

Abstract
Chronic rhinosinusitis (CRS) is a prevalent health condition characterized by sinonasal mucosal inflammation lasting at least 12 weeks. Heterogeneous in clinical presentation, histopathology, and therapeutic response, CRS represents a spectrum of disease entities with variable pathophysiology. Increased knowledge of cellular and molecular derangements in CRS suggests potential etiologies and targets for therapy. Microbial elements including fungi, staphylococcal enterotoxin, and biofilms have been implicated as inflammatory stimuli, along with airborne irritants and allergens. Defects in innate immunity have gained increased attention as contributors to the chronic inflammatory state. A combination of host susceptibility and environmental exposure is widely believed to underlie CRS, although direct evidence is lacking. Presently, without precise disease definitions and identifiable universal triggers, CRS pathogenesis is broadly described as multifactorial. Current research is beginning to unravel complex and diverse effects of chronic inflammation on sinonasal mucosal homeostasis, but dysfunctional pathways of inflammatory regulation and resolution require further elucidation.
Keywords: Chronic rhinosinusitis, Inflammation, Innate immunity, Host defense, Epithelium
Introduction
Despite its prevalence and significant health impact, the etiology of chronic rhinosinusitis (CRS) remains incompletely understood. Unlike acute bacterial sinusitis, for which the pathophysiology is well-defined, CRS is a heterogeneous condition characterized broadly by persistent inflammation of the sinonasal mucosa [1]. It is widely believed that the causes of inflammation in CRS are diverse and multifactorial, relating to overlapping host and environmental triggers. Disruption of normal epithelial function subsequent to inflammation of any origin can result in mucostasis and microbial colonization. Infection, in turn, stimulates further inflammation and exacerbates the chronic disease process. Current research in the field has attempted to elucidate the factors driving persistent sinonasal inflammation [2], highlighting the complex interplay between the environment and innate and adaptive mucosal immune mechanisms [3]. It is increasingly recognized that CRS is not a single disease entity, but instead varies widely in clinical presentation, histopathology, and response to therapy. Multiple pathophysiologic pathways likely exist that can result in the common endpoint of sinonasal mucosal inflammation (Fig. 1). Perhaps more importantly, once chronic inflammation becomes manifest, secondary activation of additional pathophysiologic pathways inevitably blurs identification of the initiating cause. The determination of which cellular and molecular features of CRS represent underlying factors inducing inflammation or merely downstream consequences remains an ongoing challenge in the growing field of CRS research. The goal of this review is to present the variety of exogenous and endogenous factors that have been implicated in CRS pathogenesis and discuss them in light of the recent literature.
Fig 1.
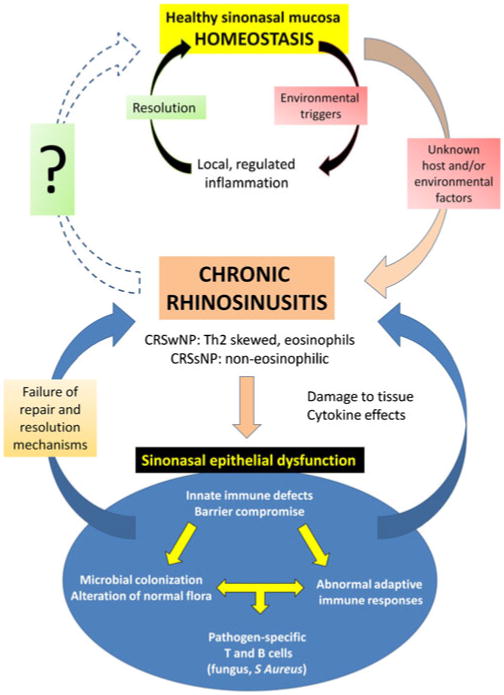
Pathophysiologic pathways of chronic rhinosinusitis. CRSwNP—chronic rhinosinusitis with nasal polyps; CRSsNP— chronic rhinosinusitis without nasal polyps.
CRS: Definition, Epidemiology, and Cellular Characteristics
Rhinosinusitis is among the most common conditions in the United States, affecting more than 31 million people annually [4]. Chronic rhinosinusitis prevalence, however, is difficult to extrapolate because of the heterogeneity of its presentation and imprecise diagnosis. Conservative estimates indicate that CRS is responsible for 18 to 22 million office visits annually, presenting a considerable economic and health care burden [5]. CRS is defined as an inflammatory process of the nose and paranasal sinus mucosa for at least 12 consecutive weeks. The diagnosis of CRS is clinical, based on subjective and objective findings, such as those recommended in guidelines published by the American Academy of Otolaryngology— Head and Neck Surgery [1]. Although these criteria are helpful in setting a framework for diagnosis and therapy, the correlation between symptoms and the degree of inflammation in CRS is not strong.
For clinical research purposes, two histopathologic subtypes of CRS have been proposed—one with nasal polyps (CRSwNP) and one without nasal polyps (CRSsNP) [2, 6]. Inflammation in both varieties of CRS displays a cellular infiltrate of neutrophils, macrophages, and lymphocytes, as well as numerous proinflammatory cytokines associated with helper T cell type 1 (Th1) inflammation. CRSwNP is distinguished by the presence of nasal polyps and an eosinophilic inflammatory infiltrate with a mixed Th1/Th2 cytokine profile that is Th2-biased [7]. CRSwNP has a high rate of recidivism, and recalcitrant forms may be associated with eosinophilic asthma [8]. The age at which nasal polyps occur is variable and not linked to any precipitating factors. Although the distinct features of CRSwNP suggest a different underlying mechanism from CRSsNP, the cause of nasal polyposis remains under investigation. Polyps are not believed to have an allergic basis, despite the apparent key role of eosinophils. Tissue eosinophilia is a consistent hallmark of CRSwNP; however, noneosinophilic polyps form in the setting of cystic fibrosis, and have also been described in CRSwNP in Asian populations [7, 9•]. With or without polyps, CRS is further characterized by poor mucociliary function, with entrapment of thickened or purulent secretions within the sinus cavities. In many cases, medical or surgical restoration of sinus outflow in CRSsNP improves or even reverses mucosal disease, suggesting that functional or anatomic obstruction can serve as a primary inflammatory stimulus [10]. In contrast, enlargement of sinus ostia does not typically improve eosinophilic inflammation in CRSwNP, although intensive medical therapy with systemic corticosteroids can dramatically, albeit transiently, reduce polyps [11, 12]. The consistent effectiveness of steroids in CRS underlines the central importance of inflammation in disease pathophysiology.
Inflammation Induced by Microbial and Environmental Factors
The sinonasal epithelium is in constant contact with the outside environment and serves as the first line of defense against inhaled pathogens and particulates. This interaction involves a complex set of innate and adaptive immune pathways at the mucosal surface driving inflammatory responses that protect the host from infection [13]. Although these mechanisms are essential to maintain homeostasis, inappropriate activation or lack of inhibition can lead to chronic inflammation that may be counterproductive and causative of symptoms. Because microbial elements are frequently observed in association with CRS, it is widely speculated that infection plays a role as an initiator of inflammation, or at least contributes to its persistence. At the same time, because the nose normally functions as a filter, the transient presence of incidental microorganisms and particulates is not necessarily pathologic. It remains unanswered whether pre-existing inflammation with impairment of mucociliary clearance is responsible for retention of microbial and environmental materials in CRS, or whether these agents in fact stimulate inflammation.
