

Abstract
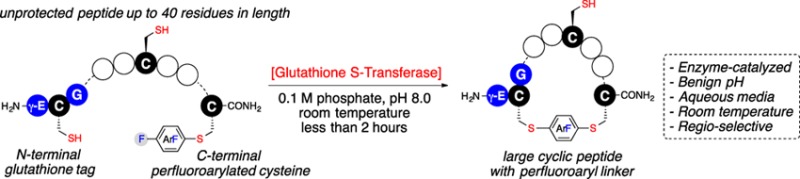
A glutathione S-transferase (GST) catalyzed macrocyclization reaction for peptides up to 40 amino acids in length is reported. GST catalyzes the selective SNAr reaction between an N-terminal glutathione (GSH, γ-Glu-Cys-Gly) tag and a C-terminal perfluoroaryl-modified cysteine on the same polypeptide chain. Cyclic peptides ranging from 9 to 24 residues were quantitatively produced within 2 h in aqueous pH = 8 buffer at room temperature. The reaction was highly selective for cyclization at the GSH tag, enabling the combination of GST-catalyzed ligation with native chemical ligation to generate a large 40-residue peptide macrocycle.
Both natural and hybrid cyclic peptides represent an important class of molecules being actively explored as potential therapeutics.1 These species are attractive because of their enhanced binding affinity, exo- and endopeptidase resistance, and in certain cases increased cell penetration compared to their linear counterparts. Chemical macrocyclization methods have been developed to cross-link two functional groups within a linear peptide substrate through head-to-tail, head-to-side chain, tail-to-side chain, or side chain-to-side chain.2 Cross-linking methods include amide bond or ester bond formation,3 thiol-based substitution with alkyl and benzyl halide electrophiles (commonly referred to as “alkylation”),4 oxidation at cysteine or selenocysteine residues to form a dichalcogenide bond,1a,5 native chemical ligation,6 intein-mediated approaches,7 Staudinger ligation,8 transition-metal-catalyzed coupling reactions,9 cycloadditions,10 coordination-based approaches,11 as well as several noncovalent strategies.12
Enzyme-catalyzed peptide macrocyclizations remain rare.13 Subtiligase was used to catalyze amide-bond formation between an N-terminal amine and a C-terminal ester to generate a 31-residue cyclic peptide with 85% yield.14 Thioesterase was successfully employed to cyclize linear peptides generated from the nonribosomal peptide synthetase system.15 Sortase was used to catalyze the cyclization of peptides, glycopeptides, and proteins.16 With the sortase-catalyzed peptide cyclization method,16a a 67% yield of cyclic peptide product was produced and the linear oligomer was the major side product.
We recently reported a facile cysteine SNAr arylation chemistry using highly activated and electrophilic perfluoroaromatic reagents.17 In particular, two cysteine residues on an unprotected polypeptide were cross-linked with bifunctional perfluoroaromatic reagents. This macrocyclization strategy was versatile enabling the cross-linking of two cysteine moieties positioned at all sites ranging from i, i+1 to i, i+14 on a 14-residue unprotected peptide.17b For longer peptides, however, we found that entropic penalty becomes large enough to render macrocyclizations inefficient. This fundamental limitation is therefore expected to be encountered with any long and unstructured polypeptide irrespective of the cyclization chemistry used. To address the challenge associated with macrocyclization of long polypeptides, we now report a strategy where the glutathione S-transferase (GST) enzyme18 is used to catalyze the SNAr reaction between cysteine and perfluoroaromatic moieties in aqueous buffer leading to large hybrid peptide macrocycles. This transformation overcomes the major length restrictions previously encountered in noncatalyzed SNAr macrocyclization reactions.
We have recently shown that GST can catalyze the conjugation of perfluoroaryl-linked biomolecules or chemical probes to polypeptides and proteins containing an N-terminal γ-Glu-Cys-Gly (GSH) tag.19 This enzymatic “click” ligation reaction enables selective modification of the cysteine in the GSH tag in the presence of other cysteines and reactive functional groups on the same polypeptide chain. Importantly, we found that GST can catalyze the cyclization of a seven-residue peptide between an N-terminal GSH tag and a C-terminal perfluoroaryl moiety; intramolecular cyclization process occurred within seconds producing cyclic peptide in quantitative yield. This result suggests that one could in principle develop a general protocol for GST-catalyzed peptide macrocyclization for potentially larger ring sizes (Figure 1).
