

Abstract
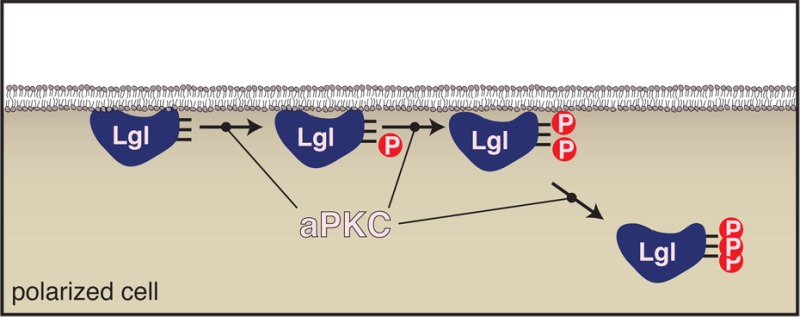
In Par complex-mediated cell polarity, phosphorylation by atypical protein kinase C (aPKC) is coupled to substrate cortical displacement. Polarized substrates often contain multiple phosphorylation sites, but the role of multisite phosphorylation in Par-mediated polarity remains unclear. Here, we have dissected the role of the three aPKC phosphorylation sites within the tumor suppressor Lethal giant larvae. Using a cultured Drosophila S2 cell cortical displacement assay, we observed that phosphorylation at any one site causes only partial displacement. Complete displacement requires that all three sites be modified. We undertook a kinetic analysis to determine if aPKC phosphorylates each site equivalently. As the sites are closely spaced, we observed not only differences in the rate of phosphorylation but also interaction between the sites. A complete description of the rates reveals a preferential order of phosphorylation. Our results provide new insights into how multiple phosphorylations and phosphorylation rates could regulate localization behaviors of fate determinants at the cortex.
Many cells organize their cortical regions into large, molecularly discrete domains. The resulting cell polarity is essential for a broad array of processes, including cytokinesis, movement, and asymmetric division.1−4 Atypical protein kinase C (aPKC), part of the Par polarity complex, has emerged as a key organizer of the cell cortex.5−7 In Par-mediated polarity, upstream components specify the location and activity of aPKC. Once at the cortex, aPKC phosphorylates substrates, displacing them into the cytoplasm, thereby ensuring that they occupy only cortical regions opposite aPKC.6,8,9 Because a key aspect of Par polarity is phosphorylation-coupled cortical displacement, we have investigated the detailed kinetics of aPKC substrate phosphorylation and the requirements for release from the cortex.
The Par complex directs polarity in diverse systems using a mechanism that relies on aPKC-mediated phosphorylation. Besides aPKC, the Par complex consists of Par-3 (Bazooka in flies) and Par-6. These proteins, along with a large number of upstream regulators, control aPKC localization and kinase activity in cells ranging from epithelia to neural stem cells.10,11 When not bound to Par-6, aPKC exists in an autoinhibited form with an internal pseudosubstrate motif bound to its kinase domain.12 Recruitment to the cortex by Par-3 and Par-6 activates aPKC such that it can efficiently phosphorylate target proteins, such as the tumor suppressor Lethal giant larvae (Lgl) and the fate determinants Numb and Miranda. Once phosphorylated, these substrates become cytoplasmic, effectively displacing them from cortical regions containing aPKC. In the absence of aPKC catalytic activity, these substrates become depolarized with severe physiological consequences.13−15
As substrate phosphorylation by aPKC appears to be a central element of polarity, we have investigated the detailed kinetics of the process. Many aPKC substrates contain numerous phosphorylation sites (e.g., three sites for Lgl,13 five sites for Mira,15 and five sites for Numb14), and nonphosphorylatable mutants are not polarized by aPKC. These studies suggest that multisite phosphorylation is an important component of polarization by aPKC, although this hypothesis has not been tested. Is phosphorylation at all sites required for cortical displacement? Are the kinetic parameters for aPKC phosphorylation the same at each site? Is there synergy between phosphorylation at each site?