Bacteria and Staphylococcus Superantigens
The role of bacteria in promoting CRS inflammation is uncertain, notwithstanding the frequent isolation of bacteria from the sinuses of CRS patients [14]. Although no single bacterial species has been proposed as the primary etiologic agent in CRS, much focus has been placed on the potential impact of Staphylococcus aureus and its enterotoxin products (Staphylococcus aureus enterotoxin [SAE]), which can behave as superantigens activating a subset of T-cells in a non–antigen-specific manner to cause inflammation [15]. The hypothesis that SAEs cause CRS is suggested by the high rate of colonizing Staphylococcus in CRSwNP, and the observation that lymphocytes from CRSwNP patients demonstrate increased responsiveness to superantigens [16–18]. It has been proposed that patients with CRSwNP are susceptible to amplification and persistence of eosinophilic inflammation as well as induction of local polyclonal IgE formation due to the effect of SAEs [19]. Although high levels of SAE-specific IgE are associated with increased interleukin (IL)-5, eosinophilic cationic protein, and comorbid asthma, the cause-and-effect relationship between Staphylococcus and CRSwNP is still not established. An alternative interpretation is that severe mucosal inflammation and high tissue IgE levels precede Staphylococcal overgrowth, and that increased exposure to multiple bacterial antigens, including SAE, leads to generation of specific IgE antibodies, even if SAE is not directly pathogenic as a superantigen [20].
Whether or not SAEs initiate inflammation in CRSwNP, it is likely that bacteria and their products can act as disease modifiers. SAEs induce both Th1 and Th2 proinflammatory responses in patients with nasal polyps and asthma in comparison to controls, perhaps relating to a basal deficiency of T regulatory cells and/or up-regulation of specific costimulatory molecules on monocytes and dendritic cell precursors [16]. Furthermore, alteration of the normal microbial flora of the nose and sinuses in CRS resulting from mucosal disruption, long-term use of antibiotics, and surgical intervention may contribute to the failure to restore homeostasis and resolve inflammation.
Biofilms and Inflammation
Biofilms are organized communities of microorganisms protected by a polysaccharide matrix, allowing enhanced survival and resistance to host defenses and antimicrobial agents. First described in CRS in 2004, biofilms are proposed to mediate chronic inflammation and recalcitrant infection [21]. Clinically, biofilms are associated with more severe disease preoperatively and persistence of postoperative symptoms, infection, and mucosal inflammation [22, 23]. Several microbes have been described in the composition of CRS biofilms including Staphylococcus aureus, Haemophilus influenzae, Pseudomonas aeruginosa, and fungus [24, 25]. S. aureus biofilms have been hypothesized to facilitate the production of superantigen toxin, as described previously, and may present with more severe disease [26]. Biofilms in CRS patients are associated with significantly elevated levels of Th1-associated inflammatory mediators and neutrophils [27]. At the same time, it has also been demonstrated that biofilms in CRS are associated with decreased levels of the antimicrobial peptide lactoferrin, which may imply that a diminished innate immune response predisposes to microbial colonization and biofilm development [28•].
Interpretation of the role of biofilms in CRS is complicated by the fact that bacteria more commonly exist in this organized form in nature, rather than as individual planktonic organisms. Additionally, biofilms can be demonstrated at the surface of healthy paranasal sinus mucosa, suggesting that they may be a normal part of the regular respiratory mucosal blanket [29]. In the absence of direct evidence that biofilms can initiate inflammation in CRS, their existence may be best viewed as a secondary effect of chronic mucosal immune and mucociliary dysfunction. It is likely that colonizing bacteria and fungi within a chronically static mucus blanket will develop naturally into biofilms. To what extent biofilms, once they occur, exacerbate preexisting inflammation and/or confer disease resistance to medical and surgical therapy is under active investigation. To date, no direct evidence exists that eradication of biofilms reverses inflammation in CRS.
Fungus
The role of fungus in CRS has been vigorously debated for the past decade. Like bacteria, fungi are transiently present in the nose under normal conditions and can be isolated from both CRS patients and normal subjects [30, 31]. A hypothesis has been strongly advanced that all cases of CRS are caused by abnormal host responses to common airborne fungi. The proposed mechanism is a non–IgE-mediated induction of T-cell responses via fungal antigens and a direct activation of eosinophils at the mucosal surface, with resulting collateral mucosal damage. The primary evidence involves hyperreactivity of peripheral blood mononuclear cells from CRS patients to high levels of fungal antigen, as well as in vitro studies of eosinophil degranulation in response to fungi [32]. After an initial period of widespread interest, the fungal hypothesis has largely been discarded as the primary cause of CRS, because of a lack of reproducibility of scientific results and the failure of systemic and topical antifungal agents to modify the disease process in double-blind, placebo-controlled trials [33, 34]. Like bacterial colonization and biofilm formation, the increased burden of fungus at the mucosal surface in CRS likely reflects decreased innate immune function and impaired mucociliary clearance. In this sense, fungus may act as a disease modifier in a subset of patients with preexisting sinonasal inflammation in whom colonization elicits an excessive host response.
Viruses, Cigarette Smoke, and Environmental Irritants
Inflammation in patients with CRS may be exacerbated by environmental factors that cause mucosal irritation or stimulate local immune responses. Although viral infections are often implicated as triggers of CRS flares, few studies have examined whether viruses contribute to the pathogenesis of mucosal inflammation. In mice, sinonasal inflammation and hyperresponsiveness, associated with significantly elevated levels of tissue T-suppressor and T-regulatory cells, persist for over a month after infection with Sendai virus [35]. In vitro studies with human sinonasal epithelial cells demonstrate that viral infection or exposure to a synthetic viral analogue, poly(I:C), causes increases in expression of innate immune effectors, including β-defensin [36] and matrix metalloproteinase (MMP) [37]. There is no direct evidence that viral infection can initiate CRS, but it is possible that abnormal host immune responses to upper respiratory viruses may play a role in some individuals. As with fungus, host defense mechanisms against virus involving expression of proteolytic enzymes may damage the epithelium and contribute to chronic mucosal inflammation [38].
In addition to infectious agents, nasal inflammation is frequently precipitated by exposure to various inhaled particulates, such as those found in cigarette smoke. In a case-control study of adults exposed to second-hand smoke, an increased risk for development of CRS was demonstrated [39], and it is broadly speculated that a predisposition to inflammatory reactions to cigarette smoke and other irritants may increase susceptibility to CRS. In addition to its cytotoxic effects, cigarette smoke also impacts the innate immune function of sinonasal epithelial cells. The combination of cigarette smoke with poly(I:C) causes exaggerated expression of the chemotactic cytokine regulated on activation normal T-cell expressed and secreted (RANTES) and the antimicrobial peptide human β-defensin 2 in CRS patients compared to controls, suggesting that cigarette smoke can augment virus-induced inflammation and exacerbate eosinophilic infiltration [40]. There is evidence in mice that smoke extract may damage the epithelium via complement activation, which is ameliorated in mice deficient in complement components C3 and factor B [41].
Inflammation Caused by Intrinsic Host Factors
The vast majority of CRS is idiopathic in origin, with only a minority of cases caused by identifiable genetic disorders (eg, cystic fibrosis, ciliary dyskinesia) or systemic inflammatory processes (eg, sarcoidosis, Wegener's granulomatosis) [15, 42, 43]. Ultimately, regardless of the cause, the defining characteristic of CRS is inflammation of the sinonasal mucosa with obstruction of sinus outflow. Current medical and surgical therapies that target inflammation and infection are effective for the majority of CRS patients, but for those in whom all treatments are ineffective or the response is not durable, various mechanisms have been invoked to explain persistent inflammation. The high incidence of radiographic and pathologic evidence of osteitis in CRS (53%) has been cited as supporting evidence that bony inflammation plays a role [44]. Increasingly, CRS may develop in the context of previous surgery, where mucosal trauma and bony exposure can be the impetus for poor healing and ongoing mucosal dysfunction. CRSwNP patients undergoing revision surgery have a significantly higher incidence of osteitis than in patients undergoing primary surgery [45], which may either imply a relationship between osteitis and disease severity, or alternatively may reflect the fact that surgery itself increases the incidence of osteitis.
