

Summary
Chemotaxis is a highly coordinated biological system where chemoattractants trigger multiple signal transduction pathways which act in concert to bring about directed migration. A signaling pathway acting through PIP3, which accumulates at the leading edge of the cell, has been extensively characterized. However, chemotaxis still remains in cells depleted of PIP3, suggesting there are PIP3-independent pathways. We have identified a pathway involving TorC2-PKBR1 as well as another containing PLA2 activity that act in parallel with PIP3. Activation of PKBR1, a myristoylated Protein Kinase B homolog, is dependent on TorC2 (Rapamycin-insensitive Tor complex 2) kinase but is completely independent of PIP3. In response to chemoattractant, PKBs rapidly phosphorylate at least eight proteins, including Talin B, PI4P 5-kinase, two RasGefs, and a RhoGap. These studies help to link the signaling pathways to specific effectors and provide a more complete understanding of chemotaxis.
Keywords: Chemotaxis, Dictyostelium discoideum, PIP3, PKB, TorC2
1. Introduction
Investigations of Dictyostelium discoideum have been extremely useful in elucidating mechanisms of chemotaxis, motility, and cytokinesis. D. discoideum cells grow in their natural soil environment by consuming bacteria and yeasts. Known as a “social amoeba,” these protozoa change from a unicellular to a multi-cellular state upon nutrient starvation. In this process, cAMP is spontaneously released from central cells every 6 min and functions as a chemoattractant for the cells to aggregate. Within 24 h the multicellular structure undergoes morphogenesis and differentiation, forming a fruiting body which contains spores that resist harsh conditions. The chemotaxis toward cAMP is easily and consistently reproducible under standard laboratory conditions. The following additional features of this organism increase its value as a model system for studies of eukaryotic chemotaxis. With a sequenced 34-Mbp genome size, the organism is genetically and biochemically tractable. For example, gene disruption by homologous recombination, restriction enzyme mediated insertional mutagenesis, and chemical mutagenesis are easily achieved. It is also simple and convenient to generate up to 3 × 1011 cells in suspension culture for biochemical assay and protein purification.
Accumulated evidence now indicates that a network of signaling pathways contribute to efficient chemotaxis (1–3) (Fig. 1). Chemoattractant binds to G-protein-coupled receptors uniformly distributed around the cell's periphery and cause activation of the heterotrimeric G-protein consisting of Gα2 and Gβγ. This activation leads to a rapid, and in most cases, transient burst of responses such as production of the second messengers PIP3, cGMP, and cAMP. PIP3 accumulates locally at the front of cells by the opposing actions of PI 3-kinase and PTEN which reversibly convert PI (4, 5) P2 to PI (3.4,5) P3. The information conveyed by the elevated PIP3 is transmitted by recruitment of proteins containing PIP3-specific PH (Pleckstrin homology) domains such as CRAC (4), PhdA (5), and PKBA (6). Although misregulation of PIP3, as occurs in cells lacking PTEN greatly impairs chemotaxis (7), PI 3-kinase activity is not essential for chemotaxis (8). The search for PIP3-independent pathways has led to the isolation of PLA2 activity as an enhancer of PIP3-dependent chemotaxis (9). PLA2 activity appears to contribute to cell motility by regulating actin polymerization in parallel with PIP3.
Fig. 1.

Model of chemoattractant signaling pathways in Dictyostelium discoideum. The chemoattractant, cAMP, binds to the cAR1 and activates the heterotrimeric G-protein. The activation conveys signals via multiple pathways to successfully achieve chemotaxis. The indicated Ras proteins thought to be locally activated at the front of cells are suggested to regulate PI3K and TorC2 activities. PIP3 production resulting from the countered reactions of PI3K and PTEN causes the recruitment of PH domain containing proteins, PhdA, Crac, and PKBA. Pla2 activity is required for the actin polymerization in parallel with PIP3 pathway. TorC2 organizes the cAMP information through PKB and other undefined activities. PKB activity is composed of PKBR1, a more predominant activity and completely dependent on TorC2 for activation, and PKBA, a minor activity dependent on recruitment to PIP3 and TorC2 (heavier or lighter arrows). Together these phosphorylate several substrates, for example, Talin B (TalB), RasGEFS (GefS), RasGEFN (GefN), PI5K, and Rho GAPQ (GacQ).