Here, we investigated the role of multiple phosphorylations using Lgl as a model aPKC substrate. First, we tested the number of phosphorylation sites needed for cortical displacement of Lgl using cultured Drosophila S2 cells. We find that Lgl requires at least two, possibly three, phosphorylations for membrane displacement. Also, we examined how aPKC phosphorylates Lgl at three Ser residues using in vitro kinase assays. These results demonstrate that the three sites are not kinetically equivalent, for aPKC shows a clear preference among them. Furthermore, we utilized phospho-mimetic mutants to test whether multiphosphorylation is dependent. We find that it is dependent, but the effects depend on the position of the phospho-mimetic mutation(s). We conclude that phosphorylation of Lgl at multiple sites by aPKC plays a vital role in the regulation of Lgl localization. Our results may be extended to the other aPKC substrates and differentiation factors to improve our understanding of the mechanism of polarization. Our findings could also provide new insight into the substrate recognition and preference of aPKC.
Experimental Procedures
S2 Lgl Cortical Localization Assay
Immunofluorescence was as previously described.16 Briefly, for S2 cell expression, Lgl fused to mCherry or eGFP was expressed using the pMT vector that places Lgl downstream of the metallothionen promoter. Lgl was visualized by direct imaging of the fluorescent protein. For conditions in which aPKC was expressed with Lgl, a constitutively active aPKC variant (A134D)12 was expressed with a modified pMT vector containing the Drosophila tubulin promoter in place of the metallothionen promoter.12 mCherry:Lgl and eGFP:Lgl coding sequences were cloned into the pMT vector using 5′-BglII and 3′-XhoI sites. Drosophila Schneider (S2) cells were maintained in Schneider’s Medium with 10% fetal bovine serum at room temperature. Approximately 2 × 106 cells were seeded per well in a six-well plate and transfected with 0.5 μg of each construct using Effectene transfection reagent according to the manufacturer’s protocol. After cells had been incubated overnight and induced with 0.5 mM CuSO4 for 24 h, 200 μL of cells was seeded on 12 mm diameter glass coverslips in a 24-well plate and allowed to adhere for 1 h. Cells were fixed for 20 min with 4% formaldehyde in phosphate-buffered saline (PBS) followed by three rinses of wash buffer (0.1% saponin in PBS) and two rinses of block buffer (0.1% saponin and 1% bovine serum albumin in PBS). Coverslips were incubated overnight at 4 °C with rabbit anti-aPKC antibody (1:1000, Santa Cruz Biotechnology). Coverslips were then rinsed three times with blocking buffer, incubated at room temperature for 2 h with a species-specific secondary antibody (1:200, Jackson Immunoresearch), rinsed three times in washing buffer, and mounted in Vectashield Hardset Mounting Medium (Vector Laboratories). Images were acquired on a confocal microscope (Radiance, Bio-Rad Laboratories) using an oil-immersion 60× 1.4 NA objective, processed with ImageJ, and assembled in Adobe Illustrator.
Quantification of Lgl Localization and Statistical Analyses
The average Lgl signal at the cell periphery from a confocal slice through the center of the cell was measured with ImageJ. Similarly, the cytoplasmic area excluding the nucleus was selected, and its mean intensity was measured for the same cell. For conditions used for the co-expression of aPKC, the mean level of expression was 7–10 times the level of endogenous aPKC, as assessed by reactivity with the anti-aPKC antibody. The average cortical:cytoplamsic ratios were calculated for each Lgl construct and plotted using GraphPad Prism. To analyze the data, we compared the average ratios of Lgl AAA, Lgl AAA with aPKC, and Lgl wt with aPKC against all the other ratios using one-way analysis of variance (ANOVA).
Purification of aPKC 259–606 (Kinase Domain)
HEK293 F cells (1 × 106 cells/mL) were transfected with pCMV His6-aPKC 259–606 and pCMV dPDK-1 (without any tag) using the 293fectin transfection reagent (Life Technologies). The cells were incubated at 37 °C for 48 h. For harvesting, the cells were resuspended with Ni2+ lysis buffer [50 mM Tris-HCl (pH 7.5), 300 mM NaCl, 1 mM MgCl, 10 mM β-ME, and 10 mM imidazole, adjusted to pH 7.5 with NaOH]. The cells were lysed by being passed through a 21-gauge needle, and the lysate was clarified by centrifugation at 15000 rpm for 30 min at 4 °C. The supernatant was incubated with Ni2+-NTA resins for 45 min at 4 °C and then washed with lysis buffer. Following elution using Ni2+ elution buffer (lysis buffer with 250 mM imidazole), the eluted proteins were dialyzed at 4 °C for 4 h against 20 mM Tris-HCl, 50 mM NaCl, and 1 mM dithiothreitol (DTT). The concentration of aPKC was determined by comparing its reactivity with an anti-aPKC antibody (Santa Cruz Biotechnology) with that of a standard at a known concentration (bacterially expressed aPKC kinase domain purified and quantified using a Bradford dye binding assay) on a Western blot.