In the final analysis, inflammation in CRS appears to arise from abnormalities of normal mucosal immune function that disrupt maintenance of homeostasis. Although several interesting observations have been made that shed light on potential inflammatory mechanisms and suggest different host-centered hypotheses of CRS pathogenesis, it must again be recognized that characterization of the active disease state does not necessarily imply causality. Just as with putative infectious triggers, many host derangements identified in a chronic state of sinonasal mucosal disequilibrium may reflect downstream effects of inflammation, rather than the basis of the disease.
Allergy and CRS
The eosinophilic inflammatory infiltrate seen in both allergy and CRSwNP has led to speculation that allergy plays a role in CRS pathogenesis [46]. Although clinical experience suggests that allergic inflammation exacerbates CRS and diminishes the response to therapy, epidemiologic and outcomes data are mixed on this topic. The prevalence of allergy in patients with CRSwNP has variably been reported to be higher or lower than the general population [2, 47, 48], and the presence of allergy has not consistently been shown to affect the extent of radiographic disease, symptom severity, or the need for surgery [49]. The prevalence of polyps among patients with allergic rhinitis is also estimated to approximate the 0.5% to 4.5% seen in the general population. In contrast, the prevalence of food allergies may be significantly higher in patients with CRSwNP (up to 81%) as compared to controls (34%) [50, 51]. Even if type I hypersensitivity cannot be clearly identified as a causative factor in CRSwNP, it is clear that local IgE, mast cell, and eosinophil numbers are prominent features of polyps, suggesting that similar mechanisms may underlie both diseases. In one subset of CRSwNP, allergic fungal sinusitis, allergy is a believed to be the main driver of inflammation.
Innate and Adaptive Mucosal Immunity
At the interface between the host and the environment, the sinonasal mucosa functions critically to provide protection against infection. Until recently, the epithelium was viewed as a passive mechanical barrier, but research has since revealed it to be a remarkably active participant in airway immunity [13]. Innate and adaptive immune components act cooperatively to identify and eliminate infectious threats from the sinonasal tract (Fig. 2). Innate mechanisms do not require prior exposure to antigens, nor do they rely on combinatorial rearrangement of receptors. Evolutionary ancient and genetically fixed innate pathways provide multiple first lines of defense, which may be constitutively active or inducible. The primary innate defense of the sinonasal tract is the mucus blanket, which serves to entrap and remove foreign materials. Secreted antimicrobials such as lysozyme, lactoferrin, β-defensins, cathelicidins, and surfactant proteins inhibit microbial growth in the mucus, and may be up-regulated in response to activation of pattern recognition receptors by bacterial, fungal, or viral proteins. In recent years, there has been an increased focus on the potential role of the sinonasal innate immune system in CRS pathogenesis. In theory, defects in the innate immune system may predispose to infection and increased antigenic exposure or may stimulate inflammation directly via interaction with adaptive immune cells. Once inflammation is present, failure of innate mechanisms to promote resolution and repair may also contribute to disease persistence.
Fig 2.
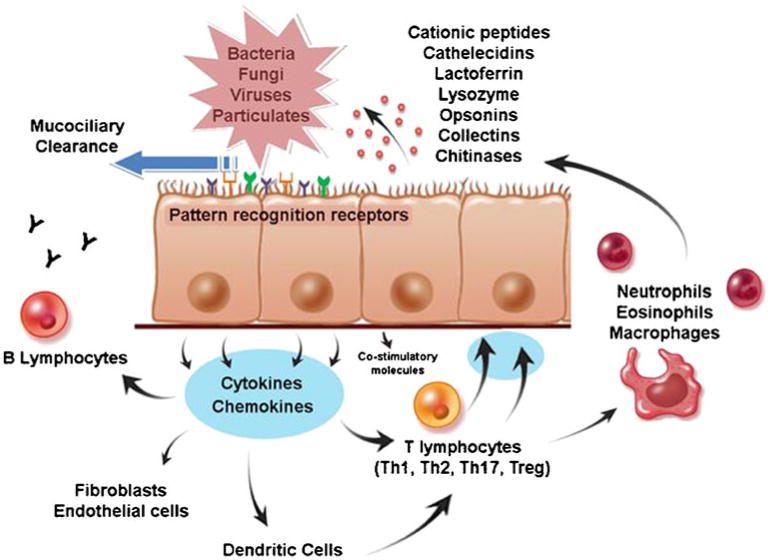
Innate immunity of the sinonasal tract. The primary mechanism of sinonasal innate immune defense is orderly mucociliary clearance. The mucus blanket, which contains many secreted antimicrobials and opsonins, is continuously propelled to the nasopharynx, providing constitutive, nonspecific protection of the sinonasal mucosal surface. In addition, sinonasal epithelial cells actively participate in innate immunity, using pattern-recognition receptors to detect luminal pathogens and responding directly with selective expression of targeted antimicrobial effectors. At the same time, epithelial cells signal to adaptive immune cells through cytokines and costimulatory molecules to coordinate a vigorous defense of the mucosal surface. Emerging evidence suggests that predominance of certain T-helper populations (Th1, Th2, and Th17) in the mucosa, as well as the presence of T-regulatory cells (Tregs) may play a role in chronic rhinosinusitis pathogenesis. Epithelial cells guide the recruitment of adaptive immune cells by producing signaling molecules that interact locally with resident dendritic cells and T cells. Cytokines produced by specific T-cell subclasses modulate the innate immune responses of epithelial cells by influencing the pattern of antimicrobial gene expression. Chronic sinonasal inflammatory disease may result from the loss of mucosal homeostasis due to dysregulation of these innate immune pathways. (From Lane [3].)
Mucociliary Dysfunction
The paranasal sinuses are lined by ciliated columnar epithelium and protected by a continuously flowing mucus blanket composed of a complex network of immunoglobulins, carbohydrates, enzymes, glycoproteins, electrolytes, and water. Impairments in mucociliary clearance contribute to mucus stasis, infection, and inflammation. In systemic diseases such as primary ciliary dyskinesia (dysfunction of ciliary movement) or cystic fibrosis (dysfunction of mucus secretion), CRS is common [52, 53]. Cytokines and other inflammatory mediators present in CRS (eg, IL-8 and IL-13) may impact ciliary function [54]. Respiratory pathogens and particulates also have been shown to impair ciliary function and may contribute to pathologic inflammation [55, 56].
Glycoproteins are important components of mucus that are responsible for determining viscosity and whose expression is altered in CRS. The genes MUC5AC and MUC5B are up-regulated in both CRSwNP and CRSsNP, contributing to secretory cell hyperplasia and metaplasia [57]. Significantly increased concentration of the glycoprotein galactose β 1,3 GalNAc are described in CRS sinonasal mucosa, perhaps contributing to the high viscosity of mucus and acting as potential innate receptors for pathogenic bacteria [58].
Adaptive Immunity
Chronic sinonasal inflammation is associated with lymphocytic infiltration and prominent expression of inflammatory cytokines [1]. Although it is recognized that CRSwNP and CRSsNP have distinct mediator profiles and cellular phenotypes, the mechanism underlying polarization into T-helper cell subtype responses is unknown. Naïve T cells differentiate into memory and effector cells upon interaction with antigen presenting cells in the context of specific co-stimulatory signals provided by cytokines and ligand-receptor interactions. Th1 cells characteristically secrete interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α), which potently activate macrophages and cytotoxic T cells, and promote B-cell antibody production. Th2 cells secrete IL-4, IL-5, IL-9, and IL-13, which promote eosinophil survival and activation, as well as production of IgE. Investigation of cytokine expression in CRS has revealed both Th1 and Th2 cytokines, reflecting crosstalk between the two inflammatory responses.