Figure 1.
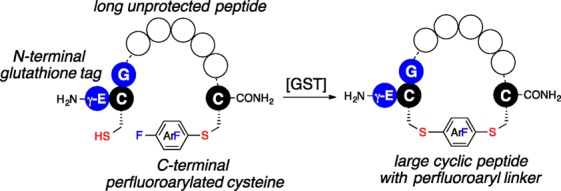
Glutathione S-transferase (GST)-catalyzed macrocyclization. The macrocyclic peptide is generated by GST-catalyzed SNAr reaction between the cysteine thiol of an N-terminal glutathione (γ-Glu-Cys-Gly) tag and a perfluoroaryl moiety linked to the C-terminal cysteine. Circles represent amino acids.
The linear substrates employed in our studies were readily prepared in two steps (Figure 2). Peptides with an N-terminal disulfide protected GSH tag and a C-terminal free cysteine were synthesized by rapid flow-based Fmoc-SPPS followed by C-terminal cysteine S-arylation with perfluoroaromatic reagents using the previously reported protocol (see the Supporting Information). The linear substrates were purified by RP-HPLC and then subjected to macrocyclization. Tris(carboxylethyl)phosphine hydrochloride (TCEP·HCl) was added to the reaction buffer in situ to deprotect the disulfide bond and generate a free cysteine in the GSH tag for GST-catalyzed macrocyclization.
Figure 2.
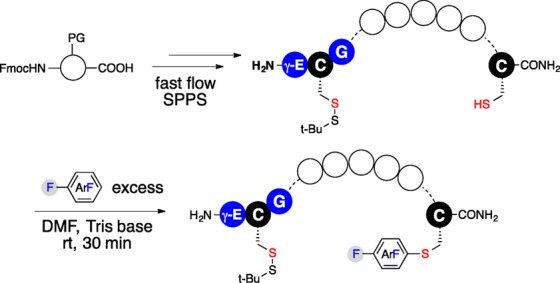
Synthesis of perfluoroaryl-linked peptides for GST-catalyzed macrocyclization. Circles represent amino acids. PG: protecting group; SPPS: solid-phase peptide synthesis.
We commenced our study by searching for the best perfluoroaromatic linker for the GST-catalyzed cyclization reaction (Figure 3). We prepared model peptides 1a–c with C-terminal cysteines S-arylated with decafluorobiphenyl, hexafluorobenzene, and pentafluorophenyl sulfide, respectively (Figure S1, Supporting Information). Upon incubation of a 0.1 mM solution of peptide 1a (Figure 3B, top chromatogram) in the presence of 10 mol % of GST (0.2 mg/mL) in pH 8.0 phosphate buffer with 20 mM TCEP·HCl, cyclic product Cyc-1a was produced in quantitative yield within 5 min at room temperature as shown by LC–MS analysis of the crude reaction mixture. We did not observe oligomeric side products (Figure 3B, bottom chromatogram). Importantly, the control reaction without added enzyme contained mostly unreacted linear peptide 1a′ with only a trace amount of cyclized product (Figure 3B, middle chromatogram). Compared with peptide 1a, peptides 1b and 1c showed significantly lower enzyme mediated cyclization yields under the same reaction conditions (Figure 3A). At extended reaction times, only 45% and 57% of cyclized products were observed for peptide 1b and 1c respectively (Figures S4 and S5, Supporting Information).
Figure 3.
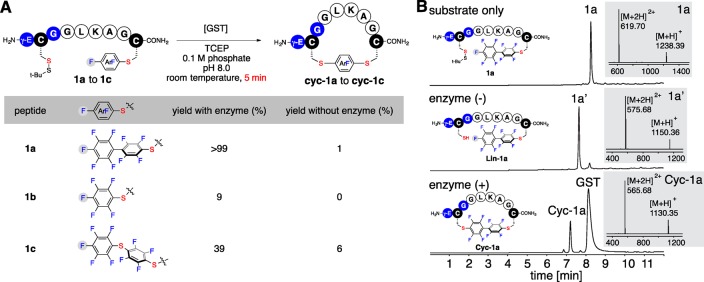
Macrocyclization of peptides with different perfluoroaromatic linkers. Reaction conditions: 0.1 mM peptide 1a–c, 0.2 mg/mL GST, 20 mM tris(carboxylethyl)phosphine hydrochloride (TCEP·HCl), 0.1 M phosphate, pH 8.0, room temperature (25 °C), 5 min. (A) Reaction yields with different linkers with or without the addition of GST. (B) LC–MS analysis of crude reaction mixtures with peptide 1a. Amino acids are shown in one-letter codes. γ-E: γ-glutamic acid. LC–MS data shown are total ion currents (TIC); mass spectra at the highest points of the TIC peaks are shown as insets.