Expression and Purification of Lgl Peptides
Residues 647–673 of Drosophila Lgl isoform A were cloned into the pMAL-C2 vector (New England BioLabs Inc.), in which a TEV protease recognition site was added following the maltose binding protein (MBP) coding sequence. For expression, Escherichia coli BL21 was transformed with the appropriate vector and induced by addition of IPTG to a final concentration of 0.4 mM followed by overnight growth at 18 °C. The bacterial lysates were incubated with amylose resins (New England BioLabs Inc.). The resins were washed with MBP lysis buffer [20 mM Tris-HCl (pH 7.5), 200 mM NaCl, 1 mM EDTA, and 1 mM DTT]. The MBP fusion proteins were eluted with MBP elution buffer (lysis buffer with 5 mM maltose) and dialyzed at 4 °C overnight in 20 mM Tris-HCl (pH 7.5), 50 mM NaCl, and 1 mM DTT.
In Vitro Kinase Activity Assay
aPKC kinase activity was measured as previously described.12 Briefly, the purified aPKC kinase domain was diluted to concentrations at which the incorporation of radiolabeled phosphate from [γ-32P]ATP into MBP-Lgl peptides was linear with respect to time and enzyme concentration. The diluted enzymes were preincubated in assay buffer [50 mM Tris-HCl (pH 7.5), 100 mM NaCl, and 10 mM MgCl2] with a wide range of MBP-Lgl peptide concentrations at 30 °C for 5 min. The reactions were initiated by adding 1 mM ATP spiked with [γ-32P]ATP (∼1.0 × 106 per nanomole of ATP). The reaction mixtures were incubated at 30 °C for 10 min. The reaction mixtures were blotted on grade P81 phosphopaper (Whatman). The reactions were quenched when the blotted P81 paper was immediately submerged in 75 mM H3PO4; 5 mL of scintillation fluid was added to measure the radioactive decays with a liquid scintillation counter. The kinetic parameters were calculated by fitting the data to the Michealis–Menten equation in GraphPad Prism.
Kinetic Modeling
Copasi was used to model aPKC phosphorylation of Lgl. The model contained empirical values of KM and kcat for each site (both with and without adjacent phosphorylations). Simulations were compared against a time course of Lgl phosphorylation to test the model.
Separation of Singly, Doubly, and Triply Phosphorylated Lgl Peptides
A reaction mixture containing 15 μM MBP-Lgl 647–673, the aPKC kinase domain, and 1 mM ATP in the reaction buffer described above was set up and incubated at 30 °C for 90 min. Small aliquots of the reaction mixture were quenched via sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) loading buffer periodically during the incubation. The quenched samples were run on Mn2+-Phos-tag SDS–PAGE (Wako USA). The phosphorylated Lgl was detected on a Western blot using mouse anti-MBP (1:1000, Santa Cruz Biotechnology) and bovine anti-mouse HRP (1:2000, Santa Cruz Biotechnology) antibodies.
Results
Lgl Cortical Displacement Requires Phosphorylation at Multiple Serines
Lgl phosphorylation by aPKC is critical for its function in cell polarity. aPKC phosphorylates Lgl at three serine residues in vitro, and nonphosphorylatable Lgl fails to be released from the cell cortex.13 The ectopic localization of nonphosphorylatable Lgl disrupts the polarity of both asymmetrically dividing neural stem cells13 and epithelia.17 Although phosphorylation plays a crucial role in Lgl localization and function, the role of Lgl’s multiple phosphorylation sites is unknown.
To determine the role of individual Lgl phosphorylation events in its localization, we expressed Lgl harboring mutations at its phosphorylation sites (Figure 1A) in cultured Drosophila S2 cells, a well-established system for assessing the effect of aPKC on Lgl cortical localization.13,15,16 In this assay, the degree to which Lgl localizes to the cortex is measured using the ratio of the Lgl signal (from the fluorescent protein signal visualized in a confocal section through the middle of the cell) at the periphery of the cell to that in the cytoplasm. When Lgl is expressed by itself in S2 cells, it is predominantly cortically localized (cortical:cytoplasmic ratio of >2:1), but co-expression of aPKC leads to its displacement into the cytoplasm (cortical:cytoplasmic ratio of ∼1:1); this effect requires the presence of the Lgl phosphorylation sites12,13,15 (Figure 1B,C). For this study, we used a constitutively active form of aPKC that contains a mutation in its pseudosubstrate (A134D) that prevents it from repressing the kinase domain.12 This high level of activity was to ensure that cells contained sufficient aPKC activity to phosphorylate all available Lgl sites. The low level of variance we observed in the localization ratio of each Lgl phospho-mutant (Figure 1C) is consistent with this scenario.