However, it is overly simplistic to view these disease subtypes only in terms of Th1 versus Th2, because there is growing evidence that regulatory T cells (Treg) and Th17 cells are important in controlling and directing mucosal immune responses. Because Tregs can suppress Th2-driven inflammation, impairment of Tregs is hypothesized to contribute to CRSwNP pathogenesis. Decreased expression of forkhead box protein 3 (Foxp3) in nasal polyp tissue has been identified in CRSwNP in comparison to CRSsNP, suggesting a local Treg deficiency or dysfunction [59••]. IgE-mediated mast cell degranulation has also been implicated as a factor reducing the expression of Treg cytokines [60]. Th17 cells—an additional subset of T-helper cells distinct from Th1 and Th2 cells—have only recently been described, and their role in chronic inflammation remains to be defined. IL-17 and other Th17 mediators produced by Th17 cells may mediate interactions between innate and adaptive pathways in the epithelium, although elevations of IL-17 in CRSwNP have not been identified. To the contrary, nasal polyps have been associated with diminished signal transducer activator transcription factor 3 (STAT3) and IL-6 activity, which has been speculated to contribute to a possible Th17 deficiency [61].
In addition to T and B cells, dendritic cells are important components of the adaptive immune system within the sinonasal mucosa. Sinonasal mucosal dendritic cells likely play a critical role in local T- and B-cell differentiation. B-cell proliferation and antigen-specific IgE are increased in polyps, and up-regulation of B-cell activators and proliferation factors are described in patients with CRSwNP in comparison to those with CRSsNP and controls [62]. Increased proliferation and maturation of B cells promotes immunoglobulin isotype switch recombination, potentially exacerbating eosinophilic inflammation in CRSwNP. A relative deficit of myeloid dendritic cell subsets in CRSwNP has been thought to favor priming of T cells to a Th2 phenotype contributing to persistent inflammation [63•]. The epithelium communicates with dendritic cells via thymic stromal lymphopoetin and other inflammatory cytokines and signaling molecules.
Sinonasal Epithelial Cell Innate Immune Function
Deficiencies in the antimicrobial activity of the sinonasal mucosa can create a permissive environment for microbial colonization. Pattern recognition receptors (PRRs), including Toll-like receptors (TLRs) allow sinonasal epithelial cells (SNEC) to detect and initiate responses against pathogens present in the airway lumen. Messenger RNA (mRNA) for all 10 TLRs have been identified in SNECs, and their function has been demonstrated by induced production of innate immune effectors such as human β-defensin 2, serum amyloid A, and surfactant proteins A and D [64]. The activity of SNEC TLRs is abnormally diminished in recalcitrant CRSwNP [65–67]. In addition, expression of the antimicrobial protein, lactoferrin, is decreased at the mRNA and protein level in the nasal mucosa of CRS patients [28•, 68]. Although decreased antimicrobials may contribute to colonization, abnormal innate immune responses of SNEC may also promote inflammation through stimulation of adaptive immune elements and effector leukocytes.
Interaction of Innate and Adaptive Immunity within the Epithelium
SNEC elaborate a wide range of mediators that interact with lymphocytes and antigen presenting cells. Although mediators induced by TLR activation tend to favor Th1 (neutrophilic) inflammation, the pathogen-associated molecule chitin, acting through an undefined PRR, induces expression of acidic mammalian chitinase and eotaxin-3, which directly promote and modify eosinophilic Th2 inflammation. Chitins are commonly found in insects, fungi, and parasitic nematodes, and chitinases produced by mammals are thought to act as innate immune effectors to target these potential pathogens. In addition, SNEC derived from recalcitrant CRSwNP patients display increased baseline expression of the pro-Th2 cytokine IL-33, which can be further increased by prolonged exposure to the TLR9 agonist CpG [69•]. CpG molecules composed of single-stranded DNA possess motifs found in the microbial genome and are capable of stimulating proinflammatory immune responses. The complement system provides another link between the innate and adaptive immune systems, regulating lymphocyte function. Increased complement factor B, C3, and C5 is reported in patients with CRS and allergic fungal rhinosinusitis compared to controls [70], suggesting that complement pathway activation may modify or exacerbate Th2-mediated inflammation.
Communication between SNEC and the adaptive immune system is bidirectional, and cytokines elaborated by infiltrating inflammatory cells can modulate the immune function of the epithelium. In vitro, the expression of multiple antimicrobial innate immune markers can be down-regulated by IL-4 or IL-13 in SNEC derived from patients with CRSwNP [71••]. At the same time, Th2 cytokines greatly stimulate expression of pro-eosinophilic mediators by SNEC. The normal balance of innate inflammatory signals in the healthy sinonasal tract seems to be neutrophilic and Th1-biased, implying that mechanisms that promote eosinophilic inflammation are normally tightly regulated or suppressed. Imbalance of this counter-regulation in the epithelium may be responsible for Th2-biased inflammation. Microarray studies have revealed increased levels of the receptor c-met, whose ligand, hepatocyte growth factor, can antagonize the effects of Th2 cytokines on SNEC in vitro [72].
Barrier Hypothesis
The broad concept of epithelial barrier dysfunction has also been invoked as an underlying cause of mucosal inflammation in CRS. It is theorized that defects in the immune barrier in CRS patients increases susceptibility to microbial colonization, with consequent sensitization of the immune system [73]. This hypothesis is derivative of similar mechanisms proposed with respect to the lower airway, intestine, and skin. The evidence for barrier dysfunction in CRS relates to decreased expression of S100 proteins, which play a role in epithelial defense and repair, as well as the serine protease inhibitor SPINK5 in CRS tissue [74]. The observation that multiple epithelial innate immune and barrier proteins are diminished in ongoing CRS again must be interpreted in light of the state of inflammation and disequilibrium. Although epithelial dysfunction is clearly a feature of active CRSwNP and CRSsNP, evidence is lacking that barrier defects are a cause rather than an effect. Because CRS varies in severity over time and anatomic location within an individual patient, it remains to be demonstrated whether barrier or innate immune dysfunction exists beyond areas of ongoing chronic inflammation. Grossly, mucosal edema and mucociliary function often improve clinically with anti-inflammatory corticosteroid therapy. Although the beneficial effects of corticosteroids in CRS have traditionally been thought to derive from immunosuppression, it is notable that steroids also enhance epithelial cell innate immunity and repair [75]. Future prospective studies in humans and the use of genetically modified animal models will be necessary to clarify the role of the epithelium as a causative factor in chronic sinonasal inflammatory disease.
Asthma and CRS
Associated comorbidities of CRS may shed light on shared pathogenetic mechanisms. Up to 50% of CRS patients have asthma, the presence of which is predictive of radiographic disease and the presence of nasal polyps [76]. It is estimated that 1% of the general population and 10% of people with asthma have Samter's triad, characterized by aspirin sensitivity, asthma, and nasal polyposis [77]. CRSwNP patients with Samter's triad have particularly high rates of symptom and polyp recurrence, but benefit from desensitization therapy [78, 79]. Long-term treatment with aspirin leads to suppression of IL-4 and down-regulation of MMP-9 [80•]. In microarray expression analysis, polyps in patients with Samter's triad are associated with up-regulation of periostin, c-met, and protein phosphatase 1 regulatory subunit 9B, and down-regulation of prolactin-induced protein and zinc α2-glycoprotein [81].