Recently, we have shown that the TorC2-PKBR1 pathway plays an important role in chemotaxis and is independent of PIP3(10). D. discoideum has two PKB (Protein Kinase B) homologs, PKBA and PKBR1 (6, 11). Both proteins are structurally very similar to the mammalian PKBs, except for the membrane-binding domain at N-terminus. Like the mammalian enzymes, PKBA has a PIP3-specific PH domain which functions to recruit it to membrane-associated PIP3, while the myr-istoylated PKBR1 is evenly distributed on the membrane. These proteins require specific phosphorylations in the activation loop and in the hydrophobic motif for the full activity. A commercial antibody specific for the phosphorylated state of PKB substrates in vivo has provided evidence that PKBR1 is the major PKB activity and that PKBA makes a minor contribution. PiaA (pianissimoA), originally isolated in a forward genetic screen for chemotaxis and early development defects, is now recognized to be a subunit of TorC2 kinase (12, 13). Work in Drosophila and mammalian cells identified TorC2 as the kinase responsible for the phosphorylation of the hydrophobic motif of PKB (14). Consistent with this, the activity of PKBR1 depends on TorC2 phosphorylation of its hydrophobic motif. The TorC2-PKBR1 pathway is insensitive to PIP3 depletion and is selectively activated at the cell's leading edge. The PIP3-independent PKBR1 and PIP3-dependent PKBA pathways converge with their overlapping substrates, including TalinB, RasGEFs, RhoGAP, PI5K, and others (10). Work is currently focused on determining which substrates are involved in signaling and cytoskeletal regulation for proper chemotaxis.
2. Materials
2.1. Cell Culture Media, Buffer, and Solutions
HL5 medium. 20 g maltose (or 10 g dextrose), 10 g proteose peptone, 5 g yeast extract, 0.51 g Na2HPO4, 0.485 g KH2PO4, H2O to 1 L. Autoclave for sterilization (see Note 1).
DB (Development buffer). 100 mL 10× phosphate buffer, 1 mL 2 M MgSO4, 0.2 mL 1 M CaCl2, H2O to 1 L.
10× Phosphate buffer. 6.8 g KH2PO4, 13.4 g Na2HPO4·7H2O, H2O to 1 L (pH should be ∼6.5 without adjustment).
100 mM caffeine. 1.94 g of caffeine, H2O to 100 mL. Store at −20°C.
PM (phosphate magnesium buffer). 100 mL 10× phosphate buffer, 1 mL 2 M MgSO4, H2O to 1 L.
Basal buffer. 20 mM Tris–HCl of pH 8.0, 2 mM MgSO4.
10 mM cAMP. 0.0369 g cAMP sodium salt monohydrate, H2O to 10 mL. Store at −20°C.
2.2. Micropipette Assay
Femtotip microinjection needles (Eppendorf).
Microinjector (Eppendorf).
Lab-Tek one-well glass chamber (Nalge Nunc, Naperville, PA).
2.3. Two-Drop Assay
1% melted agar in H2O (Difco Noble Agar, Becton Dickinson, Sparks, MD) (see Note 2).
A drawn-out Pasteur pipette. Soften a Pasteur pipette in a flame, withdraw it from the heat, and quickly pull both ends to a thin diameter, then break.
2.4. Western Blotting
Precast gels. Criterion Tris–HCl 4–15% gels (Bio-Rad, Hercules, CA).
PVDF membrane (Immobilon-P, Millipore, Bedford, MA) (see Note 3).
3-MM Chr chromatography paper (Whatman, Maidstone, U.K).
Transfer Buffer. 25 mM Tris base, 190 mM glycine, 20% (v/v) methanol.
TBST (Tris-buffered saline with Tween). 20 mM Tris-HCl of pH 7.5, 137 mM NaCl, 0.1% Tween-20.
Blocking buffer. 5% (w/v) nonfat dry milk in TBST.
Secondary antibody. Antirabbit or antimouse IgG-conjugated HRP (horse radish peroxidase) (GE-Healthcare, Piscataway, NJ).
ECL (enhanced chemiluminescent) reagent (GE Healthcare).
2.5. Primary Antibodies for Western Blotting
All phosphospecific antibodies from Cell Signaling Technology, Danvers, MA.
Rabbit antiphospho PKB substrate monoclonal antibody (110B7). Use 1:2,500 dilution in TBST containing 5% (w/v) BSA; detect with antirabbit IgG-HRP.
Mouse antiphospho PDK docking motif monoclonal antibody (18A2). Use 1:2,000 dilution in TBST containing 5% (w/v) BSA; detect with antimouse IgG-HRP.
Rabbit antiphospho PKC (pan) monoclonal antibody (190D10). Use 1:2,000 dilution in TBST containing 5% (w/v) BSA; detect with antirabbit IgG-HRP.
2.6. Indirect Immunofluorescence
Plasmid. pJK1-R1-AKT-HA transfected cells are selected with G418. (see Note 4).