Figure 1.

Phosphorylation at all three Lgl aPKC sites is required for complete cortical displacement. (A) Lgl domain structure and aPKC phospho-site positions. (B) Localization of Lgl (wild type and aPKC phospho-site mutants) in cultured Drosophila S2 cells. Images represent confocal slices through the center of the cell using the fluorescence of the attached mCherry or eGFP fluorophore. (C) Quantification of Lgl cortical and cytoplasmic localization (n = 50 for each condition). The cortical signal was measured as the mean signal from the periphery of the cell, whereas cytoplasmic was the mean from the non-nuclear interior. ANOVA statistical analysis reveals that cortical recruitment of wild-type Lgl is indistinguishable from that of singly phosphorylated Lgl (SAA, ASA, and AAS), and Lgl variants with two phosphorylatable sites (ASS, SAS, and SSA) can be distinguished from the wild type. Error bars represent the standard error.
We tested all possible combinations of Lgl phospho-site mutations (serine to alanine) to determine the effect of multiple mutations on Lgl localization behavior. The resulting data set would allow us to determine if phosphorylation on a particular site is required for cortical displacement or if the total number of phosphorylations is the determining factor. We observed remarkably uniform behavior for Lgl proteins of a particular category (i.e., one or two phospho-sites), indicating that localization “information” is distributed among each of the sites (Figure 1B,C). Singly phosphorylated Lgls localized in a manner indistinguishable from that of the wild type, and while two phosphorylations had a detectable displacement into the cytoplasm, three phosphorylations had the lowest cortical:cytoplasmic ratio (Figure 1B,C). We conclude that no individual phosphorylation is sufficient to release Lgl from the cortex and that all three phosphorylations are required for full displacement.
aPKC Preferentially Phosphorylates Lgl at S656 and S664
The localization assay demonstrated the necessity of multiple phosphorylations for Lgl displacement. This raises the question of how Lgl becomes phosphorylated at all sites. In particular, we sought to determine if intrinsic kinetic differences among the three sites could be important for Lgl localization and function, and if phosphorylation at one site influenced the kinetic parameters at others. The proximity of the Lgl phospho-sites, and the positively charged character of the aPKC recognition sequence (Figure 2A),18 suggested that interactions between sites are highly likely. While each satisfies the requirement at positions P–5, P–2, and P+1, they all fail to match at positions P–4 and P+2 (the positions of these sites on the aPKC kinase domain structure18 are shown in Figure 2B). Also, there are more variations at N-terminal residues away from each phosphorylation site. Therefore, comparison of each of the Lgl subsite sequences with the consensus sequence further suggests that aPKC could have different kinetic parameters for each of the three sites.
Figure 2.

Nonequivalent phosphorylation of the three Lgl phospho-sites by aPKC. (A) Amino acid sequence alignment of Lgl phosphorylation sites. The phopho-accepting serines (P0) are labeled with red asterisks. The optimal consensus sequence is shown at the bottom.25 (B) Structure of a Par-3 peptide bound to the kinase domain of human aPKCι.18 The substrate’s residue positions are labeled with respect to the phospho-accepting residue (P0). The residues positioned N-terminal and C-terminal to P0 are negatively and positively numbered, respectively. Protein Data Bank entry 4DC2. (C) Kinetic analyses of each singly phosphorylated Lgl peptide, showing differences in KM and kcat. Error bars represent the standard error. (D) Possible pathways to phosphorylate all three serine residues. Phosphorylated residues are colored red. If the phosphorylation events are independent, on the basis of the results from panel B, preferential phosphorylation occurs in the following order: S664 → S656 → S660.
