholar]
- Pevarello P.; Bonsignori A.; Dostert P.; Heidempergher F.; Pinciroli V.; Colombo M.; McArthur R. A.; Salvati P.; Post C.; Fariello R. G.; Varasi M. (1998) Synthesis and anticonvulsant activity of a new class of 2-[(arylalkyl)amino]alkanamide derivatives. J. Med. Chem. 41, 579–590. [DOI] [PubMed] [Google Scholar]
- Perucca E.; Yasothan U.; Clincke G.; Kirkpatrick P. (2008) Lacosamide. Nat. Rev. Drug Discovery 7, 973–974. [DOI] [PubMed] [Google Scholar]
- Fariello R. G.; McArthur R. A.; Bonsignori A.; Cervini M. A.; Maj R.; Marrari P.; Pevarello P.; Wolf H. H.; Woodhead J. W.; White H. S.; Varasi M.; Salvati P.; Post C. (1998) Preclinical evaluation of PNU-151774E as a novel anticonvulsant. J. Pharm. Exp. Therap. 285, 397–403. [PubMed] [Google Scholar]
- Schapira A. H.; Stocchi F.; Borgohain R.; Onofrj M.; Bhatt M.; Lorenzana P.; Lucini V.; Giuliani R.; Anand R. (2013) Long-term efficacy and safety of safinamide as add-on therapy in early Parkinson’s disease. Eur. J. Neurol. 20, 271–280. [DOI] [PubMed] [Google Scholar]
- Errington A. C.; Stohr T.; Heers C.; Lees G. (2008) The investigational anticonvulsant lacosamide selectively enhances slow inactivation of voltage-gated sodium channels. Mol. Pharmacol. 73, 157–169. [DOI] [PubMed] [Google Scholar]
- Sheets P. L.; Heers C.; Stoehr T.; Cummins T. R. (2008) Differential block of sensory neuronal voltage-gated sodium channels by lacosamide, lidocaine and carbamazepine. J. Pharmacol. Exp. Ther. 326, 89–99. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Park K. D.; Salome C.; Wilson S. M.; Stables J. P.; Liu R.; Khanna R.; Kohn H. (2011) Development and characterization of novel derivatives of the antiepileptic drug lacosamide that exhibit far greater enhancement in slow inactivation of voltage-gated sodium channels. ACS Chem. Neurosci. 2, 90–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvati P.; Maj R.; Caccia C.; Cervini M. A.; Fornaretto M. G.; Lamberti E.; Pevarello P.; Skeen G. A.; White H. S.; Wolf H. H.; Faravelli L.; Mazzanti M.; Mancinelli M.; Varasi M.; Fariello R. G. (1999) Biochemical and electrophysiological studies on the mechanism of action of PNU-151774E, a novel antiepileptic compound. J. Pharmacol. Exp. Ther. 288, 1151–1159. [PubMed] [Google Scholar]
- Wang Y.; Wilson S. M.; Brittain J. M.; Ripsch M. S.; Salome C.; Park K. D.; White F. A.; Khanna R.; Kohn H. (2011) Merging structural motifs of functionalized amino acids and α-aminoamides results in novel anticonvulsant compounds with significant effects on slow and fast inactivation of voltage-gated sodium channels and in the treatment of neuropathic pain. ACS Chem. Neurosci. 2, 317–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White H. S., Woodhead J. H., Franklin M. R., Swinyard E. A., and Wolf H. H. (1995) General principles: Experimental selection, quantification, and evaluation of antiepileptic drugs. In Antiepileptic Drugs (Levy R. H., Mattson R. H., and Meldrum B. S., Eds.), 4th ed., pp 99–110, Raven Press, New York. [Google Scholar]
- Stoehr T.; Kupferberg H. J.; Stables J. P.; Choi D.; Harris R. H.; Kohn H.; Walton N.; White H. S. (2007) Lacosamide, a novel anticonvulsant drug, shows efficacy with a wide safety margin in rodent models for epilepsy. Epilepsy Res. 74, 147–154. [DOI] [PubMed] [Google Scholar]
- Lee B. H.; Won R.; Balk E. J.; Lee S. H.; Moon C. H. (2000) An animal model of neuropathic pain employing injury to the sciatic nerve branches. NeuroReport 11, 657–661. [DOI] [PubMed] [Google Scholar]
- Wang H.; Oxford G. S. (2000) Voltage-dependent ion channels in CAD cells: A catecholaminergic neuronal line that exhibits inducible differentiation. J. Neurophysiol. 84, 2888–2895. [DOI] [PubMed] [Google Scholar]
- Barton M. E.; Klein B. D.; Wolff H. H.; White H. S. (2001) Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res. 47, 217–227. [DOI] [PubMed] [Google Scholar]
- Tjolsen A.; Berge O.-G.; Hunskaar S.; Rosland J. H.; Hole K. (1992) The formalin test: An evaluation of the method. Pain 51, 5–17. [DOI] [PubMed] [Google Scholar]
- Anderson G. W.; Zimmerman J. E.; Callahan F. M. (1967) A reinvestigation of the mixed anhydride method of peptide synthesis. J. Am. Chem. Soc. 89, 5012–5017. [DOI] [PubMed] [Google Scholar]