Genetic Contribution to CRS
Genetic predisposition is frequently cited as a contributing factor in CRS pathogenesis, although direct evidence is lacking. CRS appears to occur sporadically, and, with the exception of certain systemic diseases with sinonasal manifestations, patterns of heritability are not described. Nevertheless, attempts to define the role of genetics have used genotyping and analysis of polymorphisms. Single-nucleotide polymorphisms (SNPs) in the TNF-α [82] and MMP-9 [83] genes have been associated with increased susceptibility to CRSwNP, whereas polymorphisms in IL-13 [84], IL-33 [85], and IL-1A [86] genes have also been described in relation to CRS populations. One genome-wide screen for CRS susceptibility identified a locus on 7q31.1–7q32.1, with the largest linkage signal near the CFTR gene [87], which is intriguing because of the known increased proportion of CFTR mutations in CRS patients compared to controls [52]. Alterations in the 6p22, 22q13, and 1q23 chromosomal regions have been demonstrated in both CRS and aspirin-sensitive asthma phenotypes [81]. Genome-wide surveys are potentially powerful screening methods for detecting gene transcription changes in CRS that have produced interesting but heterogeneous results, likely reflecting differences in study populations. The challenge remains to obtain and interpret meaningful information from large volumes of data derived from small sample sizes.
Conclusions
CRS is a commonly diagnosed health condition with a significant impact on quality of life, but the highly heterogenous nature of the disease confounds identification of an underlying cause. Numerous potential etiologic and disease-modifying factors have been proposed in CRS, leading to a belief that the chronic inflammation must be multifactorial. Certainly, a complex interplay normally exists between the host and the environment at the boundary surfaces of the sinonasal tract, and homeostasis requires a properly functioning mucosal immune system. Episodic inflammatory responses must be precisely regulated and terminated to control infectious threats without damaging the host. In CRS, when inflammation is perpetually unresolved, homeostatic balance cannot be restored, resulting in a state of immune disequilibrium. Study of chronically inflamed CRS tissue reveals numerous cellular and molecular derangements of host defense and repair mechanisms, which in turn are associated with alterations in microbial flora. Abnormalities in the adaptive immune response likely contribute importantly to the chronicity of the disease process. At the current time, the initial drivers of inflammation in CRS remain unclear, but the local features of the chronically inflamed epithelium are becoming better characterized. Although CRS is currently perceived as multifactorial in origin, a more complete understanding of the pathophysiology of active CRS will allow differentiation among disease initiators, disease modifiers, and unrelated epiphenomena. Ultimately, identification of the etiologic factors responsible for initiating inflammation in CRS may be less important than elucidation of deficient pathways responsible for its down-regulation and resolution.
Footnotes
Disclosure Conflicts of Interest: S. Lee—None; A.P. Lane—None.
Contributor Information
Stella Lee, Department of Otolaryngology—Head and Neck Surgery, Johns Hopkins University School of Medicine, Baltimore, MD, USA.
Andrew P. Lane, Email: alane3@jhmi.edu, Department of Otolaryngology—Head and Neck Surgery, Johns Hopkins University School of Medicine, Baltimore, MD, USA; Department of Otolaryngology—Head and Neck Surgery, Johns Hopkins Outpatient Center, 601 North Caroline Street, Sixth Floor, 6250, Baltimore, MD 21287-0910, USA.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Benninger MS, Ferguson BJ, Hadley JA, et al. Adult chronic rhinosinusitis: definitions, diagnosis, epidemiology, and pathophysiology. Otolaryngol Head Neck Surg. 2003;129(3 Suppl):S1–S32. doi: 10.1016/s0194-5998(03)01397-4. [DOI] [PubMed] [Google Scholar]
- 2.Fokkens W, Lund V, Mullol J. European position paper on rhinosinusitis and nasal polyps 2007. Rhinol Suppl. 2007;20:1–136. [PubMed] [Google Scholar]
- 3.Lane AP. The role of innate immunity in the pathogenesis of chronic rhinosinusitis. Curr Allergy Asthma Rep. 2009;9(3):205–12. doi: 10.1007/s11882-009-0030-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lethbridge-Cejku M, Schiller JS, Bernadel L. Summary health statistics for U.S. adults: National Health Interview Survey, 2002. Vital Health Stat. 2004 Jul;10(222):1–151. [PubMed] [Google Scholar]
- 5.Benninger MS, Sedory Holzer SE, Lau J. Diagnosis and treatment of uncomplicated acute bacterial rhinosinusitis: summary of the Agency for Health Care Policy and Research evidence-based report. Otolaryngol Head Neck Surg. 2000;122(1):1–7. doi: 10.1016/S0194-5998(00)70135-5. [DOI] [PubMed] [Google Scholar]
- 6.Meltzer EO, Hamilos DL, Hadley JA, et al. Rhinosinusitis: establishing definitions for clinical research and patient care. Otolaryngol Head Neck Surg. 2004;131(6 Suppl):S1–S62. doi: 10.1016/j.otohns.2004.09.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Zele T, Claeys S, Gevaert P, et al. Differentiation of chronic sinus diseases by measurement of inflammatory mediators. Allergy. 2006;61(11):1280–9. doi: 10.1111/j.1398-9995.2006.01225.x. [DOI] [PubMed] [Google Scholar]
- 8.ten Brinke A, Grootendorst DC, Schmidt JT, et al. Chronic sinusitis in severe asthma is related to sputum eosinophilia. J Allergy Clin Immunol. 2002;109(4):621–6. doi: 10.1067/mai.2002.122458. [DOI] [PubMed] [Google Scholar]
- 9•.Zhang N, Van Zele T, Perez-Novo C, et al. Different types of T-effector cells orchestrate mucosal inflammation in chronic sinus disease. J Allergy Clin Immunol. 2008;122(5):961–68. doi: 10.1016/j.jaci.2008.07.008. This paper evaluated the role of Th17 cells and outlined the differences in T-cell polarizations in polyps to explain the neutrophilic bias in Asian populations in contrast to an eosinophilic bias in European populations. [DOI] [PubMed] [Google Scholar]
- 10.Caughey RJ, Jameson MJ, Gross CW, Han JK. Anatomic risk factors for sinus disease: fact or fiction? Am J Rhinol. 2005;19(4):334–9. [PubMed] [Google Scholar]
- 11.Hissaria P, Smith W, Wormald PJ, et al. Short course of systemic corticosteroids in sinonasal polyposis: a double-blind, randomized, placebo-controlled trial with evaluation of outcome measures. J Allergy Clin Immunol. 2006;118(1):128–33. doi: 10.1016/j.jaci.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 12.Benitez P, Alobid I, de Haro J, et al. A short course of oral prednisone followed by intranasal budesonide is an effective treatment of severe nasal polyps. Laryngoscope. 2006;116(5):770–5. doi: 10.1097/01.mlg.0000205218.37514.0f. [DOI] [PubMed] [Google Scholar]
