

Abstract
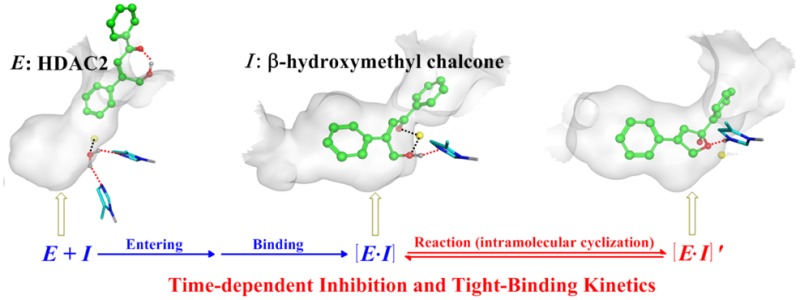
Development of isoform-selective histone deacetylase (HDAC) inhibitors is of great biological and medical interest. Among 11 zinc-dependent HDAC isoforms, it is particularly challenging to achieve isoform inhibition selectivity between HDAC1 and HDAC2 due to their very high structural similarities. In this work, by developing and applying a novel de novo reaction-mechanism-based inhibitor design strategy to exploit the reactivity difference, we have discovered the first HDAC2-selective inhibitor, β-hydroxymethyl chalcone. Our bioassay experiments show that this new compound has a unique time-dependent selective inhibition on HDAC2, leading to about 20-fold isoform-selectivity against HDAC1. Furthermore, our ab initio QM/MM molecular dynamics simulations, a state-of-the-art approach to study reactions in biological systems, have elucidated how the β-hydroxymethyl chalcone can achieve the distinct time-dependent inhibition toward HDAC2.
Inhibition of histone deacetylases (HDAC) has emerged as a highly promising strategy for the development of new therapeutics against cancer and various other human disorders.1−3 Up to now, many HDAC inhibitors (HDACi) have been designed based on the reactant, intermediate, and product states along the deacetylation reaction (as shown in Figure S1)4−7 and more than a dozen HDACi in advanced clinical testing have been reported to inhibit cell growth and induce terminal differentiation in tumor cells.8,9 Although promising, a number of phase I/II trials have shown that the unselective inhibition of HDAC leads to a variety of side effects since HDACs also play essential roles in normal cell functioning.9,10 Therefore, it is of great interest and medical importance to develop isoform specific HDAC modulators.11−13
Among 11 zinc-dependent HDAC isoforms, it is particularly challenging to achieve isoform inhibition selectivity between HDAC1 and HDAC2, since they share a very high sequence similarity (97.8%, see Figure S2), have the same conserved residues around the catalytic pocket (see Figure 1), and the RMSD of two aligned protein crystal structures is only 0.7 Å (see Figure S2). As a result, it has been very difficult to develop an HDAC2 selective inhibitor by employing conventional structure-based or ligand-based design approaches. However, recent structural studies14,15 indicate that the metal ion located about 7 Å from the Zn2+ is different (K+ in HDAC1 but Ca2+ in HDAC2), as shown in Figure 1. Moreover, our previous ab initio quantum mechanics/molecular mechanics (QM/MM) simulations16 of HDAC8 had demonstrated that this metal ion (K+ in HDAC8) has a large influence on the substrate reactivity. Thus, if a designer inhibitor could undergo further reaction after its binding to the catalytic Zn2+ ion, an isoform selective inhibitor might be achieved since the reactivity of the designed compound could be distinguished between HDAC1 and HDAC2.
Figure 1.
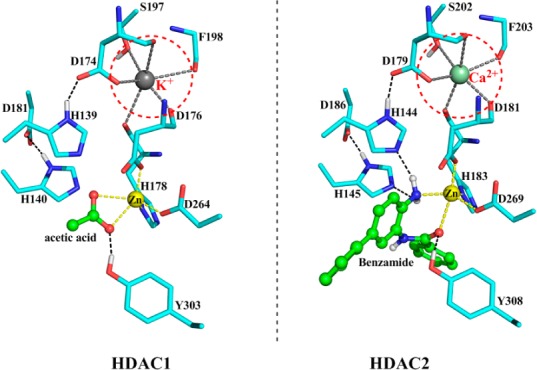
Comparison of the active site in HDAC1/2 crystal structures (PDB code: 4BKX and 3MAX, respectively).