To determine whether the three serine residues are equivalent, we generated singly phoshorylatable Lgl peptides and performed in vitro kinase assays. We used the kinase domain as full length aPKC is repressed by a pseudosubstrate contained within its sequence. Using Michaelis–Menten analyses, we determined the catalytic efficiency (kcat/KM) for phosphorylation at each subsite. As shown in Figure 2C, KM values for phosphorylation at each site were 7.8 ± 1.5, 11.4 ± 5.8, and 3.8 ± 0.6 μM and kcat values were 3.2 ± 0.2, 1.5 ± 0.3, and 4.3 ± 0.2 s–1 for SAA, ASA, and ASA, respectively. The catalytic efficiency values were 0.40 ± 0.06, 0.13 ± 0.01, and 1.14 ± 0.40 μM–1 s–1, respectively. There are six possible pathways to phosphorylate all three Lgl serine residues (Figure 2D), and if the sites were independent, our data would indicate the following preferential order: S664 > S656 > S660.
aPKC Phosphorylation of Lgl Is Cooperative
As each serine residue in Lgl is separated by only three residues, it is likely that phosphorylation events interact with one another, with phosphorylation at one site influencing the kinetic parameters of others. To investigate whether Lgl phosphorylation by aPKC is cooperative, we utilized phospho-mimetic mutants of Lgl peptides and tested them in kinase activity assays. We have used phospho-mimetic mutants of aPKC substrates in previous studies and found that they are indistinguishable from phosphorylated substrates.12
First, we tested how phosphorylation of a serine residue affects phosphorylation of directly neighboring sites (Figure 3A). The values of KM for SDA, DSA, ASD, and ADS were 14.4 ± 4.0, 5.9 ± 2.2, 7.9 ± 5.3, and 6.2 ± 1.4 μM and the values of kcat 3.3 ± 0.4, 1.8 ± 0.2, 0.7 ± 0.2, and 4.82 ± 0.4 s–1, respectively. The catalytic efficiencies were 0.23 ± 0.02, 0.31 ± 0.03, 0.09 ± 0.01, and 0.77 ± 0.22 μM–1 s–1, respectively. Compared to SAA, SDA showed a 50% reduction in catalytic efficiency. The reduction was attributed to a KM increase. Both DSA and ASD showed a decrease in KM values relative to that of ASA. However, the replacement of S656 with an Asp increased kcat and the catalytic efficiency, while the replacement of S664 decreased both values. Finally, ADS showed an increase in its KM value, reducing the phosphorylation rate compared to that of AAS. These data indicate that prior phosphorylations on the direct neighboring sites reduce the phosphorylation rates of S656 and S664. In contrast, the presence of the Asp residue at the adjacent site had a favorable effect for S660, which may indicate that the negative charges may be needed for its efficient phosphorylation.
Figure 3.

aPKC phosphorylation of Lgl is cooperative. Phosphorylation rates of individual Lgl sites are affected by the phosphorylation state of neighboring sites (as assessed by phospho-mimetic residues). Error bars represent the standard error. Panel A shows the effect of modification at directly neighboring sites, while panel B shows the effect of more distant modifications. Finally, panel C demonstrates the effect of modifications at both of the other sites. The data show the phosphorylation rates are cooperative but the effects differ depending on the positions of the phospho-accepting residue and modified site(s). On the basis of the results shown in panels A–C, phosphorylation primarily occurs in the following order: S664 → S660 → S656.
We next examined how a phosphate group influences the more distant site using SAD and DAS peptides (Figure 3B). Initial rate analyses showed that KM values were 27.4 ± 9.5 and 3.7 ± 0.4 μM and the kcat values were 4.4 ± 0.9 and 3.4 ± 0.1 s–1, respectively. The catalytic efficiencies were 0.16 ± 0.02 and 0.91 ± 0.23 μM–1 s–1, respectively. The effect of the Asp residue at position P+8 was more pronounced than that at position P+4 for S656, for the KM value of SDA was 2 times higher than that of SAA and the effect on kcat was minimal while the KM value of SAD was 3 times higher and the kcat 40% higher than those of SAA. For DAS, on the other hand, the effect was more subtle, in which kcat was reduced from the value of AAS by 20% and there was almost no change in KM. Combined, the data show that a phospho-mimetic residue at position P+8 influenced kcat, but KM was changed with a phospho-mimetic residue at position P+4. These data indicate that proximal phosphorylation at position P–8 or P+8 reduces catalytic efficiency.