- 13.Ramanathan M, Jr, Lane AP. Innate immunity of the sinonasal cavity and its role in chronic rhinosinusitis. Otolaryngol Head Neck Surg. 2007;136(3):348–56. doi: 10.1016/j.otohns.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 14.Stephenson MF, Mfuna L, Dowd SE, et al. Molecular characterization of the polymicrobial flora in chronic rhinosinusitis. J Otolaryngol Head Neck Surg. 2010;39(2):182–7. [PubMed] [Google Scholar]
- 15.Seiberling KA, Conley DB, Tripathi A, et al. Superantigens and chronic rhinosinusitis: detection of staphylococcal exotoxins in nasal polyps. Laryngoscope. 2005;115(9):1580–5. doi: 10.1097/01.mlg.0000168111.11802.9c. [DOI] [PubMed] [Google Scholar]
- 16.Perez Novo CA, Jedrzejczak-Czechowicz M, Lewandowska-Polak A, et al. T cell inflammatory response, Foxp3 and TNFRS18-L regulation of peripheral blood mononuclear cells from patients with nasal polyps-asthma after staphylococcal superantigen stimulation. Clin Exp Allergy. 2010;40(9):1323–32. doi: 10.1111/j.1365-2222.2010.03577.x. [DOI] [PubMed] [Google Scholar]
- 17.Patou J, Gevaert P, Van Zele T, et al. Staphylococcus aureus enterotoxin B, protein A, and lipoteichoic acid stimulations in nasal polyps. J Allergy Clin Immunol. 2008;121(1):110–5. doi: 10.1016/j.jaci.2007.08.059. [DOI] [PubMed] [Google Scholar]
- 18.Conley DB, Tripathi A, Seiberling KA, et al. Superantigens and chronic rhinosinusitis: skewing of T-cell receptor V beta-distributions in polyp-derived CD4+ and CD8+ T cells. Am J Rhinol. 2006;20(5):534–9. doi: 10.2500/ajr.2006.20.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bachert C, Claeys SE, Tomassen P, et al. Rhinosinusitis and asthma: a link for asthma severity. Curr Allergy Asthma Rep. 2010;10(3):194–201. doi: 10.1007/s11882-010-0096-0. [DOI] [PubMed] [Google Scholar]
- 20.Pratt E, Collins AM, Sewell WA, Harvey RJ. Antigen selection in IgE antibodies from individuals with chronic rhinosinusitis with nasal polyps. Am J Rhinol Allergy. 2010;24(6):416–21. doi: 10.2500/ajra.2010.24.3538. [DOI] [PubMed] [Google Scholar]
- 21.Cryer J, Schipor I, Perloff JR, Palmer JN. Evidence of bacterial biofilms in human chronic sinusitis. ORL J Otorhinolaryngol Relat Spec. 2004;66(3):155–8. doi: 10.1159/000079994. [DOI] [PubMed] [Google Scholar]
- 22.Psaltis AJ, Weitzel EK, Ha KR, Wormald PJ. The effect of bacterial biofilms on post-sinus surgical outcomes. Am J Rhinol. 2008;22(1):1–6. doi: 10.2500/ajr.2008.22.3119. [DOI] [PubMed] [Google Scholar]
- 23.Singhal D, Psaltis AJ, Foreman A, Wormald PJ. The impact of biofilms on outcomes after endoscopic sinus surgery. Am J Rhinol Allergy. 2010;24(3):169–74. doi: 10.2500/ajra.2010.24.3462. [DOI] [PubMed] [Google Scholar]
- 24.Prince AA, Steiger JD, Khalid AN, et al. Prevalence of biofilm-forming bacteria in chronic rhinosinusitis. Am J Rhinol. 2008;22(3):239–45. doi: 10.2500/ajr.2008.22.3180. [DOI] [PubMed] [Google Scholar]
- 25.Foreman A, Psaltis AJ, Tan LW, Wormald PJ. Characterization of bacterial and fungal biofilms in chronic rhinosinusitis. Am J Rhinol Allergy. 2009;23(6):556–61. doi: 10.2500/ajra.2009.23.3413. [DOI] [PubMed] [Google Scholar]
- 26.Foreman A, Wormald PJ. Different biofilms, different disease? A clinical outcomes study. Laryngoscope. 2010;120(8):1701–6. doi: 10.1002/lary.21024. [DOI] [PubMed] [Google Scholar]
- 27.Hekiert AM, Kofonow JM, Doghramji L, et al. Biofilms correlate with TH1 inflammation in the sinonasal tissue of patients with chronic rhinosinusitis. Otolaryngol Head Neck Surg. 2009;141(4):448–53. doi: 10.1016/j.otohns.2009.06.090. [DOI] [PubMed] [Google Scholar]
- 28•.Psaltis AJ, Wormald PJ, Ha KR, Tan LW. Reduced levels of lactoferrin in biofilm-associated chronic rhinosinusitis. Laryngoscope. 2008;118(5):895–901. doi: 10.1097/MLG.0b013e31816381d4. This paper demonstrates that the antimicrobial peptide lactoferrin is reduced in CRS patients with biofilms, potentially contributing to disease pathogenesis. [DOI] [PubMed] [Google Scholar]
- 29.Mladina R, Skitarelic N, Music S, Ristic M. A biofilm exists on healthy mucosa of the paranasal sinuses: a prospectively performed, blinded, scanning electron microscope study. Clin Otolaryngol. 2010;35(2):104–10. doi: 10.1111/j.1749-4486.2010.02097.x. [DOI] [PubMed] [Google Scholar]
- 30.Scheuller MC, Murr AH, Goldberg AN, et al. Quantitative analysis of fungal DNA in chronic rhinosinusitis. Laryngoscope. 2004;114(3):467–71. doi: 10.1097/00005537-200403000-00015. [DOI] [PubMed] [Google Scholar]
- 31.Gosepath J, Brieger J, Vlachtsis K, Mann WJ. Fungal DNA is present in tissue specimens of patients with chronic rhinosinusitis. Am J Rhinol. 2004;18(1):9–13. [PubMed] [Google Scholar]
- 32.Shin SH, Ponikau JU, Sherris DA, et al. Chronic rhinosinusitis: an enhanced immune response to ubiquitous airborne fungi. J Allergy Clin Immunol. 2004;114(6):1369–75. doi: 10.1016/j.jaci.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 33.Weschta M, Rimek D, Formanek M, et al. Effect of nasal antifungal therapy on nasal cell activation markers in chronic rhinosinusitis. Arch Otolaryngol Head Neck Surg. 2006;132(7):743–7. doi: 10.1001/archotol.132.7.743. [DOI] [PubMed] [Google Scholar]
- 34.Weschta M, Rimek D, Formanek M, et al. Topical antifungal treatment of chronic rhinosinusitis with nasal polyps: a randomized, double-blind clinical trial. J Allergy Clin Immunol. 2004;113(6):1122–8. doi: 10.1016/j.jaci.2004.03.038. [DOI] [PubMed] [Google Scholar]
- 35.Klemens JJ, Thompson K, Langerman A, Naclerio RM. Persistent inflammation and hyperresponsiveness following viral rhinosinusitis. Laryngoscope. 2006;116(7):1236–40. doi: 10.1097/01.mlg.0000224526.43698.52. [DOI] [PubMed] [Google Scholar]
- 36.Watanabe S, Wang J, Matsukura S, Suzaki H. Expression of antiviral molecular genes in nasal polyp-derived cultured epithelial cells. Acta Otolaryngol Suppl. 2009;562:101–4. doi: 10.1080/00016480902912001. [DOI] [PubMed] [Google Scholar]
- 37.Wang JH, Kwon HJ, Jang YJ. Rhinovirus upregulates matrix metalloproteinase-2, matrix metalloproteinase-9, and vascular endothelial growth factor expression in nasal polyp fibroblasts. Laryngoscope. 2009;119(9):1834–8. doi: 10.1002/lary.20574. [DOI] [PubMed] [Google Scholar]