18-mm coverslips and slideglasses (Fisher, Pittsburgh, PA).
Fix solution (see Note 5). Mix 0.2 g paraformaldehyde (see Note 6) and 5 mL of 20 mM PIPES Buffer (pH 6.0) in a 50-mL disposable tube, microwave in brief pulses until dissolved, and then cool quickly on ice to room temperature. Finally add 3.25 mL H2O, 0.25 mL 2.5 M sucrose, and then 1.5 mL saturated picric acid solution (Fluka).
Quenching solution. 100 mM glycine in PBS.
PBS (Phosphate-buffered saline). 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4 (pH 7.4).
Blocking solution. 2% (w/v) BSA in TBS-TX100.
TBS-TX100 (Tris-buffered saline with Triton X100). 20 mM Tris-HCl of pH 7.5, 137 mM NaCl, 0.1% Triton X100.
Primary antibodies. Rabbit anti phospho AKT(S473) polyclonal antibody (Cell Signaling Technology), diluted 1:50 in TBS-TX100. Anti HA polyclonal antibody (Zymed, South San Francisco, CA), diluted 1:100 in TBS-TX100.
Secondary antibody. Antirabbit IgG conjugated with rhodamine (Santa Cruz Biotechnology, Santa Cruz, CA), diluted 1:100 in TBS-TX100.
Mounting medium. Vectashield (Vector Laboratories Inc., Burlingame, CA).
2.7. Immunopurification of PKB Substrates
2× NP-40 lysis buffer. 80 mM HEPES (pH 7.5), 100 mM NaF, 4 mM Na3VO4, 50 mM sodium pyrophosphate, 1% NP-40, 2× protease inhibitor complete EDTA free (Roche, Manheim, Germany), 2% protease inhibitor cocktail (Sigma Aldrich, St. Louis, MO).
Protein G-Sepharose (GE-Healthcare); wash twice with PBS containing 5 mg/mL BSA before use.
Antibody. Antiphospho PKB substrate antibody (Cell Signaling Technology).
1× RIPA buffer. Tris-HCl of pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 1× protease inhibitor complete EDTA free (Roche), 1% protease inhibitor cocktail (Sigma Aldrich).
150 × 25 mm of tissue culture dish from (Becton Dickinson).
2.8. PIP3 Detection Biochemically
Nucleopore Track-Etch membrane, 5 μm pore size (Whatman).
Syringes for cell lysis. For each, fold a Nucleopore membrane in half and place it between the needle and barrel of a 1-mL disposable syringe.
2.9. Genes and Dictyostelium Data Base (DDB) Numbers
All genes referred to in these protocols are listed in Table 1 together with their DDB numbers, which can be used to search dictybase (http://www.dictybase.org) to access a knowledge base for each gene, including sequence information, expression properties, relevant literature, and available resources.
Table 1. Genes and DDB (Dictyostelium Data Base) numbers.
| GENE | PKBA | PKBR1 | PIAA | RIP3 | TALB | GEFN |
|---|---|---|---|---|---|---|
| DDB # | DDB0191195 | DDB0191365 | DDB0185055 | DDB0201626 | DDB0191526 | DDB0167277 |
| GENE | GEFS | GACQ | PI5K | PDKA | PDKB | PI3K1 |
| DDB # | DDB0191324 | DDB0233774 | DDB0234212 | DDB0216243 | DDB0216246 | DDB0214949 |
| GENE | PI3K2 | PI3K4 | PI3K5 | PTEN | CRAC | PHDA |
| DDB # | DDB0191474 | DDB0235157 | DDB0235158 | DDB0191093 | DDB0191434 | DDB0191446 |
| GENE | PLA2A | CAR1 | gα2 (GPA2) | gβ (GPBA) | gγ (GPGA) | RASC |
| DDB # | DDB0235269 | DDB0185024 | DDB0191237 | DDB0252679 | DDB0185201 | DDB0214827 |
| GENE | RASG | GEFA(ALEA) | GEFR | |||
| DDB # | DDB0201663 | DDB0191187 | DDB0185198 |
3. Methods
3.1. Preparation of Chemotactically Competent Cells
D. discoideum shows reliable chemotaxis to cAMP at the early development stage, after 5-h starvation. These cells are typically used for chemotaxis analysis. This section describes the method for preparing the cells.
3.1.1. Starvation of Dictyostelium discoideum Cells
Culture axenically in HL5 medium at 22°C to a density of less than 5 × 106 cells/mL.
Centrifuge 1 × 108 cells for 5 min at 500 × g and remove the supernatant. Wash with 50 mL of DB buffer twice.