Herein, guided by our previously characterized HDAC reaction mechanism,16 we have developed a de novo reaction-mechanism-based inhibitor design strategy, as shown in Figure 2: first, the intermolecular nucleophilic attack reaction is translated to an intramolecular reaction (namely cyclization) by linking with an allyl group; then the hydroxyl is replaced by an amino based on the bioisosteres theory, leading to the basic skeletons with two substitutional groups R1 and R2. Our working hypothesis is that a desired inhibitor should be stable in solution while it should react intramolecularly after binding to the HDAC active site and thus mimics the enzymatic transition state. To examine how R1/R2 and the enzyme environment would modulate the reactivity of the intramolecular nucleophilic attack reaction, theoretical calculations on several nonenzyme and corresponding enzyme models have been carried out (see Supporting Information for details, Figures S3–S7), and the primary results are summarized in Table S1. We can see that among all molecules that we calculated the most promising candidates are two designed β-substituted chalcones (as highlighted in red in Figure 2). As seen in Table 1, the calculated reaction barriers indicate that each of them would be stable at the nonenzyme environment, while the intramolecular nucleophilic attack reaction would occur after it binds to the HDAC1/2 active site. Furthermore, either for the β-aminomethyl or β-hydroxymethyl chalcone, its reactivity is higher in HDAC2 than that in HDAC1. Especially for the β-hydroxymethyl chalcone, it would be stable in a nonenzyme environment (∼38 kcal/mol barrier) and undergo further reaction as well as present distinct reactivity in HDAC1/2 (21.2 and 12.1 kcal/mol, respectively).
Figure 2.
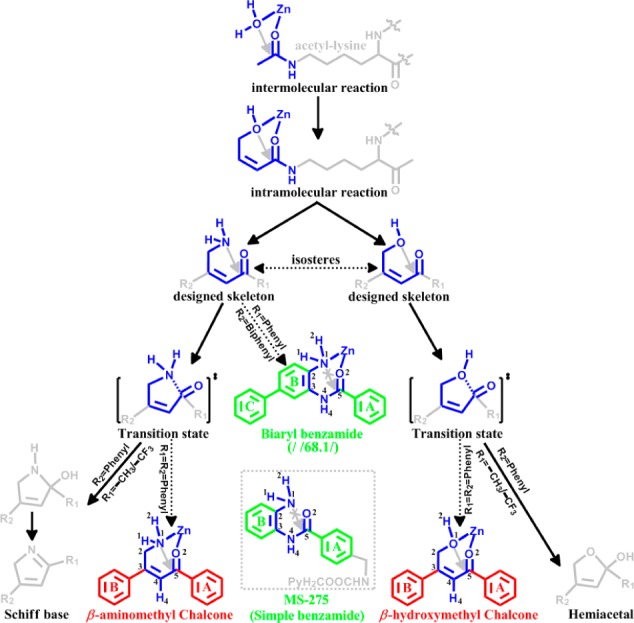
Reaction-mechanism-based HDAC inhibitor design strategy.
Table 1. Intramolecular Nucleophilic-Attack Reaction Barriers of the Designed Chalcones (Shown in Figure 2, a Lower Reaction Barrier Indicates the Higher Reactivity)a.
| relative reaction barrier
(unit: kcal/mol) |
||
|---|---|---|
| simulation models | β-am-chalcone | β-hm-chalcone |
| gas-phase | 33.5 | 38.8 |
| solvent | 28.0 | 38.0 |
| HDAC2 | 23.7 | 12.1 |
| HDAC1 | 27.7 | 21.2 |
All computational details refer to Figures S3–S7. β-am-chalcone: β-aminomethyl chalcone. β-hm-chalcone: β-hydroxymethyl chalcone.
Since the two designed β-substituted chalcones were most promising based on our calculations, they were synthesized as shown in Figure S8, and an HDAC inhibitory activity assay was performed. All the HDAC dose–response curves are shown in Figure S9, and the IC50 values are summarized in Table 2. We can see that the β-substituted chalcones preferentially inhibit HDAC1 and HDAC2 compared with HDAC3, while MS-275, a promising class-I specific inhibitor in a phase-II clinical trial, shows nearly equal inhibitory activity on HDAC1–3. Moreover, a distinct time-dependent inhibition phenomenon for the β-hydroxymethyl chalcone toward HDAC2 is found. As shown in Table 2, it is about a 54-fold increase on time-dependent inhibitory activity of HDAC2, while almost no such time-dependent inhibition enhancement exists for either HDAC1 or HDAC3. This time-dependent inhibitory effect leads to selective inhibition of HDAC2 against HDAC1 (about 20-fold). In contrast, no distinguishable time-dependent inhibitory phenomenon was observed for either MS-275 or the β-aminomethyl chalcone for HDAC1–3 (see Table 2).