Finally, we examined how two phosphate groups affect the phosphorylation of the remaining Ser residues using SDD, DSD, and DDS Lgl peptides (Figure 3C). The KM values were 26.4 ± 6.2, 2.5 ± 1.5, and 6.4 ± 1.1 μM and kcat values 2.0 ± 0.3, 0.5 ± 0.1, and 2.5 ± 0.14 s–1, respectively. The catalytic efficiencies were 0.08 ± 0.01, 0.22 ± 0.01, and 0.40 ± 0.05 μM–1 s–1, respectively. These data show that the presence of two Asp residues decreases the rates of phosphorylation of S656 and S664 and increases that of S660. These results indicate that S660 is more likely to be phosphorylated after S656 and S664 are phosphorylated.
All the kinetic parameters measured showed that the replacement of S660 with Asp resulted in increased KM values for S656 and S664. On the other hand, the presence of a phospho-mimetic residue(s) was beneficial to S660. Taken together, the primary order of phosphorylation is likely to be S664, S660, and then S656 followed by a secondary pathway of S656, S664, and then S660.
L661F and E663K Mutations Together Reduce the KM of the ASA Peptide
Considering how similar the sequences of the three Lgl phosphorylation subsites are, it was surprising how aPKC presented clear preferences among them. We reasoned that the residues in each subsite that do not match the consensus sequence may not contribute significantly to the observed activity differences. In this case, the differences in kinetic parameters between S660 and S664 could originate from the residues surrounding the two subsites.
To investigate the basis for the subsite preference within Lgl, we examined a series of mutations in the context of Lgl ASA (Figure 4A). First, we noticed that ASA peptide had a Glu residue at position P+3, while SAA and AAS peptides had a Lys at the same position. The values of KM and kcat for the ASA E663K mutant were 12.4 ± 3.6 μM and 1.7 ± 0.2 μM–1 s–1, respectively, indicating that replacement of Glu with Lys at position P+3 does not affect the rate of ASA phosphorylation. We also tested the ASA L661F mutant. Although the ASA mutant satisfies the hydrophobic residue requirement at position P+1, both SAA and AAS peptides had a Phe residue. The values for KM and kcat for ASA L661F were 10.7 ± 2.0 μM and 2.5 ± 0.2 μM–1 s–1, respectively, suggesting that the substitution of Lue with Phe at position P+1 did not affect KM but led to a 66% increase in kcat, compared to that of ASA. When the ASA L661F/E663K double mutant was tested, the kcat value was 1.7 ± 0.1 μM–1 s–1 and comparable to that of ASA, but the KM value was reduced to 1.9 ± 0.7 μM. This was 6 times lower than that of ASA and 2 times lower than that of AAS, implying that the those two residues may play roles in binding affinity.
Figure 4.

Ordered phosphorylations of aPKC. (A) Critical recognition positions that determine the KM for aPKC phosphorylation. The L661F and E663K mutations together converted the KM of ASA to that of AAS. Error bars represent the standard error. (B) Time course of wild-type Lgl phosphorylation as determined by phospho-resolving gel electrophoresis. (C) Modeling of Lgl phosphorylation by aPKC. The degree of phosphorylation was quantified using the data from panel B overlaid with a simulation of wild-type Lgl phosphorylation (solid lines) using the data measured in the previous sections.
We followed the concentration of singly, doubly, and triply phosphorylated Lgl to determine how well the kinetic parameters from each site could predict bulk phosphorylation behavior. Using SDS–PAGE gels containing Mn2+ and a Phos tag, we were able to resolve each of the phosphorylated species (Figure 4B)
Discussion
Phosphorylation-coupled release of aPKC substrates from the plasma membrane is a central element of Par-mediated polarization. The mechanism by which substrates are released from cortex, and in particular the role of multisite phosphorylation, has been unclear, however. In the case of the tumor suppressor Lgl, phosphorylation by aPKC is crucial for not only asymmetric cell division but also epithelial cell polarity and cell migration.19−21 In this work, we have found that aPKC phosphorylation of Lgl at each of its three phospho-sites is required for full displacement from the cortex in a cultured S2 cell model system. Because multisite phosphorylation is essential for cortical release, and therefore polarization, we investigated the kinetics at each site and how phosphorylation at one site influences others. We observed striking differences between the kinetic parameters for each subsite. A mathematical model for Lgl phosphorylation based on these parameters was able to predict the overall rates of Lgl phosphorylation.