Resuspend cells in 5 mL of DB at 2 × 107 cells/mL and rotate in a 50-mL flask for 1 h at 110 rpm.
Pulse with 60 nM of cAMP every 6 min for the next 4 h.
3.1.2. “Basalation” of Cells
Add caffeine to 5 mM.
Shake at 200 rpm for 30 min.
Centrifuge the cells for 5 min at 500 × g at 4°C and remove the supernatant. Wash with 30 ml of ice-cold DB twice.
Resuspend at 2 × 107 cells/mL in DB and keep on ice before assay.
3.2. Under Buffer Assay
This section describes a method to assess cell aggregation in response to starvation with or without an inhibitor, such as LY294002 (see Note 7). This aggregation process includes chemotaxis and other cellular events. The method can also be applied to assess the defect in a mutant cell line.
Plate 1 × 105 cells in HL5 medium in wells of a 96-well plate.
Let them to adhere on the plate for 30–60 min.
Aspirate the medium.
Add 200 μL of DB with or without a drug.
Incubate at 22°C for about 18 h to form aggregates.
Take a picture using a dissection microscope (see Fig. 2a).
Fig. 2.

Chemotaxis assays (a) Under buffer assay. In the upper panel, the indicated cells are starved in DB and allowed to make aggregates (dark spots in a picture). WT (wild-type), Pla2A− (plaA−), and PI3K1−/2− (pi3K1−/2−) cells can aggregate, but the triple mutant cells (plaA−/pi3K1−/2−) cannot, suggesting that both Pla2A and PI3K function in parallel. The lower panel shows the effects of LY294002 (LY) as an inhibitor of PI3K. Consistent with the upper panel, LY prevents aggregation of only pla2A− cells. (b) Micropipette assay. cAMP gradients are formed from the tip of the micropipette and the responses of WT and piaA− cells are shown before (left panel) and 30 min after (right panel) the needle is placed. WT cells move toward the higher concentration of cAMP with a typically polarized morphology, but piaA− cells abrogate chemotaxis as well as polarity. (c) Two-drop assay. The responses of WT (upper panel) and piaA- cells (lower panel) are shown 30 min after the assay. A cAMP (not shown) and cell droplet are juxtaposed to each other. (Panels A and B reproduced from refs. 9 and 10, respectively, with permission from Elsevier Science.).
3.3. Chemotaxis Assays
This section describes two different methods to evaluate chemotaxis. These two methods are not necessarily identical in the degree of chemotaxis activity. It might be important to carry out several different assays to document subtle defects.
3.4. Micropipette Assay
Dilute competent cells to around 1–4 × 105 cells/mL in DB.
Disperse cells by pipetting several times or vortexing weakly.
Spot 20 μL on a chamber cover glass.
Wait for 10–15 min for cells to adhere; then gently add 3 mL of DB.
Load a Femtotip microinjection needle with 10 μM cAMP, connect the needle to a microinjector, lower the needle to touch the coverslip using a micromanipulator, and apply positive pressure (25 psi) with the microinjector.
Take photographs at 30-s intervals for 30 min (see Note 8) (see Fig. 2b).
3.4.1. Two-Drop Assay
Pour 10 ml of 1% melted agar in a 90-mm Petri dish 1 h before assay (see Note 9).
Dilute competent cells to about 1 × 106 cells/mL in DB.
Disperse cells by pipetting several times or vortexing weakly.
Spot a 3-mm-diameter drop of cell suspension (∼200–500 cells/drop) on the agar surface using the fine tip of a drawn-out Pasteur pipette and capillary action. Spots of DB with or without 0.01, 0.1, or 1 μM cAMP are placed 3 mm from the cell spots. Wait for 30–60 min.
Evaluate positive chemotaxis under a microscope by viewing each spot. Positive chemotaxis is scored when cells move to the edge of the drop toward the nearby cAMP and away from the far edge (see Note 10) (see Fig. 2c).
3.5. Detection of PIP3 Production
This section describes the detection of PIP3 products, the output of the PI3K pathway, by imaging and by biochemical analysis. A PIP3-specific PH domain from either CRAC or PKBA is typically used as a biosensor.
3.5.1. PIP3 Detection by a Biosensor, PH-GFP (Fluorescent Microscope)
PIP3 production following addition of uniform chemoattractant or spatial localization in cells under a variety of conditions is detected by one of the PIP3-specific PH domain fused with a fluorescent protein (6, 7).
Starve cells expressing this biosensor to chemotactic competence and uniformly stimulated with cAMP at a final concentration of 1 μM.
Capture images at 2-s intervals for 1–2 min.