Table 2. The Time-Dependent Inhibition upon Inhibitors against HDAC1–3a.
| IC50 (μM) |
|||
|---|---|---|---|
| compound | HDAC1 | HDAC2 | HDAC3 |
| β-hm-chalcone | 3.68 (2.74) | 9.19 (0.17) | ∼50 (∼50) |
| β-am-chalcone | 5.96 (8.65) | 19.64 (26.44) | >50 (>50) |
| MS-275 | 2.26 (0.80) | 6.42 (4.13) | 9.37 (7.95) |
Data order: 1 h (24 h); the detailed curves refer to Figure S9. β-am-chalcone: β-aminomethyl chalcone. β-hm-chalcone: β-hydroxymethyl chalcone.
Considering the above experimental results, the first intriguing question is how the β-substituted chalcones can achieve a better isoform-selectivity (inhibiting HDAC1/2 not HDAC3) than a phase II clinical class-I specific inhibitor MS-275 (inhibiting HDAC 1/2/3). In comparison, with HDAC1–2 and HDAC3, which are all class-I HDACs, one known difference in their active sites is that the foot pocket of HDAC3 is smaller than the other two, due to the existence of the Tyr96 in HDAC3 while the corresponding residue in HDAC1/2 is Ser113/118, respectively.4,5,17 Meanwhile, a key structural distinction between the two designed β-substituted chalcones and MS-275 is that the B-ring (ABC ring, refer to Figure 2) in two designed β-substituted chalcones is more extended out than that in MS-275. Thus, it would be reasonable to suggest that the observed selective inhibitory effects may also originate from steric hindrance, which had been previously proposed to account for the HDAC inhibition selectivity of the C-ring-containing biaryl benzamide.17−20 In order to computationally examine this hypothesis, we have carried out ab initio QM/MM MD simulations for the MS-275-like simple benzamide in HDAC3 as well as the β-hydroxymethyl chalcone in HDAC1/2/3. As illustrated in Figure 3, the simple benzamide can be well accommodated by HDAC3 (Figure 3a). However, the Tyr96 needs to be rotated out in HDAC3 to accommodate the extended B ring of the β-hydroxymethyl-chalcone (see Figure 3b), which indicates the steric effect in the binding site. Meanwhile, as shown in Figure 3c and d, due to the larger foot pocket of HDAC1/2, the extended B-ring of β-substituted chalcone can be well accommodated without changing the side chain orientation of Ser113/118. Thus, these computational results further support the steric hindrance hypothesis to account for our observed inhibition selectivity difference between the C-ring-absent MS-275 and the two designed chalcones.
Figure 3.
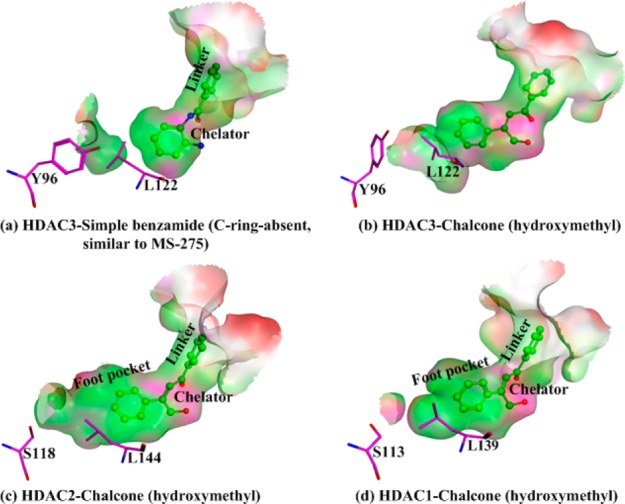
Comparison of the active pocket in HDAC1/2/3 with different ligands (red means exposed region; pink, polar region; green, hydrophobic region).