The highest cyclization rate under GST catalysis was achieved with peptide 1a as determined by LC–MS time course studies (Figures S3 and S9, Supporting Information). The cyclization rate of peptide 1a under enzyme-catalysis was ∼900-fold faster than without enzyme (Figure S10, Supporting Information). This high cyclization efficiency under enzyme catalysis permitted macrocyclization at increased concentrations of linear peptide: unlike other enzyme-catalyzed macrocyclization methods where increasing the concentrations of linear substrates can lead to the formation of oligomeric side products via competing intermolecular reaction pathways.16a We found that increasing the concentration of peptide 1a to 10 mM with only 0.1 mol % of GST still produced the cyclized product quantitatively within 3 h (Figure S11, Supporting Information); no oligomeric byproducts were observed. The unique preference and high efficiency of peptide 1a for GST-catalyzed cyclization indicates the perfluorobiphenyl moiety is highly suited for this chemistry.
To probe macrocyclization of peptides with increasing lengths, we prepared peptides 2a–4a with variable numbers (2–4) of repeating GLKAG pentapeptide sequences positioned between the GSH tag and the perfluoroaryl-linked C-terminal cysteine (Figure S2, Supporting Information). Although decreased cyclization rates were observed as the peptide lengths increased (Figure S10, Supporting Information), cyclization of these three peptides under reaction conditions similar to those described in Figure 3 yielded cyclized products Cyc-2a–Cyc-4a nearly quantitatively within 2 h (Figure 4). Only 5% of cyclic products were observed for reactions without GST present (Figure 4A and Figures S6–S8, Supporting Information).
Figure 4.
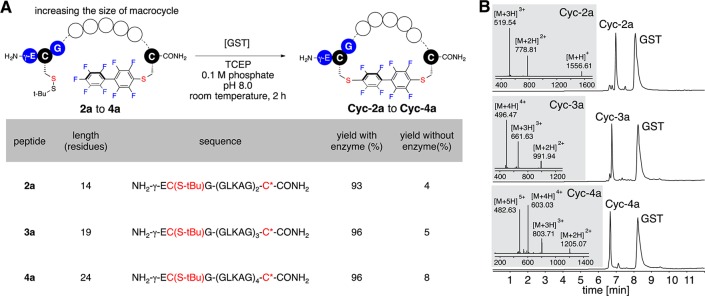
Macrocyclization of peptides with increasing lengths. Reaction conditions: 0.1 mM peptide 2a–4a, 0.2 mg/mL GST, 20 mM TCEP·HCl, 0.1 M phosphate, pH 8.0, room temperature (25 °C), 2 h. (A) Reaction yields of cyclization reactions with and without the addition of GST (C(S-tBu): tert-butylthio-protected cysteine; C*: perfluoroaryl-modified cysteine). The two cysteines that were cross-linked together are highlighted in red. (B) LC–MS analysis of reactions of peptide 2a to 4a with the addition of GST. LC–MS data shown are total ion currents (TIC); mass spectra at the highest points of the TIC peaks are shown as insets.
To compare the GST-catalyzed macrocyclization in water with the uncatalyzed macrocyclization in organic solvent side-by-side, we prepared and purified peptides 2a′–4a′ by reducing the disulfide moiety in peptides 2a–4a and purified them by RP-HPLC (Figure S12, Supporting Information). Adding DMF with Tris base to peptides 2a′–4a′ initiated their intramolecular cyclization reaction and produced cyclized products Cyc-2a–Cyc-4a. The highest reaction yield was observed for the shortest peptide 2a′ with 39% of cyclized product generated within 2 h as measured by LC–MS analysis of the crude reaction mixture (Figure S12B, Supporting Information). Significant amounts of oligo- and polymeric side products were observed for the macrocyclizations of longer peptides 3a′ and 4a′; desired cyclic products were minor products identified in the LC–MS chromatograms (Figure S12, Supporting Information).