To visualize the localized accumulation of PIP3, cells are placed on the chamber coverslip and observed under fluorescence microscope doing chemotaxis toward spontaneously secreted cAMP or toward a gradient from a micropipette.
3.5.2. PIP3 Detection by a Biosensor, PH-GFP (Biochemically)
Starve cells expressing PH-GFP at 2 × 107 cells/mL in 5 mL and basalate with caffeine as described in Subheading 3.1.1.
Wash cells with 30 mL of ice-cold PM twice.
Suspend cells in 1.25 mL of PM (i.e., 8 × 107 cells/mL).
Transfer the cells to a 5-ml disposable plastic beaker and add 100 μM of cAMP to a final concentration of 1 μM.
Take 100 μL of cells at time points of 0, 5, 20, 60 s. (see Note 11)
Transfer into a 1-mL of disposable syringe containing 100 μL of basal buffer and lyse through 5-μm membrane into a micro-centrifuge tube containing 1 mL PM on ice.
Microcentrifuge at full speed for 1 min at 4°C and aspirate the supernatant.
Suspend the pellet in 50 μL of 1× SDS sample buffer and boil them for 5 min.
Run SDS PAGE followed by western blotting with a-GFP antibody as a primary antibody.
3.6. Detection of PKB and TorC2 Activity
This section describes methods to biochemically and cytologically assess PKB and TorC2 activity using the following phospho-specific antibodies. First, a phospho PKB substrate antibody used to detect the phosphorylated state of pp350, pp200, pp180 (GefS), pp110 (GefN and PI5K), and pp65/67 (GacQ) and other substrates of PKB. Second, a phospho PDK docking motif antibody used to detect the phosphorylated state of the hydrophobic motif of PKBR1 (T470). For PKBR1 the extent of this phosphorylation correlates strongly with the activation state of TorC2. Third, a phospho PKC (pan) antibody used to detect the phosphorylation state of the activation loops of PKBR1 (T309) and PKBA (T278). Evidence from other model systems suggests that these phosphorylations would be catalyzed by a PDK homolog and essential for their activities. Two PDK homologs are present in D. discoideum. Fourth, antiphospho AKT (S473) can be used to detect activation of TorC2 in cells expressing a chimeric protein where the PH domain of human AKT is replaced with the myristoylated N-terminal of PKBR1.
3.6.1. cAMP Stimulation and Sample Preparation
Transfer 0.5-mL competent cells on ice (see Note 12) at a density of 2 × 107 cells/mL to a 5-mL disposable plastic beaker shaking at 150 rpm (see Note 13). Within 2 min, add 100 μM of cAMP to final concentration of 1 μM.
Transfer 40 μL of cells at time points of 10, 20, 30, 60, 120, 180 s into microcentrifuge tubes containing 10 μL of 5× SDS sample buffer.
Quickly move tube to a hot-block at 95°C for 5 min.
3.6.2. Western Blotting
Load wells of precast gels with 2.5 ml of sample (4 × 104 cells) for antiphospho PKB substrate antibody and 5 μl (8 × 104 cells) for antiphospho PDK docking motif antibody and antiphospho PKC (pan) antibody.
Run gels by electrophoresis at 150 V for 85 min.
To transfer proteins to a PVDF membrane, place a pad and two sheets of 3-MM paper wetted with the transfer buffer on the black (cathode -) side of the cassette holder. Place the gel on the 3-MM paper and lay the PVDF membrane on top of it. Remove bubbles between the gel and the PVDF and put two more sheets of 3-MM paper and a pad.
Place the sandwich into the transfer tank such that the PVDF membrane is between the gel and the anode (+) and fill the cold transfer buffer.
Turn on the system for 80 min at 75 mV in the cold room.
After the transfer, rinse the PVDF membrane with TBST twice quickly.
Incubate the membrane in 50 ml of blocking buffer for 1 h at room temperature.
Discard the blocking buffer and rinse the membrane with TBST twice quickly.
Incubate the membrane with a primary antibody at 4°C overnight.
Remove the primary antibody, rinse the membrane with TBST twice quickly, and further wash with TBST for 10 min, 5 min, and 5 min at room temperature.
Incubate the membrane with a secondary antibody for 1 h at room temperature.
Remove the secondary antibody, rinse the membrane with TBST twice quickly, followed by one 10 min and four 5 min washes with TBST at room temperature.
Prepare the ECL reagent and incubate the membrane with it for 1 min.
Remove the ECL reagent and wrap the membrane with plastic wrap.
Expose the membrane to X-ray film in the cassette for a suitable time.
To confirm the loading of proteins, stain the membrane in CBB solution for couple of minutes and destain it in 50% methanol for suitable time, typically a few minutes. Place the membrane on a bench to let it dry at room temperature.