The next essential question is the origin of distinct time-dependent inhibition phenomenon for the β-hydroxymethyl chalcone toward HDAC2, which leads to a 54-fold increase in inhibitory activity and about 20-fold isoform-selectivity against HDAC1. It should be noted that time dependent inhibitory phenomena had been observed in previous studies of HDAC inhibitors, and three factors contributing to such time-dependent binding kinetics have been proposed:4,5,17,21−32 (A) the ligand exchange, (B) the hydrogen-bond rearrangement, and (C) the local-conformation change. As shown in Figure 4, all three factors focus on the formation of [E•I] complex, which would take the so-called “target-resident time” to reach the optimal binding modes in the HDAC active site.21,22,33,34 However, considering these factors alone cannot account for β-hydroxymethyl chalcone’s unique selective inhibitory effect on HDAC2: since HDAC1/2 have a highly similar pocket shape and share the same conserved residues around the active site, it would be expected that two β-substituted chalcones yield similar binding kinetics toward either HDAC1 or HDAC2; on the other hand, our bioassay experiments indicated that only the β-hydroxymethyl chalcone demonstrates the distinct and unique slow tight-binding kinetics on HDAC2 but no time-dependent effect for β-aminomethyl chalcone toward either HDAC1 or HDAC2.
Figure 4.
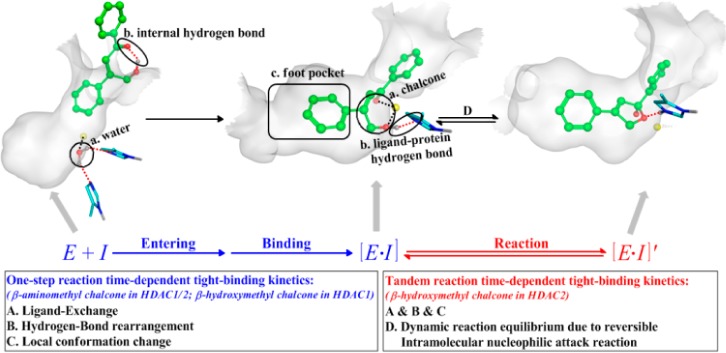
Proposed contributing factors lead to the different kinds of time-dependent tight-binding kinetics of HDAC inhibitors (E, enzyme; I, inhibitor).
Here, we suggest the subsequent dynamic equilibrium of the intramolecular nucleophilic attack reaction (namely the formation of [E•I]′ complex) as another contributing factor (D) to account for the time-dependent inhibition phenomenon, which can be termed as the “tandem reaction time-dependent tight-binding kinetics” mechanism (see Figure 4). On the basis of our ab initio QM/MM simulations, as shown in Figure 5 and Table S1, the intramolecular nucleophilic attack reactivity of β-hydroxymethyl chalcone in HDAC2 (a reaction barrier of 14.6 kcal/mol) is much higher than that in HDAC1 (with a higher barrier of 21.2 kcal/mol) as well as β-aminomethyl chalcone in either HDAC1 or HDAC2 (with a reaction barrier of 27.7 kcal/mol or 23.7 kcal/mol, respectively). Thus, our observed distinct time-dependent inhibition of β-hydroxymethyl chalcone toward HDAC2 can be attributed to the “tandem reaction time-dependent tight-binding kinetics” mechanism, while such a time-dependent inhibition effect is much smaller for β-hydroxymethyl chalcone in HDAC1, and no time-dependent effect is observed for β-aminomethyl chalcone toward either HDAC1 or HDAC2. As discussed above, the higher reactivity in HDAC2 against that in HDAC1 is mainly due to the presence of Ca2+ in HDAC2 instead of K+ in HDAC1, which increased the charge on the catalytic zinc ion by the remote ES effect. As shown in Figure S11, the charge of the zinc ion in HDAC2 is more positive than that in HDAC1 (1.24 vs 0.88), to serve as a stronger Lewis acid to activate the nucleophilic-attack reaction. Meanwhile, the charge of the O1 atom in HDAC2 is more negative (−0.57 vs −0.32) while that of the C5 atom in HDAC2 is more positive (0.68 vs 0.51), leading to the higher intromolecular nucleophilic-attack ability in HDAC2. In addition, the remote ES effect on regulating the reactivity is further confirmed by additional simulations on artificial modes (see Figure S12). Furthermore, we have performed 20 ns additional classical MD simulations on the cyclic product state of the β-hydroxymethyl chalcone in HDAC2 (see Figure 5), which indicate that the final cyclic product state [E•I]′ can be further stabilized through a conformation change. As shown in Figure S13a and S14, the active pocket channel would become narrower due to the formation of the π–π stacking between F155 and F210, which is also inconsistent with the results that several water molecules would gradually leave the pocket during classical MD simulations (see Figure S13b). As a result, it can lead to unique slow tight-binding kinetics for the β-hydroxymethyl chalcone in HDAC2 but not in HDAC1, and the inhibitory activity and selectivity are increased due to the longer target-resident time in HDAC2.