- Lebreton L.; Curet O.; Gueddari S.; Mazouz F.; Bernard S.; Burstein C.; Milcent R. (1995) Selective and potent monoamine oxidase type B inhibitors: 2-substituted 5-aryltetrazole derivatives. J. Med. Chem. 38, 4786–4792. [DOI] [PubMed] [Google Scholar]
- Lee H.; Gold A. S.; Yang X.-F.; Khanna R.; Kohn H. (2013) Benzyloxybenzylammonium chlorides: Simple amine salts that display anticonvulsant activity. Bioorg. Med. Chem. 21, 7655–7662. [DOI] [PubMed] [Google Scholar]
- For a comparable procedure for resolving stereoisomers, see:Weisman G. R.Nuclear magnetic resonance analysis using chiral solvating agents. (1983) In Asymmetric Synthesis-Analytical Methods (Morrison J. D., Ed.), Vol. 1, Chapter 8, pp 153–172, Academic Press, New York. [Google Scholar]
- Errington A. C.; Stohr T.; Lees G. (2005) Voltage gated ion channels: Targets for anticonvulsant drugs. Curr. Top. Med. Chem. 5, 15–30. [DOI] [PubMed] [Google Scholar]
- Lee H.; Park K. D.; Torregrosa R.; Yang X.-F.; Dustrude E. T.; Wang Y.; Wilson S. M.; Barbosa C.; Xiao Y.; Cummins T. R.; Khanna R.; Kohn H. (2014) Substituted N-(Biphenyl-4′-yl)methyl (R)-2-acetamido-3-methoxypropionamides: Potent anticonvulsants that affect frequency (use)-dependence and slow inactivation of sodium channels. J. Med. Chem. 57, 6165–6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Brittain J. M.; Jarecki B. W.; Park K. D.; Wilson S. M.; Wang B.; Hale R.; Meroueh S. O.; Cummins T. R.; Khanna R. (2010) In silico docking and electrophysiological characterization of lacosamide binding sites on collapsin response mediator protein 2 (CRMP-2) identifies a pocket important in modulating sodium channel slow inactivation. J. Biol. Chem. 285, 25296–25307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King A. M.; Yang X.-F.; Wang Y.; Dustrude E. T.; Barbosa C.; Due M. R.; Piekarz A. D.; Wilson S. M.; White F. A.; Salome C.; Cummins T. R.; Khanna R.; Kohn H. (2012) Identification of the benzyloxyphenyl pharmacophore: A structural unit that promotes sodium channel slow inactivation. ACS Chem. Neurosci. 3, 1037–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib-Hajj S. D.; Yang Y.; Black J. A.; Waxman S. G. (2013) The NaV 1.7 sodium channel from molecule to man. Nat. Rev. Neurosci. 14, 49–62. [DOI] [PubMed] [Google Scholar]
- Goldin A. L. (2001) Resurgence of sodium channel research. Annu. Rev. Physiol. 63, 871–894. [DOI] [PubMed] [Google Scholar]
- Catterall W. A.; Goldin A. L.; Waxman S. G. (2005) International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharm. Rev. 57, 397–409. [DOI] [PubMed] [Google Scholar]
- Kuo C.-C.; Bean B. P. (1994) Slow binding of phenytoin to inactivated sodium channels in rat hippocampal neurons. Mol. Pharmacol. 46, 716–725. [PubMed] [Google Scholar]
- Kahlig K. K.; Hirakawa R.; Liu L.; George A. L. Jr.; Belardinelli L.; Rajamani S. (2014) Ranolazine reduces neuronal hyperexcitability by interacting with inactivated states of brain sodium channels. Mol. Pharmacol. 85(1), 162–174. [DOI] [PubMed] [Google Scholar]
- Hodgkin A. L.; Huxley A. F. (1952) The dual effect of membrane potential on sodium conductance in the giant axon of Loligo. J. Physiol. 116, 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy B. (1978) Slow inactivation of the sodium conductance in squid giant axons. Pronase resistance. J. Physiol. 283, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean B. P. (2007) The action potential in mammalian central neurons. Nat. Rev. Neurosci. 8, 451–465. [DOI] [PubMed] [Google Scholar]
- Do M. T. H.; Bean B. P. (2003) Subthreshold sodium currents and pacemaking of subthalamic neurons: Modulation by slow inactivation. Neuron 39, 109–120. [DOI] [PubMed] [Google Scholar]
- Vilin Y. Y.; Ruben P. C. (2001) Slow inactivation in voltage-gated sodium channels: Molecular substrates and contributions to channelopathies. Cell Biochem. Biophys. 35, 171–190. [DOI] [PubMed] [Google Scholar]