As shown in Fig. 3a, the phospho PKB substrate antibody stains few bands in competent cells prior to addition of cAMP. Following cAMP stimulation, about ten prominent bands rapidly appear and display characteristic time courses, eventually subsiding over the next several minutes. Seven or eight of these bands are specific PKB substrates which are greatly decreased in cells lacking PKB activity such as pkbR1−, pkbR1−/pkbA−, and pia−. These bands reappear when the appropriate proteins are expressed in the null mutants. A few of the bands (pp250, pp30, and pp23) are not dependent on PKB activity and are presumably substrates for other kinases with the consensus motif RXRXXS/T.
Fig. 3.
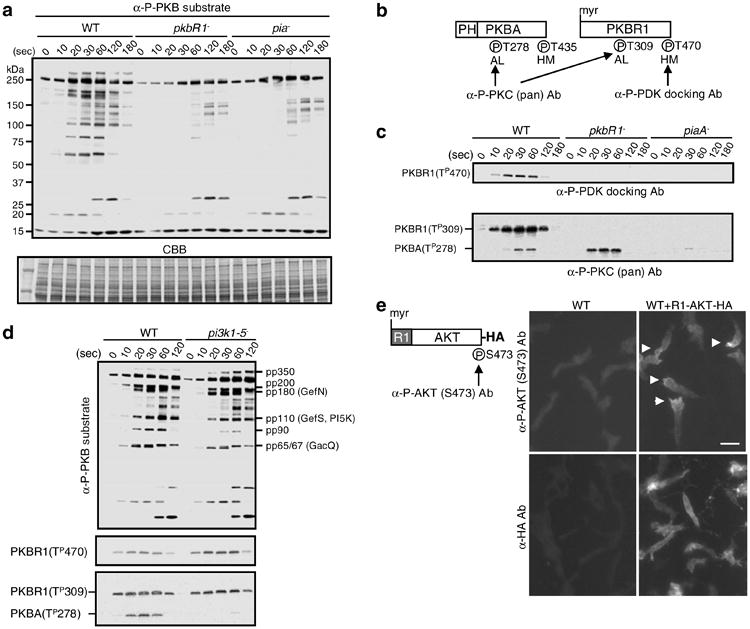
Assays to detect TorC2 and PKB activities. (a) The in vivo PKB activity is evaluated by antiphospho PKB substrate antibody (α-P-PKB substrate). The bands of pp350, pp200, pp180, pp110, pp90, and pp65/67 in WT cells are dependent on the activities of PKBR1 and PiaA, a subunit of TorC2. CBB (coomassie brilliant blue) staining is for the loading control. (b) The schematic structures of PKBA and PKBR1 are shown. The phosphorylation sites (P) can be detected by the antibodies, antiphospho PDK docking motif antibody (α-P-PDK docking Ab), and antiphospho PKC (pan) antibody (α-P-PKC (pan) Ab). The labels AL and HM refer to activation loop and hydrophobic motif. (c) The upper panel shows the phosphorylation at T470 of PKBR1 in WT, pkbR1−, piaA− cells by α-P-PDK docking Ab. The lower panel shows the phosphorylations at T278 of PKBA and T309 of PKBR1 in WT, pkbR1−, piaA− cells by α-P-PKC (pan) Ab. (d) The PKB activity, the phosphorylation of T470 of PKBR1 by α-P-PDK docking Ab, and the phosphorylations of T278 of PKBA and T309 of PKBR1 by α-P-PKC (pan) Ab are compared in between WT and pi3k1−-5− cells. (e) The schematic structure of R1-AKT-HA is shown with the phosphorylation sites (P) that are detected by the antiphospho AKT (S473) antibody (α-P-AKT (S473) Ab). In the right panel, chemotaxing cells are stained with α-P-AKT (S473) Ab to detect the phosphorylation of S473 or α-HA Ab for the localization of R1-AKT-HA. Arrow heads show localized staining. The scale bar represents 10 μm. (Reproduced from ref. 10 with permission from Elsevier Science.).
As shown in Fig. 3b, c, the phospho PDK docking motif antibody, very specifically detects the phosphorylation state of the hydrophobic motif of the PKBR1. This phosphorylation is completely absent in competent cells prior to stimulation. Following addition of cAMP, it increases rapidly, peaks at 30 s, and disappears within 2 min. This phosphorylation is completely absent in cells lacking PiaA and substantially decreased in cells lacking Rip3 (data not shown), indicating that it is a TorC2 substrate.