Multisite Phosphorylation Is Coupled to the Cortical Release of Lgl
We first addressed the relationship between the number of phosphorylations and cortical localization of Lgl by expressing various Lgl constructs in cultured Drosophila S2 cells. We observed a general trend of an increasing level of cytoplasmic Lgl with increasing levels of phosphorylation states, indicating an additive effect of multisite phosphorylation on membrane interaction. Similar behavior has been observed with another aPKC substrate, the myristoylated alanine rich C kinase substrate (MARCKS). Phosphorylation of three serine residues in MARCKS weakens its electrostatic attraction for the membrane, resulting in translocation of MARCKS into the cytoplasm.22−24 Multisite phosphorylation of aPKC substrates could be a general mechanism for altering the electrostatic charges of the substrates to regulate their membrane interactions in the cell. It is of interest to determine whether multisite phosphorylation exhibits the same effects on other aPKC substrates such as Miranda and Numb.
Phosphorylation Order as a Regulatory Element for Cortical Displacement
We examined whether phosphorylation of each serine residue was equivalent and found that aPKC preferentially phosphorylates Lgl in the following order: S664 > S656 > S660. This indicates that the phospho-sites are not equivalent. We also tested if phospho-sites interact with one another and found that the effects of phosphorylation (using phospho-mimetic residues as a proxy) depend on their position relative to the target serine. For example, the presence of Asp at S656 increases the KM of adjacent sites. On the other hand, the phospho-mimetic residue(s) decreased the KM for S660. Our data indicate the primary pathway for Lgl phosphorylation is S664 → S660 → S656 while the secondary pathway is S656 → S664 → S660. The differences in kinetic parameters could determine how well fate determinants interact with the plasma membrane (i.e., the association and dissociation rates). Additionally, the order of phosphorylation may play a role in the availability of the subsites when Lgl is bound to the membrane, as phosphorylation could cooperatively release the membrane-bound subsite that otherwise is not accessible to aPKC. The kinetic analyses of other aPKC substrates, combined with localization assays, will be useful for a more generalized understanding of the relationship between the number of phosphorylations and cortical release behaviors.
Flexibility in the aPKC Substrate Recognition Sequence
Substrate recognition is an important aspect of substrate binding and specificity. Though the three serine residues and the surrounding amino acid sequences are similar to one another, aPKC showed a clear preference among them, suggesting that the subtle difference in sequence could have a large impact on kinetic parameters and substrate preference. Lgl phosphorylation sites showed some deviation from the known consensus amino acid sequence of aPKC substrates.18,25,26 All Lgl peptides satisfied the requirement at positions P–5, P–2, and P+1, while they all failed to match at positions P–4 and P+2. Our data demonstrate that the mutation of Leu to Phe at position P+1 slightly increased Vmax, suggesting that the larger hydrophobic residue interacts more favorably with the kinase interaction surface. In contrast, mutation of Glu to Lys at position P+3, thought to be critical for recognition, did not influence the phosphorylation rate of S660. Taken together, our findings suggest that the collective properties of the phospho-accepting residue regions play a key role in preferential binding and efficient catalysis. This could explain the range of amino acid requirements seen in a single recognition site. Our study emphasizes the complexity of substrate specificity and the flexibility of aPKC’s substrate recognition mechanism.
Acknowledgments
We thank Matt Bailey and Lyle McPherson for their generous help during the revision process and all members of the Prehoda lab for valuable feedback on this work.
The authors declare no competing financial interest.
This study was supported by National Institutes of Health Grant GM068032 (K.E.P.).