Figure 5.
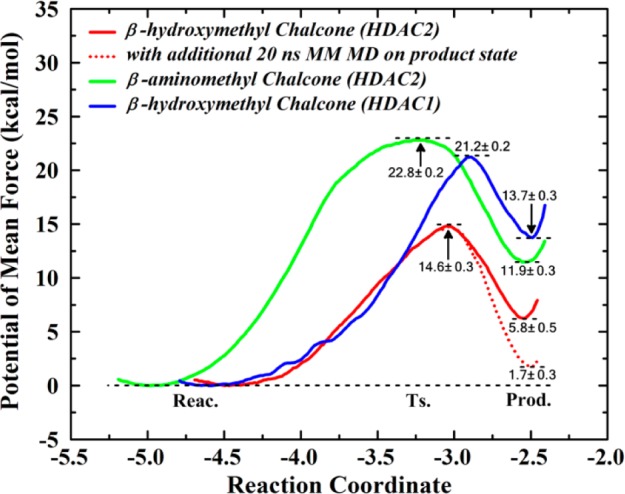
The comparison of the free energy profiles for the intramolecular nucleophilic attack reaction in each model. The statistical error is estimated by averaging the free energy difference between 5 and 13 and 13 and 20 ps. And the corresponding active structure of each reaction state for the β-substitutional chalcone in HDAC2 is shown in Figure S10c–h.
As we know, the drug efficacy is not only associated with its thermodynamics but also related to the binding kinetics,32,33,35−37 that is the so-called “drug-target resident time.” In recent years, the importance of binding kinetics has been increasingly recognized and appreciated in drug discovery by many groups.37−40 In our current work, due to the much higher reactivity in HDAC2 with the existence of Ca2+, as well as the further local conformation rearrangement to achieve the final tandem reaction equilibrium, the β-hydroxymethyl chalcone shows unique slow tight-binding kinetics and brings a longer drug-target resident time, which leads to a better inhibitory activity and higher selectivity on HDAC2 against the highly similar HDAC1.
In summary, by developing and applying a de novo reaction-mechanism-based HDAC inhibitor design strategy, we have discovered a novel HDAC inhibitor, β-hydroxymethyl chalcone, which has a unique time-dependent (∼24 h) selective inhibition on HDAC2 against the other two class-I HDAC enzymes: HDAC1 and HDAC3. To the best of our knowledge, this is the “first” time-dependent HDAC2-selective inhibitor. It should be noted that since the designer β-hydroxymethyl chalcone has very good structural stability and many potential substitute-modification sites on A/B rings, it provides extensive possibilities to further increase its isoform-selectivity and inhibitory activities. Moreover, the present proposed strategy opens an alternative avenue toward the inhibitor design, especially for enzymes with highly similar structures and sequences.
Experimental and Computational Section
The HDAC inhibition activities of chemicals were determined by the popular two-step fluorogenic assay method,9,41,42 and the protocols of the QM/MM MD simulations are similar to our previous study of HDAC.16,43,44 The more experimental and computational details are presented in the Supporting Information.
Acknowledgments
This work was supported by the National Science Foundation of China (21203257 and 21272289). Y.Z. would like to thank the support of National Institute of Health (R01-GM079223, R21-GM097530). We thank the National Supercomputing Center in Shenzhen and Guangzhou for providing the computational resources. We also thank Prof. Y. Cheng at University of North Texas for his helpful suggestions.