As shown in Fig. 3c, the phospho PKC (pan) antibody specifically detects the activation loop phosphorylations of PKBA and PKBR1. The time courses of these phosphorylations closely follow those of the hydrophobic motif (see Fig. 3c). The phosphorylation of PKBA (T278) is abolished in cells lacking PIP3 production such as pi3k1−/5− cells (see Fig. 3d) and substantially reduced in piaA− cells (see Fig. 3c). Interestingly, however, the phosphorylation of PKBR1 (T309) is unaffected by the absence of PIP3 (see Fig. 3d). It does absolutely require a prerequisite phosphorylation at T470 since T309 is not phosphorylated in T470A versions of PKBR1 or in piaA− cells (see Fig. 3c). These observations show that the activation of PKBR1 (T309) depends on TorC2 and not PIP3, while activation of PKBA (T278) depends on both TorC2 and PIP3. This blotting with the activation loop antibody can be used on any cell line to rapidly assess activation of TorC2 as well as PI3K. The antibody is also effective in detecting TorC2 activity cytologically in cells overexpressing PKBR1.
3.6.3. Indirect Immunofluorescence
Place 18-mm coverslips on parafilm.
Put 5 × 105 competent cells expressing R1-AKT-HA in 500 μL DB on the coverslip and allow cells to initiate chemotaxis for 20–30 min at room temperature.
Place 500 μL fix solution on the parafilm and transfer the coverslip into this solution followed by 30-min incubation at room temperature.
Transfer the coverslip into 500 μL of quenching solution on the parafilm followed by 10-min incubation at room temperature.
Wash the coverslip with PBS twice.
Permeabilize and block cells by incubation in the blocking solution for 30 min at room temperature.
Remove the blocking solution and place the coverslip cells facing down on 100 μL of the primary antibody solution, either anti Phospho AKT(S473) or anti HA, on the parafilm followed by overnight incubation in the moist chamber at 4°C.
Wash the coverslip in 3 mL of TBS using 6-well plate for 5 min three times.
Incubate with 100 μL of the secondary antibody (as was done for the primary antibody step) for 1 h at room temperature in the dark.
Wash the coverslip in 3 mL of TBS using 6-well plate for 5 min three times.
Dip the coverslip once in deionized water.
Invert the coverslip cells attached side down and put into a drop of mounting medium on a slide glass (see Note 14).
Observe the sample under the fluorescence microscope by excitation at 543 nm. (see Fig. 3e)
The TorC2-PKBR1 pathway is selectively activated at the leading edge of chemotaxing cells. The R1-AKT-HA chimeric protein, where the PH domain of human AKT is replaced with N-terminus PKBR1 containing the myristoylation site, is phosphorylated at S473 in the hydrophobic motif in response to cAMP stimulation. This response is completely dependent on TorC2 activity and does not occur in cells lacking PiaA. As shown in Fig. 3e, indirect immunofluorescence of a cell expressing the R1-AKT-HA by antiphospho AKT(S473) antibody can be used to visualize the activation of TorC2 cytologically. Prior to stimulation there is little or no staining of cells. In highly polarized cells the staining is found selectively at the leading edge.
3.7. Purification of Substrates of PKB
This section describes the purification of PKB substrates.
After basalating (see Subheading 3.1.1), cells are washed with 15 mL of ice-cold PM buffer twice.
Stimulate cells with or without 1 μM cAMP for 30 s.
After stimulation, lyse cells with an equal volume of 2× NP-40 lysis buffer on ice for 5 min.
Centrifuge the cell extracts for 5 min at full speed in micro-centrifuge at 4°C.
Transfer the supernatant to a new microcentrifuge tube.
Incubate the supernatant with prewashed protein G-Sepharose (see Subheading 2.7, item 2) for 1 h at 4°C.
Centrifuge at 500 × g for 1 min and transfer the supernatant into a new microcentrifuge tube.
Add antiphospho PKB substrate antibody (110B7) (1% volume of total cell extracts) and incubate it overnight at 4°C.
Add prewashed protein G-Sepharose and incubate for further 1 h at 4°C.
Centrifuge at 500 × g for 1 min and wash the beads with 1× NP-40 buffer four times (twice quickly and twice for 15 min).
Repeat step 10 using 1× RIPA buffer.
Elute the proteins by boiling beads in 1× SDS sample buffer.
Separate proteins by SDS-PAGE on 18-well precast gels.
Visualize proteins by silver staining (Silver Quest Silver-staining Kit; Invitrogen) in a 150 × 25 mm of tissue culture dish.
Acknowledgments
The authors wish to thank Dr. Stacey Willard for sharing unpublished data. This work was supported by NIH GM 28007 and NIH GM 34933 to P.N.D. and by the Uehara Memorial Foundation to Y.K.