Funding Statement
National Institutes of Health, United States
References
- Knoblich J. A. (2010) Asymmetric cell division: Recent developments and their implications for tumour biology. Nat. Rev. Mol. Cell Biol. 11(12), 849–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prehoda K. E. (2009) Polarization of Drosophila neuroblasts during asymmetric division. Cold Spring Harbor Perspect. Biol. 1(2), a001388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstralh D. T.; Haack T.; St Johnston D. (2013) Epithelial polarity and spindle orientation: Intersecting pathways. Philos. Trans. R. Soc., B 368(1629), 20130291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S. (2008) Polarity proteins in migration and invasion. Oncogene 27(55), 6970–6980. [DOI] [PubMed] [Google Scholar]
- Suzuki A.; Ohno S. (2006) The PAR-aPKC system: Lessons in polarity. J. Cell Sci. 119(Part 6), 979–987. [DOI] [PubMed] [Google Scholar]
- Rolls M. M.; et al. (2003) Drosophila aPKC regulates cell polarity and cell proliferation in neuroblasts and epithelia. J. Cell Biol. 163(5), 1089–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein B.; Macara I. G. (2007) The PAR proteins: Fundamental players in animal cell polarization. Dev. Cell 13(5), 609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. Y.; Robinson K. J.; Doe C. Q. (2006) Lgl, Pins and aPKC regulate neuroblast self-renewal versus differentiation. Nature 439(7076), 594–598. [DOI] [PubMed] [Google Scholar]
- Jan Y. N.; Jan L. Y. (2001) Asymmetric cell division in the Drosophila nervous system. Nat. Rev. Neurosci. 2(11), 772–779. [DOI] [PubMed] [Google Scholar]
- Gonczy P. (2008) Mechanisms of asymmetric cell division: Flies and worms pave the way. Nat. Rev. Mol. Cell Biol. 9(5), 355–366. [DOI] [PubMed] [Google Scholar]
- Macara I. G. (2004) Parsing the polarity code. Nat. Rev. Mol. Cell Biol. 5(3), 220–231. [DOI] [PubMed] [Google Scholar]
- Graybill C.; et al. (2012) Partitioning-defective Protein 6 (Par-6) Activates Atypical Protein Kinase C (aPKC) by Pseudosubstrate Displacement. J. Biol. Chem. 287(25), 21003–21011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betschinger J.; Mechtler K.; Knoblich J. A. (2003) The Par complex directs asymmetric cell division by phosphorylating the cytoskeletal protein Lgl. Nature 422(6929), 326–330. [DOI] [PubMed] [Google Scholar]
- Smith C. A.; et al. (2007) aPKC-mediated phosphorylation regulates asymmetric membrane localization of the cell fate determinant Numb. EMBO J. 26(2), 468–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood S. X.; Prehoda K. E. (2009) aPKC phosphorylates Miranda to polarize fate determinants during neuroblast asymmetric cell division. Curr. Biol. 19(9), 723–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee B.; et al. (2011) Canoe binds RanGTP to promote Pins(TPR)/Mud-mediated spindle orientation. J. Cell Biol. 195(3), 369–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutterer A.; et al. (2004) Sequential roles of Cdc42, Par-6, aPKC, and Lgl in the establishment of epithelial polarity during Drosophila embryogenesis. Dev. Cell 6(6), 845–854. [DOI] [PubMed] [Google Scholar]
- Wang C.; et al. (2012) Substrate recognition mechanism of atypical protein kinase Cs revealed by the structure of PKCι in complex with a substrate peptide from Par-3. Structure 20(5), 791–801. [DOI] [PubMed] [Google Scholar]
- Yamanaka T.; et al. (2003) Mammalian Lgl forms a protein complex with PAR-6 and aPKC independently of PAR-3 to regulate epithelial cell polarity. Curr. Biol. 13(9), 734–743. [DOI] [PubMed] [Google Scholar]
- Yamanaka T.; et al. (2006) Lgl mediates apical domain disassembly by suppressing the PAR-3-aPKC-PAR-6 complex to orient apical membrane polarity. J. Cell Sci. 119(Part 10), 2107–2118. [DOI] [PubMed] [Google Scholar]
- Parsons L. M.; et al. (2010) Lgl/aPKC and Crb regulate the Salvador/Warts/Hippo pathway. Fly 4(4), 288–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmori S.; et al. (2000) Importance of protein kinase C targeting for the phosphorylation of its substrate, myristoylated alanine-rich C-kinase substrate. J. Biol. Chem. 275(34), 26449–26457. [DOI] [PubMed] [Google Scholar]
- McLaughlin S.; Murray D. (2005) Plasma membrane phosphoinositide organization by protein electrostatics. Nature 438(7068), 605–611. [DOI] [PubMed] [Google Scholar]
- Herget T.; et al. (1995) The myristoylated alanine-rich C-kinase substrate (MARCKS) is sequentially phosphorylated by conventional, novel and atypical isotypes of protein kinase C. Eur. J. Biochem. 233(2), 448–457. [DOI] [PubMed] [Google Scholar]
- Kang J. H.; et al. (2012) Protein kinase C (PKC) isozyme-specific substrates and their design. Biotechnol. Adv. 30(6), 1662–1672. [DOI] [PubMed] [Google Scholar]
- Nishikawa K.; et al. (1997) Determination of the specific substrate sequence motifs of protein kinase C isozymes. J. Biol. Chem. 272(2), 952–960. [DOI] [PubMed] [Google Scholar]