- 38.Wang J, Watanabe S, Matsukura S, Suzaki H. Double-stranded RNA poly(I:C) enhances matrix metalloproteinase mRNA expression in human nasal polyp epithelial cells. Acta Otolaryngol Suppl. 2009;562:105–9. doi: 10.1080/00016480902911979. [DOI] [PubMed] [Google Scholar]
- 39.Reh DD, Lin SY, Clipp SL, et al. Secondhand tobacco smoke exposure and chronic rhinosinusitis: a population-based case-control study. Am J Rhinol Allergy. 2009;23(6):562–7. doi: 10.2500/ajra.2009.23.3377. [DOI] [PubMed] [Google Scholar]
- 40.Yamin M, Holbrook EH, Gray ST, et al. Cigarette smoke combined with Toll-like receptor 3 signaling triggers exaggerated epithelial regulated upon activation, normal T-cell expressed and secreted/ CCL5 expression in chronic rhinosinusitis. J Allergy Clin Immunol. 2008;122(6):1145–1153 e1143. doi: 10.1016/j.jaci.2008.09.033. [DOI] [PubMed] [Google Scholar]
- 41.Davis KS, Casey SE, Mulligan JK, et al. Murine complement deficiency ameliorates acute cigarette smoke-induced nasal damage. Otolaryngol Head Neck Surg. 2010;143(1):152–8. doi: 10.1016/j.otohns.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reed J, deShazo RD, Houle TT, et al. Clinical features of sarcoid rhinosinusitis. Am J Med. 2010;123(9):856–62. doi: 10.1016/j.amjmed.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 43.Cannady SB, Batra PS, Koening C, et al. Sinonasal Wegener granulomatosis: a single-institution experience with 120 cases. Laryngoscope. 2009;119(4):757–61. doi: 10.1002/lary.20161. [DOI] [PubMed] [Google Scholar]
- 44.Lee JT, Kennedy DW, Palmer JN, et al. The incidence of concurrent osteitis in patients with chronic rhinosinusitis: a clinicopathological study. Am J Rhinol. 2006;20(3):278–82. doi: 10.2500/ajr.2006.20.2857. [DOI] [PubMed] [Google Scholar]
- 45.Telmesani LM, Al-Shawarby M. Osteitis in chronic rhinosinusitis with nasal polyps: a comparative study between primary and recurrent cases. Eur Arch Otorhinolaryngol. 2010;267(5):721–4. doi: 10.1007/s00405-009-1146-x. [DOI] [PubMed] [Google Scholar]
- 46.Daines SM, Orlandi RR. Inflammatory cytokines in allergy and rhinosinusitis. Curr Opin Otolaryngol Head Neck Surg. 2010;18(3):187–90. doi: 10.1097/MOO.0b013e328338206a. [DOI] [PubMed] [Google Scholar]
- 47.Emanuel IA, Shah SB. Chronic rhinosinusitis: allergy and sinus computed tomography relationships. Otolaryngol Head Neck Surg. 2000;123(6):687–91. doi: 10.1067/mhn.2000.110961. [DOI] [PubMed] [Google Scholar]
- 48.Houser SM, Keen KJ. The role of allergy and smoking in chronic rhinosinusitis and polyposis. Laryngoscope. 2008;118(9):1521–7. doi: 10.1097/MLG.0b013e31817d01b8. [DOI] [PubMed] [Google Scholar]
- 49.Robinson S, Douglas R, Wormald PJ. The relationship between atopy and chronic rhinosinusitis. Am J Rhinol. 2006;20(6):625–8. doi: 10.2500/ajr.2006.20.2907. [DOI] [PubMed] [Google Scholar]
- 50.Collins MM, Loughran S, Davidson P, Wilson JA. Nasal polyposis: prevalence of positive food and inhalant skin tests. Otolaryngol Head Neck Surg. 2006;135(5):680–3. doi: 10.1016/j.otohns.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 51.Pang YT, Eskici O, Wilson JA. Nasal polyposis: role of subclinical delayed food hypersensitivity. Otolaryngol Head Neck Surg. 2000;122(2):298–301. doi: 10.1016/S0194-5998(00)70259-2. [DOI] [PubMed] [Google Scholar]
- 52.Wang X, Moylan B, Leopold DA, et al. Mutation in the gene responsible for cystic fibrosis and predisposition to chronic rhinosinusitis in the general population. Jama. 2000;284(14):1814–9. doi: 10.1001/jama.284.14.1814. [DOI] [PubMed] [Google Scholar]
- 53.Ostrowski LE, Yin W, Rogers TD, et al. Conditional deletion of dnaic1 in a murine model of primary ciliary dyskinesia causes chronic rhinosinusitis. Am J Respir Cell Mol Biol. 2010;43(1):55–63. doi: 10.1165/rcmb.2009-0118OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gomperts BN, Kim LJ, Flaherty SA, Hackett BP. IL-13 regulates cilia loss and foxj1 expression in human airway epithelium. Am J Respir Cell Mol Biol. 2007;37(3):339–46. doi: 10.1165/rcmb.2006-0400OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Min YG, Oh SJ, Won TB, et al. Effects of staphylococcal enterotoxin on ciliary activity and histology of the sinus mucosa. Acta Otolaryngol. 2006;126(9):941–7. doi: 10.1080/00016480500469016. [DOI] [PubMed] [Google Scholar]
- 56.Tamashiro E, Xiong G, Anselmo-Lima WT, et al. Cigarette smoke exposure impairs respiratory epithelial ciliogenesis. Am J Rhinol Allergy. 2009;23(2):117–22. doi: 10.2500/ajra.2009.23.3280. [DOI] [PubMed] [Google Scholar]
- 57.Ding GQ, Zheng CQ. The expression of MUC5AC and MUC5B mucin genes in the mucosa of chronic rhinosinusitis and nasal polyposis. Am J Rhinol. 2007;21(3):359–66. doi: 10.2500/ajr.2007.21.3037. [DOI] [PubMed] [Google Scholar]
- 58.Berger G, Kogan T, Ophir D, et al. Glycoconjugate expression of sinus mucosa in chronic rhinosinusitis: a lectin histochemical study. Am J Rhinol. 2008;22(4):349–55. doi: 10.2500/ajr.2008.22.3185. [DOI] [PubMed] [Google Scholar]
- 59••.Van Bruaene N, Perez-Novo CA, Basinski TM, et al. T-cell regulation in chronic paranasal sinus disease. J Allergy Clin Immunol. 2008;121(6):1435–1441. 1441 e1431–1433. doi: 10.1016/j.jaci.2008.02.018. This study points toward a deficient Treg function in CRSwNP, but not in CRSsNP, based on tissue expression of transcription factor markers associated with T-cell subpopulations. [DOI] [PubMed] [Google Scholar]
- 60.de Vries VC, Wasiuk A, Bennett KA, et al. Mast cell degranulation breaks peripheral tolerance. Am J Transplant. 2009;9(10):2270–80. doi: 10.1111/j.1600-6143.2009.02755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peters AT, Kato A, Zhang N, et al. Evidence for altered activity of the IL-6 pathway in chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2010;125(2):397–403 e310. doi: 10.1016/j.jaci.2009.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kato A, Peters A, Suh L, et al. Evidence of a role for B cell-activating factor of the TNF family in the pathogenesis of chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2008;121(6):1385–1392. 1392 e1381–1382. doi: 10.1016/j.jaci.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63•.Kirsche H, Niederfuhr A, Deutschle T, et al. Ratio of myeloid and plasmacytoid dendritic cells and TH2 skew in CRS with nasal polyps. Allergy. 2010;65(1):24–31. doi: 10.1111/j.1398-9995.2009.02174.x. Supporting the idea that CRSwNP and CRSsNP are pathophysiologically different diseases based on cellular makeup this study specifically looked at the ratio of myeloid and plasmacytoid dendritic cells and its role in T-cell differentiation. [DOI] [PubMed] [Google Scholar]