To further compare the GST-catalyzed cyclization with our previously reported cross-linking chemistry,17b we synthesized peptides 2b–4b with free cysteines at both the GSH tag and the C-terminus. Peptides 2b–4b could be cyclized by the reaction with decafluorobiphenyl to produce Cyc-2a–Cyc-4a. Similar to peptide 2a′–4a′, we observed decreased cyclization yields as the peptide lengths increased. 37% of desired product was formed with the shortest peptide 2b, but no detectable cyclic product resulted with longer peptides 3b and 4b (Figure S14 and S13, Supporting Information). We previously found reducing the concentration of linear substrate could improve the reaction yield of the cyclization reaction for small model peptides in DMF; however, cyclization of 3b and 4b at diluted conditions still resulted in mixtures of byproducts (Figure S13F,I, Supporting Information).
In contrast to the GST-catalyzed cyclization where increasing the peptide length had no effects on the cyclization yield, all cyclization reactions in DMF showed significantly diminished yields as the peptide lengths increased (Figure S14, Supporting Information). The significantly lowered reaction efficiencies and yields for macrocyclization in DMF compared to macrocyclization under GST catalysis in water highlight the effectiveness of the GST-catalyzed macrocyclization process which compensates the entropic penalty derived from the increased size of the peptide substrate.
Our previous studies have shown that the GST-catalyzed perfluoroarylation reaction was highly selective toward the cysteine residue within the GSH moiety.19 The desired GSH S-arylated product could be selectively and quantitatively formed in the presence of another cysteine in the same polypeptide chain. We reasoned this highly selective nature of the GST-catalyzed reaction could permit the combination of GST-catalyzed ligation with native chemical ligation (NCL) to generate large cyclic peptides from linear substrate synthesized by NCL reaction (Figure 5). GST selectively recognizes the GSH tag and only catalyzes the arylation reaction at the GSH thiol in the presence of the free cysteine generated from NCL reaction, thus allowing NCL product to be cyclized by GST-catalyzed ligation (Figure 5).
Figure 5.
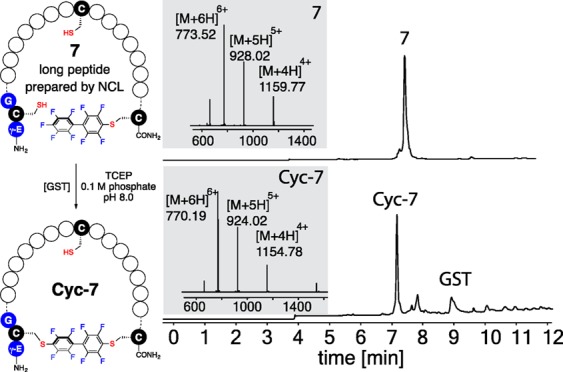
Macrocyclization of a 40-residue peptide 7 prepared from native chemical ligation. Reaction conditions: 0.1 mM peptide 7, 0.2 mg/mL GST, 0.2 M phosphate, 20 mM TCEP·HCl, pH 8.0, room temperature, 2 h. LC–MS data shown are total ion currents of starting material (top chromatogram) and crude reaction mixture (bottom chromatogram); mass spectra at the highest points of the TIC peaks are shown as insets.
We synthesized and purified a large 40-residue linear peptide 7 obtained from the hydrazide-based NCL reaction21 (Figure S15, Supporting Information). Cyclization of peptide 7 under GST catalysis produced desired cyclized product Cyc-7 with 70% yield as measured by LC–MS analysis of the crude reaction mixture. Trypsin digestion and MS/MS analysis confirmed that the cysteine at the NCL ligation site remained unmodifed after the GST-catalyzed cyclization reaction (Figure S18, Supporting Information). Reaction without enzyme showed only a trace amount of product formation and no regioselectivity (Figure S17, Supporting Information). In addition, we further showed that the native chemical ligation and GST-catalyzed ligation could be done in one pot with the desired macrocyclic peptide as the major product (Figure S16, Supporting Information). In most cases, the macrocyclic product ring size is restricted to lengths of peptides that can be accessed from SPPS and the efficiency of the cyclization reactions. In our method, combining GST-catalyzed ligation and native chemical ligation offers a method to overcome these challenges for the synthesis of large macrocyclic peptides.
We have shown how glutathione S-transferase (GST) can efficiently mediate the macrocyclization reaction of long peptides by catalyzing a SNAr process between cysteine and perfluoroaromatic moieties. This transformation operates under mild aqueous conditions, at ambient temperature, and takes only a few hours to complete. The reaction was shown to be highly efficient and selective, especially for the generation of macrocyclic peptides with large ring sizes. To demonstrate the unique selectivity of our GST-catalyzed reaction, a 40-residue macrocyclic peptide was prepared by combining GST-catalyzed ligation with native chemical ligation.