Supporting Information Available
Benchmark test on the non-enzyme and corresponding enzyme models, structure determination of the β-aminomethyl and β-hydroxymethyl chalcone, experimental and computational details, references S8–S10, Figures S1–S14, and Table S1. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Gottesfeld J. M.; Rusche J. R.; Pandolfo M. (2013) Increasing frataxin gene expression with histone deacetylase inhibitors as a therapeutic approach for Friedreich’s ataxia. J. Neurochem. 126, 147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helquist P.; Maxfield F. R.; Wiech N. L.; Wiest O. (2013) Treatment of Niemann--pick type C disease by histone deacetylase inhibitors. Neurotherapeutics 10, 688–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. Y.; Tsai A. C.; Chen M. C.; Shen P. J.; Cheng Y. C.; Kuo C. C.; Pan S. L.; Liu Y. M.; Liu J. F.; Yeh T. K.; Wang J. C.; Chang C. Y.; Chang J. Y.; Liou J. P. (2014) Azaindolylsulfonamides, with a More Selective Inhibitory Effect on Histone Deacetylase 6 Activity, Exhibit Antitumor Activity in Colorectal Cancer HCT116 Cells. J. Med. Chem. 57, 4009–l4022ll. [DOI] [PubMed] [Google Scholar]
- Bertrand P. (2010) Inside HDAC with HDAC inhibitors. Eur. J. Med. Chem. 45, 2095–2116. [DOI] [PubMed] [Google Scholar]
- Bressi J. C.; Jennings A. J.; Skene R.; Wu Y.; Melkus R.; De Jong R.; O’Connell S.; Grimshaw C.; Navre M.; Gangloff A. R. (2010) Exploration of the HDAC2 foot pocket: Synthesis and SAR of substituted N-(2-aminophenyl)benzamides. Bioorg. Med. Chem. Lett. 20, 3142–3145. [DOI] [PubMed] [Google Scholar]
- Suzuki N.; Suzuki T.; Ota Y.; Nakano T.; Kurihara M.; Okuda H.; Yamori T.; Tsumoto H.; Nakagawa H.; Miyata N. (2009) Design, Synthesis, and Biological Activity of Boronic Acid-Based Histone Deacetylase Inhibitors. J. Med. Chem. 52, 2909–2922. [DOI] [PubMed] [Google Scholar]
- Duvic M.; Talpur R.; Ni X.; Zhang C.; Hazarika P.; Kelly C.; Chiao J. H.; Reilly J. F.; Ricker J. L.; Richon V. M.; Frankel S. R. (2007) Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 109, 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones A. P.; Haynes R.; Harvey I. M.; Jewell T. (2012) Road traffic crashes and the protective effect of road curvature over small areas. Health Place 18, 315–320. [DOI] [PubMed] [Google Scholar]
- Wang C.; Flemming C. J.; Cheng Y. Q. (2012) Discovery and activity profiling of thailandepsins A through F, potent histone deacetylase inhibitors, from Burkholderia thailandensis E264. MedChemComm. 3, 976–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrowsmith C. H.; Bountra C.; Fish P. V.; Lee K.; Schapira M. (2012) Epigenetic protein families: a new frontier for drug discovery. Nat. Rev. Drug Discovery 11, 384–400. [DOI] [PubMed] [Google Scholar]
- Frumm S. M.; Fan Z. P.; Ross K. N.; Duvall J. R.; Gupta S.; VerPlank L.; Suh B. C.; Holson E.; Wagner F. F.; Smith W. B.; Paranal R. M.; Bassil C. F.; Qi J.; Roti G.; Kung A. L.; Bradner J. E.; Tolliday N.; Stegmaier K. (2013) Selective HDAC1/HDAC2 inhibitors induce neuroblastoma differentiation. Chem. Biol. 20, 713–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey T. A. (2011) Isoform-selective HDAC inhibitors: closing in on translational medicine for the heart. J. Mol. Cell Cardiol. 51, 491–496. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Fang H.; Feng J.; Jia Y.; Wang X.; Xu W. (2011) Discovery of a tetrahydroisoquinoline-based hydroxamic acid derivative (ZYJ-34c) as histone deacetylase inhibitor with potent oral antitumor activities. J. Med. Chem. 54, 5532–5539. [DOI] [PubMed] [Google Scholar]
- Paris M.; Porcelloni M.; Binaschi M.; Fattori D. (2008) Histone Deacetylase Inhibitors: From Bench to Clinic. J. Med. Chem. 51, 1505–1529. [DOI] [PubMed] [Google Scholar]