- 64.Woodworth BA, Lathers D, Neal JG, et al. Immunolocalization of surfactant protein A and D in sinonasal mucosa. Am J Rhinol. 2006;20(4):461–5. doi: 10.2500/ajr.2006.20.2892. [DOI] [PubMed] [Google Scholar]
- 65.Wang J, Matsukura S, Watanabe S, et al. Involvement of Toll-like receptors in the immune response of nasal polyp epithelial cells. Clin Immunol. 2007;124(3):345–52. doi: 10.1016/j.clim.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 66.Vandermeer J, Sha Q, Lane AP, Schleimer RP. Innate immunity of the sinonasal cavity: expression of messenger RNA for complement cascade components and toll-like receptors. Arch Otolaryngol Head Neck Surg. 2004;130(12):1374–80. doi: 10.1001/archotol.130.12.1374. [DOI] [PubMed] [Google Scholar]
- 67.Lin CF, Tsai CH, Cheng CH, et al. Expression of Toll-like receptors in cultured nasal epithelial cells. Acta Otolaryngol. 2007;127(4):395–402. doi: 10.1080/00016480601089416. [DOI] [PubMed] [Google Scholar]
- 68.Psaltis AJ, Bruhn MA, Ooi EH, et al. Nasal mucosa expression of lactoferrin in patients with chronic rhinosinusitis. Laryngoscope. 2007;117(11):2030–5. doi: 10.1097/MLG.0b013e31812e01ab. [DOI] [PubMed] [Google Scholar]
- 69•.Reh DD, Wang Y, Ramanathan M, Jr, Lane AP. Treatment-recalcitrant chronic rhinosinusitis with polyps is associated with altered epithelial cell expression of interleukin-33. Am J Rhinol Allergy. 2010;24(2):105–9. doi: 10.2500/ajra.2010.24.3446. This paper demonstrated production of IL-33 by sinonasal epithelial cells, with enhanced expression after TLR stimulation in those derived from nasal polyps, suggesting a role of epithelial cells in modulating Th2 adaptive immunity severe forms of CRSwNP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schlosser RJ, Mulligan RM, Casey SE, et al. Alterations in gene expression of complement components in chronic rhinosinusitis. Am J Rhinol Allergy. 2010;24(1):21–5. doi: 10.2500/ajra.2010.24.3399. [DOI] [PubMed] [Google Scholar]
- 71••.Ramanathan M, Jr, Lee WK, Spannhake EW, Lane AP. Th2 cytokines associated with chronic rhinosinusitis with polyps down-regulate the antimicrobial immune function of human sinonasal epithelial cells. Am J Rhinol. 2008;22(2):115–121. doi: 10.2500/ajr.2008.22.3136. This study shows that expression of multiple innate genes in sinonasal epithelial cells is reduced in CRSwNP and demonstrates a mechanism by which direct effects of leukocyte-derived Th2 cytokines may contribute to disease pathogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Reh DD, Ramanathan M, Jr, Sultan B, et al. The role of hepatocyte growth factor/c-Met in chronic rhinosinusitis with nasal polyps. Am J Rhinol Allergy. 2010;24(4):266–70. doi: 10.2500/ajra.2010.24.3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tieu DD, Kern RC, Schleimer RP. Alterations in epithelial barrier function and host defense responses in chronic rhinosinusitis. J Allergy Clin Immunol. 2009;124(1):37–42. doi: 10.1016/j.jaci.2009.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tieu DD, Peters AT, Carter RT, et al. Evidence for diminished levels of epithelial psoriasin and calprotectin in chronic rhinosinusitis. J Allergy Clin Immunol. 2010;125(3):667–75. doi: 10.1016/j.jaci.2009.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang N, Truong-Tran QA, Tancowny B, et al. Glucocorticoids enhance or spare innate immunity: effects in airway epithelium are mediated by CCAAT/enhancer binding proteins. J Immunol. 2007;179(1):578–89. doi: 10.4049/jimmunol.179.1.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Senior BA, Kennedy DW, Tanabodee J, et al. Long-term impact of functional endoscopic sinus surgery on asthma. Otolaryngol Head Neck Surg. 1999;121(1):66–8. doi: 10.1016/S0194-5998(99)70127-0. [DOI] [PubMed] [Google Scholar]
- 77.Szczeklik A, Stevenson DD. Aspirin-induced asthma: advances in pathogenesis, diagnosis, and management. J Allergy Clin Immunol. 2003;111(5):913–21. doi: 10.1067/mai.2003.1487. [DOI] [PubMed] [Google Scholar]
- 78.Kim JE, Kountakis SE. The prevalence of Samter's triad in patients undergoing functional endoscopic sinus surgery. Ear Nose Throat J. 2007;86(7):396–9. [PubMed] [Google Scholar]
- 79.McMains KC, Kountakis SE. Medical and surgical considerations in patients with Samter's triad. Am J Rhinol. 2006;20(6):573–6. doi: 10.2500/ajr.2006.20.2913. [DOI] [PubMed] [Google Scholar]
- 80•.Katial RK, Strand M, Prasertsuntarasai T, et al. The effect of aspirin desensitization on novel biomarkers in aspirin-exacerbated respiratory diseases. J Allergy Clin Immunol. 2010;126(4):738–744. doi: 10.1016/j.jaci.2010.06.036. This paper explores the mechanism underlying aspirin desensitization and characterizes the airway inflammatory response to desensitization, including the role of mast cell degranulation acutely and decrease in IL-4 levels with treatment. [DOI] [PubMed] [Google Scholar]
- 81.Stankovic KM, Goldsztein H, Reh DD, et al. Gene expression profiling of nasal polyps associated with chronic sinusitis and aspirin-sensitive asthma. Laryngoscope. 2008;118(5):881–9. doi: 10.1097/MLG.0b013e31816b4b6f. [DOI] [PubMed] [Google Scholar]
- 82.Bernstein JM, Anon JB, Rontal M, et al. Genetic polymorphisms in chronic hyperplastic sinusitis with nasal polyposis. Laryngoscope. 2009;119(7):1258–64. doi: 10.1002/lary.20239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang LF, Chien CY, Tai CF, et al. Matrix metalloproteinase-9 gene polymorphisms in nasal polyposis. BMC Med Genet. 2010;11:85. doi: 10.1186/1471-2350-11-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Palikhe NS, Kim SH, Cho BY, et al. IL-13 gene polymorphisms are associated with rhinosinusitis and eosinophilic inflammation in aspirin intolerant asthma. Allergy Asthma Immunol Res. 2010;2(2):134–40. doi: 10.4168/aair.2010.2.2.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Buysschaert ID, Grulois V, Eloy P, et al. Genetic evidence for a role of IL33 in nasal polyposis. Allergy. 2010;65(5):616–22. doi: 10.1111/j.1398-9995.2009.02227.x. [DOI] [PubMed] [Google Scholar]
- 86.Mfuna Endam L, Cormier C, Bosse Y, et al. Association of IL1A, IL1B, and TNF gene polymorphisms with chronic rhinosinusitis with and without nasal polyposis: a replication study. Arch Otolaryngol Head Neck Surg. 2010;136(2):187–92. doi: 10.1001/archoto.2009.219. [DOI] [PubMed] [Google Scholar]
- 87.Pinto JM, Hayes MG, Schneider D, et al. A genomewide screen for chronic rhinosinusitis genes identifies a locus on chromosome 7q. Laryngoscope. 2008;118(11):2067–72. doi: 10.1097/MLG.0b013e3181805147. [DOI] [PMC free article] [PubMed] [Google Scholar]