Acknowledgments
This work was generously sponsored by the National Institute of Health (GM101762 for A.M.S.) and also supported by an MIT start-up fund, a Damon Runyon Cancer Research Foundation Award, and the Sontag Foundation Distinguished Scientist Award for B.L.P. C.Z. is a recipient of an Amgen Summer Graduate Research Fellowship. We thank Mr. Mark Simon (MIT) for providing technical assistance for peptide synthesis and Prof. R. John Collier (Harvard) for contributing select laboratory equipment used in this study.
Supporting Information Available
Full experimental details and data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Craik D. J.; Cemazar M.; Wang C. K. L.; Daly N. L. Biopolymers 2006, 84, 250. [DOI] [PubMed] [Google Scholar]; b Baeriswyl V.; Heinis C. ChemMedChem 2013, 8, 377. [DOI] [PubMed] [Google Scholar]; c Smith J. M.; Frost J. R.; Fasan R. J. Org. Chem. 2013, 78, 3525. [DOI] [PubMed] [Google Scholar]
- a White C. J.; Yudin A. K. Nat. Chem. 2011, 3, 509. [DOI] [PubMed] [Google Scholar]; b Moretto A.; Crisma M.; Formaggio F.; Toniolo C. Biopolymers 2010, 94, 721. [DOI] [PubMed] [Google Scholar]; c Davies J. S. J. Pept. Sci. 2003, 9, 471. [DOI] [PubMed] [Google Scholar]; d Jiang S.; Li Z.; Ding K.; Roller P. P. Curr. Org. Chem. 2008, 12, 1502. [Google Scholar]; e Lambert J. N.; Mitchell J. P.; Roberts K. D. J. Chem. Soc., Perkin Trans. 1 2001, 471. [Google Scholar]
- a Montalbetti C. A. G. N.; Falque V. Tetrahedron 2005, 61, 10827. [Google Scholar]; b Parenty A.; Moreau X.; Campagne J. M. Chem. Rev. 2006, 106, 911. [DOI] [PubMed] [Google Scholar]
- a Timmerman P.; Beld J.; Puijk W. C.; Meloen R. H. ChemBioChem 2005, 6, 821. [DOI] [PubMed] [Google Scholar]; b Smeenk L. E. J.; Dailly N.; Hiemstra H.; van Maarseveen J. H.; Timmerman P. Org. Lett. 2012, 14, 1194. [DOI] [PubMed] [Google Scholar]; c Muppidi A.; Doi K.; Edwardraja S.; Drake E. J.; Gulick A. M.; Wang H.-G.; Lin Q. J. Am. Chem. Soc. 2012, 134, 14734. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Heinis C.; Rutherford T.; Freund S.; Winter G. Nat. Chem. Biol. 2009, 5, 502. [DOI] [PubMed] [Google Scholar]; e Jo H.; Meinhardt N.; Wu Y.; Kulkarni S.; Hu X.; Low K. E.; Davies P. L.; DeGrado W. F.; Greenbaum D. C. J. Am. Chem. Soc. 2012, 134, 17704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besse D.; Moroder L. J. Pept. Sci. 1997, 3, 442. [DOI] [PubMed] [Google Scholar]
- a Camarero J. A.; Muir T. W. Chem. Commun. 1997, 1369. [Google Scholar]; b Tulla-Puche J.; Barany G. J. Org. Chem. 2004, 69, 4101. [DOI] [PubMed] [Google Scholar]; c Tam J. P.; Lu Y.-A.; Yu Q. J. Am. Chem. Soc. 1999, 121, 4316. [Google Scholar]; d Yan L. Z.; Dawson P. E. J. Am. Chem. Soc. 2001, 123, 526. [DOI] [PubMed] [Google Scholar]; e Shao Y.; Lu W.; Kent S. B. H. Tetrahedron Lett. 1998, 39, 3911. [Google Scholar]
- a Tavassoli A.; Benkovic S. J. Nat. Protoc. 2007, 2, 1126. [DOI] [PubMed] [Google Scholar]; b Smith J. M.; Vitali F.; Archer S. A.; Fasan R. Angew. Chem., Int. Ed. 2011, 50, 5075. [DOI] [PubMed] [Google Scholar]
- Kleineweischede R.; Hackenberger C. P. R. Angew. Chem., Int. Ed. 2008, 47, 5984. [DOI] [PubMed] [Google Scholar]