- Rasheed W. K.; Johnstone R. W.; Prince H. M. (2007) Histone deacetylase inhibitors in cancer therapy. Expert Opin. Invest. Drugs 16, 659–678. [DOI] [PubMed] [Google Scholar]
- Wu R.; Wang S.; Zhou N.; Cao Z.; Zhang Y. (2010) A proton-shuttle reaction mechanism for histone deacetylase 8 and the catalytic role of metal ions. J. Am. Chem. Soc. 132, 9471–9479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methot J. L.; Chakravarty P. K.; Chenard M.; Close J.; Cruz J. C.; Dahlberg W. K.; Fleming J.; Hamblett C. L.; Hamill J. E.; Harrington P.; Harsch A.; Heidebrecht R.; Hughes B.; Jung J.; Kenific C. M.; Kral A. M.; Meinke P. T.; Middleton R. E.; Ozerova N.; Sloman D. L.; Stanton M. G.; Szewczak A. A.; Tyagarajan S.; Witter D. J.; Secrist J. P.; Miller T. A. (2008) Exploration of the internal cavity of histone deacetylase (HDAC) with selective HDAC1/HDAC2 inhibitors (SHI-1:2). Bioorg. Med. Chem. Lett. 18, 973–978. [DOI] [PubMed] [Google Scholar]
- Hamblett C. L.; Methot J. L.; Mampreian D. M.; Sloman D. L.; Stanton M. G.; Kral A. M.; Fleming J. C.; Cruz J. C.; Chenard M.; Ozerova N.; Hitz A. M.; Wang H.; Deshmukh S. V.; Nazef N.; Harsch A.; Hughes B.; Dahlberg W. K.; Szewczak A. A.; Middleton R. E.; Mosley R. T.; Secrist J. P.; Miller T. A. (2007) The discovery of 6-amino nicotinamides as potent and selective histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 17, 5300–5309. [DOI] [PubMed] [Google Scholar]
- Methot J. L.; Hamblett C. L.; Mampreian D. M.; Jung J.; Harsch A.; Szewczak A. A.; Dahlberg W. K.; Middleton R. E.; Hughes B.; Fleming J. C.; Wang H.; Kral A. M.; Ozerova N.; Cruz J. C.; Haines B.; Chenard M.; Kenific C. M.; Secrist J. P.; Miller T. A. (2008) SAR profiles of spirocyclic nicotinamide derived selective HDAC1/HDAC2 inhibitors (SHI-1:2). Bioorg. Med. Chem. Lett. 18, 6104–6109. [DOI] [PubMed] [Google Scholar]
- Moradei O. M.; Mallais T. C.; Frechette S.; Paquin I.; Tessier P. E.; Leit S. M.; Fournel M.; Bonfils C.; Trachy-Bourget M. C.; Liu J.; Yan T. P.; Lu A.; Rahil J.; Wang J.; Lefebvre S.; Li Z.; Vaisburg A. F.; Besterman J. M. (2007) Novel aminophenyl benzamide-type histone deacetylase inhibitors with enhanced potency and selectivity. J. Med. Chem. 50, 5543–5546. [DOI] [PubMed] [Google Scholar]
- Lauffer B. E.; Mintzer R.; Fong R.; Mukund S.; Tam C.; Zilberleyb I.; Flicke B.; Ritscher A.; Fedorowicz G.; Vallero R.; Ortwine D. F.; Gunzner J.; Modrusan Z.; Neumann L.; Koth C.; Lupardus P. J.; Kaminker J. S.; Heise C. E.; Steiner P. (2013) Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J. Biol. Chem. 288, 26926–26943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C. J.; Herman D.; Gottesfeld J. M. (2008) Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases. J. Biol. Chem. 283, 35402–35409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazian M. D.; Grunstein M. (2007) Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 76, 75–100. [DOI] [PubMed] [Google Scholar]
- Bradner J. E.; West N.; Grachan M. L.; Greenberg E. F.; Haggarty S. J.; Warnow T.; Mazitschek R. (2010) Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 6, 238–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavan A. V.; Somani R. R. (2010) HDAC inhibitors - new generation of target specific treatment. Mini. Rev. Med. Chem. 10, 1263–1276. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L.-W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta.Crystallogr., Sect. D: Biol. Crystallogr. 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindarajan N.; Agis-Balboa R. C.; Walter J.; Sananbenesi F.; Fischer A. (2011) Sodium butyrate improves memory function in an Alzheimer’s disease mouse model when administered at an advanced stage of disease progression. J. Alzheimers Dis. 26, 187–197. [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. (2007) Phasercrystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricobaraza A.; Cuadrado-Tejedor M.; Marco S.; Pérez-Otaño I.; Garcia-Osta A. (2012) Phenylbutyrate rescues dendritic spine loss associated with memory deficits in a mouse model of Alzheimer disease. Hippocampus. 22, 1040–1050. [DOI] [PubMed] [Google Scholar]