- Lee H.; Park K. D.; Yang X.-F.; Dustrude E. T.; Wilson S. M.; Khanna R.; Kohn H. (2013) (Biphenyl-4-yl)methylammonium chlorides: Potent anticonvulsants that modulate Na+ currents. J. Med. Chem. 56, 5931–5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stables J. P., and Kupferberg H. G.. The NIH Anticonvulsant Drug Development (ADD) program: Preclinical anticonvulsant screening project. (1997) In Molecular and Cellular Targets for Antiepileptic Drugs (Avanzini G., Regesta G., Tanganelli P., and Avoli M., Eds.), Chapter 16, pp 191–198, John Libbey, London. [Google Scholar]
- Porter R. J.; Cereghino J. J.; Gladding G. D.; Hessie B. J.; Kupferberg H. J.; Scoville B.; White B. G. (1984) Antiepileptic drug development program. Cleveland Clin. Q. 51, 293–305. [DOI] [PubMed] [Google Scholar]
- Dunham N. W.; Miya T.-S. (1957) A note on a simple apparatus for detecting neurological deficit in rats and mice. J. Am. Pharm. Assoc. 46, 208–209. [DOI] [PubMed] [Google Scholar]
- White H. S., Woodhead J. H., Wilcox K. S., Stables J. P., Kupferberg H. J., and Wolf H. H. (2002) General Principles: Discovery and Preclinical Development of Antiepileptic Drugs. In Antiepileptic Drugs (Levy R. H., Mattson R. H., Meldrum B. S., and Perruca E., Eds.), 5th ed., pp 36–48, Lippincott, Williams and Wilkins, Philadelphia, PA, . [Google Scholar]
- Swinyard E. A. (1969) Laboratory evaluation of antiepileptic drugs: Review of laboratory methods. Epilepsia 10, 107–119. [DOI] [PubMed] [Google Scholar]
- Salome C.; Salome-Grosjean E.; Park K. D.; Morieux P.; Swendiman R.; DeMarco E.; Stables J. P.; Kohn H. (2010) Synthesis and anticonvulsant activities of (R)-N-(4′-substituted)benzyl 2-acetamido-3-methoxypropionamides. J. Med. Chem. 53, 1288–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubuisson D.; Dennis S. G. (1977) The formalin test: A quantitative study of the analgesic effects of morphine, meperidine, and brain stem stimulation in rats and cats. Pain 4, 161–177. [DOI] [PubMed] [Google Scholar]
- Wheeler-Aceto H.; Cowan A. (1991) Standardization of the rat paw formalin test for the evaluation of analgesics. Psychopharmacology 104, 35–44. [DOI] [PubMed] [Google Scholar]
- Abbott F. V.; Franklin K. B.; Westbrook R. F. (1995) The formalin test: Scoring properties of the first and second phases of the pain response in rats. Pain 60, 91–102. [DOI] [PubMed] [Google Scholar]
- Stohr T.; Krause E.; Selve N. (2006) Lacosamide displays potent antinociceptive effects in animal models for inflammatory pain. Eur. J. Pain. 10, 241–249. [DOI] [PubMed] [Google Scholar]
- Stummann T. C.; Salvati P.; Fariello R. G.; Faravelli L. (2005) The anti-nociceptive agent ralfinamide inhibits tetrodotoxin-resistant and tetrodotoxin-sensitive Na+ currents in dorsal root ganglion neurons. Eur. J. Pharmacol. 510, 197–208. [DOI] [PubMed] [Google Scholar]
- Feldman P.; Due M. R.; Ripsch M. S.; Khanna R.; White F. A. (2012) The persistent release of HMGB1 contributes to tactile hyperalgesia in a rodent model of neuropathic pain. J. Neuroinflammation 9, 180. 10.1186/1742-2094-9-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju W.; Li Q.; Allette Y. M.; Ripsch M. S.; White F. A.; Khanna R. (2012) Suppression of pain-related behavior in two distinct rodent models of peripheral neuropathy by a homopolyarginine-conjugated CRMP2 peptide. J. Neurochem. 124, 869–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhangoo S.; Ren D.; Miller R. J.; Henry K. J.; Lineswala J.; Hamdouchi C.; Li B.; Monahan P. E.; Chan D. M.; Ripsch M. S.; White F. A. (2007) Delayed functional expression of neuronal chemokine receptors following focal nerve demyelination in the rat: a mechanism for the development of chronic sensitization of peripheral nociceptors. Mol. Pain 3, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brittain J. M.; Piekarz A. D.; Wang Y.; Kondo T.; Cummins T. R.; Khanna R. (2009) An atypical role for collapsing response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated Ca2+ channels. J. Biol. Chem. 284, 31375–31390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brittain J. M.; Wang Y.; Eruvwetere O.; Khanna R. (2012) Cdk5-mediated phosphorylation of CRMP-2 enhances its interaction with CaV2.2. FEBS Lett. 586, 3813–3818. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.