- a Kim Y.-W.; Grossmann T. N.; Verdine G. L. Nat. Protoc. 2011, 6, 761. [DOI] [PubMed] [Google Scholar]; b Blackwell H. E.; Grubbs R. H. Angew. Chem., Int. Ed. 1998, 37, 3281. [DOI] [PubMed] [Google Scholar]
- a Bock V. D.; Speijer D.; Hiemstra H.; Van Maarseveen J. H. Org. Biomol. Chem. 2007, 5, 971. [DOI] [PubMed] [Google Scholar]; b Bock V. D.; Perciaccante R.; Jansen T. P.; Hiemstra H.; Van Maarseveen J. H. Org. Lett. 2006, 8, 919. [DOI] [PubMed] [Google Scholar]; c Turner R. A.; Oliver A. G.; Lokey R. S. Org. Lett. 2007, 9, 5011. [DOI] [PubMed] [Google Scholar]; d Madden M. M.; Muppidi A.; Li Z.; Li X.; Chen J.; Lin Q. Bioorg. Med. Chem. Lett. 2011, 21, 1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ghadiri M. R.; Choi C. J. Am. Chem. Soc. 1990, 112, 1630. [Google Scholar]; b Smith S. J.; Du K.; Radford R. J.; Tezcan F. A. Chem. Sci. 2013, 4, 3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Marqusee S.; Baldwin R. L. Proc. Natl. Acad. Sci. U.S.A. 1987, 84, 8898. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dolain C.; Hatakeyama Y.; Sawada T.; Tashiro S.; Fujita M. J. Am. Chem. Soc. 2010, 132, 5564. [DOI] [PubMed] [Google Scholar]
- a Aboye T. L.; Camarero J. A. J. Biol. Chem. 2012, 287, 27026. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gruenewald J.; Marahiel M. A. Microbiol. Mol. Biol. Rev. 2006, 70, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson D. Y.; Burnier J. P.; Wells J. A. J. Am. Chem. Soc. 1995, 117, 819. [Google Scholar]
- a Kohli R. M.; Walsh C. T.; Burkart M. D. Nature 2002, 418, 658. [DOI] [PubMed] [Google Scholar]; b Trauger J. W.; Kohli R. M.; Mootz H. D.; Marahiel M. A.; Walsh C. T. Nature 2000, 407, 215. [DOI] [PubMed] [Google Scholar]
- a Wu Z.; Guo X.; Guo Z. Chem. Commun. 2011, 47, 9218. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bolscher J. G. M.; Oudhoff M. J.; Nazmi K.; Antos J. M.; Guimaraes C. P.; Spooner E.; Haney E. F.; Garcia Vallejo J. J.; Vogel H. J.; van’t Hof W.; Ploegh H. L.; Veerman E. C. I. FASEB J. 2011, 25, 2650. [DOI] [PubMed] [Google Scholar]; c Antos J. M.; Popp M. W.-L.; Ernst R.; Chew G.-L.; Spooner E.; Ploegh H. L. J. Biol. Chem. 2009, 284, 16028. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Dasgupta S.; Samantaray S.; Sahal D.; Roy R. P. J. Biol. Chem. 2011, 286, 23996. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Jia X.; Kwon S.; Wang C.-I. A.; Huang Y.-H.; Chan L. Y.; Tan C. C.; Rosengren K. J.; Mulvenna J. P.; Schroeder C. I.; Craik D. J. J. Biol. Chem. 2014, 289, 6627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Spokoyny A. M.; Zou Y.; Ling J. J.; Yu H.; Lin Y.-S.; Pentelute B. L. J. Am. Chem. Soc. 2013, 135, 5946. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zou Y.; Spokoyny A. M.; Zhang C.; Simon M. D.; Yu H.; Lin Y.-S.; Pentelute B. L. Org. Biomol. Chem. 2014, 12, 566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong R. N. Chem. Res. Toxicol. 1997, 10, 2. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Spokoyny A. M.; Zou Y.; Simon M. D.; Pentelute B. L. Angew. Chem., Int. Ed. 2013, 52, 14001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J.-S.; Tang S.; Qi Y.-K.; Wang Z.-P.; Liu L. Nat. Protoc. 2013, 8, 2483. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.