- Ricobaraza A.; Cuadrado-Tejedor M.; Pérez-Mediavilla A.; Frechilla D.; Del Río J.; Garcia-Osta A. (2009) Phenylbutyrate Ameliorates Cognitive Deficit and Reduces Tau Pathology in an Alzheimer’s Disease Mouse Model. Neuropsychopharmacology 34, 1721–1732. [DOI] [PubMed] [Google Scholar]
- Ryu H.; Smith K.; Camelo S. I.; Carreras I.; Lee J.; Iglesias A. H.; Dangond F.; Cormier K. A.; Cudkowicz M. E.; H Brown R. Jr.; Ferrante R. J. (2005) Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J. Neurochem. 93, 1087–1098. [DOI] [PubMed] [Google Scholar]
- Kral A. M.; Ozerova N.; Close J.; Jung J.; Chenard M.; Fleming J.; Haines B. B.; Harrington P.; Maclean J.; Miller T. A.; Secrist P.; Wang H.; Heidebrecht R. J. (2014) Divergent Kinetics Differentiate the Mechanism of Action of Two HDAC Inhibitors. Biochemistry 53, 725–734. [DOI] [PubMed] [Google Scholar]
- Bai F.; Xu Y.; Chen J.; Liu Q.; Gu J.; Wang X.; Ma J.; Li H.; Onuchic J. N.; Jiang H. (2013) Free energy landscape for the binding process of Huperzine A to acetylcholinesterase. Proc. Natl. Acad. Sci. U.S.A. 110, 4273–4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett S.; Mohr P. G.; Schmidt P. M.; McKimm-Breschkin J. L. (2011) Real time enzyme inhibition assays provide insights into differences in binding of neuraminidase inhibitors to wild type and mutant influenza viruses. PLoS. One 6, e23627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R.; Monsma F. (2010) Binding kinetics and mechanism of action: Toward the discovery and development of better and best in class drugs. Expert Opin. Drug Discovery 5, 1023–1029. [DOI] [PubMed] [Google Scholar]
- Kenakin T.; Jenkinson S.; Watson C. (2006) Determining the potency and molecular mechanism of action of insurmountable antagonists. J. Pharmacol. Exp. Ther. 319, 710–723. [DOI] [PubMed] [Google Scholar]
- Swinney D. C. (2004) Biochemical mechanisms of drug action: What does it take for success. Nat. Rev. Drug. Discovery 3, 801–808. [DOI] [PubMed] [Google Scholar]
- Buch I.; Giorgino T.; De Fabritiis G. (2011) Complete reconstruction of an enzymeinhibitor binding process by molecular dynamics simulations. Proc. Natl. Acad. Sci. U.S.A. 108, 10184–10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D.; Caflisch A. (2011) The free energy landscape of small molecule unbinding. PLOS Comput. Biol. 7, e1002002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Held M.; Metzner P.; Prinz J.-H.; Noé F. (2011) Mechanisms of protein-ligand association and its modulation by protein mutations. Biophys. J. 100, 701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Henkes L. M.; Doughty L. B.; He M.; Wang D.; Meyer-Almes F. J.; Cheng Y. Q. (2011) Thailandepsins: bacterial products with potent histone deacetylase inhibitory activities and broad-spectrum antiproliferative activities. J. Nat. Prod. 74, 2031–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegener D.; Hildmann C.; Riester D.; Schwienhorst A. (2003) Improved fluorogenic histone deacetylase assay for high-throughput-screening applications. Anal. Biochem. 321, 202–208. [DOI] [PubMed] [Google Scholar]
- Wu R.; Hu P.; Wang S.; Cao Z.; Zhang Y. (2010) Flexibility of Catalytic Zinc Coordination in Thermolysin and HDAC8: A Born-Oppenheimer ab initio QM/MM Molecular Dynamics Study. J. Chem. Theory Comput. 6, 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R.; Lu Z.; Cao Z.; Zhang Y. (2011) Zinc chelation with hydroxamate in histone deacetylases modulated by water access to the linker binding channel. J. Am. Chem. Soc. 133, 6110–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.