Footnotes
Antibiotics, such as streptomycin, may be added to prevent contamination.
Noble agar can be substituted for hydrophobic agar used in the original protocol (15).
The PVDF membrane is quickly prewetted with 100% methanol followed by 2-min soak in H2O and equilibrated for at least 5 min in the transfer buffer.
For AX3 strains, 20 μg/mL of G418 is used.
Fixative solution should be prepared freshly.
Measure in the hood. Do not inhale.
LY294002 is dissolved in DMSO (Dimethyl sulfoxide). Since DMSO affects cell differentiation, the comparable concentration of DMSO is used as a control.
For a large field, a 10× phase objective lens is used.
Do not dry the plate too much; otherwise, a droplet cannot be maintained during assay.
Score at least eight spots per test concentration.
Take duplicate 0-s samples for basal activity.
If cells are not maintained on ice, they spontaneously secrete and respond to cAMP within 7 min.
A styrofoam rack for 50-mL tubes makes a convenient holder of multiple beakers.
The sample is stable at 4°C for at least 1 month.
References
- 1.Franca-Koh J, Kamimura Y, Devreotes PN. Navigating signaling networks: Chemotaxis in Dictyostelium discoideum. Curr Opin Genet Dev. 2006;16:333–338. doi: 10.1016/j.gde.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 2.Stephens L, Milne L, Hawkins P. Moving towards a better understanding of chemotaxis. Curr Biol. 2008;18:R485–494. doi: 10.1016/j.cub.2008.04.048. [DOI] [PubMed] [Google Scholar]
- 3.Kay RR, Langridge P, Traynor D, Hoeller O. Changing directions in the study of chemotaxis. Nat Rev Mol Cell Biol. 2008;9:455–463. doi: 10.1038/nrm2419. [DOI] [PubMed] [Google Scholar]
- 4.Parent C, Blacklock B, Froelich W, Murphy D, Devreotes PN. G protein signaling events are activated at the leading edge of chemotactic cells. Cell. 1998;95:81–91. doi: 10.1016/s0092-8674(00)81784-5. [DOI] [PubMed] [Google Scholar]
- 5.Funamoto S, Milan K, Meili R, Firtel RA. Role of phosphatidylinositol 3′ kinase and a downstream pleckstrin homology domain-containing protein in controlling chemotaxis in Dictyostelium. J Cell Biol. 2001;153:795–810. doi: 10.1083/jcb.153.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meili R, Ellsworth C, Lee S, Reddy TBK, Ma H, Firtel RA. Chemoattractant-mediated transient activation and membrane localization of Akt/PKB is required for efficient chemotaxis to cAMP in Dictyostelium. EMBO J. 1999;18:2092–2105. doi: 10.1093/emboj/18.8.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iijima M, Devreotes PN. Tumor suppressor PTEN mediates sensing of chemoattractant gradients. Cell. 2002;109:599–610. doi: 10.1016/s0092-8674(02)00745-6. [DOI] [PubMed] [Google Scholar]
- 8.Hoeller O, Kay RR. Chemotaxis in the absence of PIP3 gradients. Curr Biol. 2007;17:813–817. doi: 10.1016/j.cub.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 9.Chen L, Iijima M, Tang M, Landree MA, Huang YE, Xiong Y, Iglesias PA, Devreotes PN. PLA2 and Pi3K/PTEN pathways act in parallel to mediate chemotaxis. Dev Cell. 2007;12:603–614. doi: 10.1016/j.devcel.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kamimura Y, Xiong Y, Iglesias PA, Hoeller O, Bolourani P, Devreotes PN. PIP3-independent activation of TorC2 and PKB at the cell's leading edge mediates chemotaxis. Curr Biol. 2008;18:1034–1043. doi: 10.1016/j.cub.2008.06.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meili R, Ellsworth C, Firtel RA. A novel Akt/PKB-related kinase is essential for morphogenesis in Dictyostelium. Curr Biol. 2000;10:708–717. doi: 10.1016/s0960-9822(00)00536-4. [DOI] [PubMed] [Google Scholar]
- 12.Chen MY, Long Y, Devreotes PN. A novel cytosolic regulator, Pianissimo, is required for chemoattractant receptor and G protein-mediated activation of the 12 transmembrane domain adenylyl cyclase in Dictyostelium. Genes Dev. 1997;11:3218–3231. doi: 10.1101/gad.11.23.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee S, Comer FI, Sasaki A, McLeod IX, Duong Y, Okumura K, Yates JR, 3rd, Parent CA, Firtel RA. TOR complex 2 integrates cell movement during chemotaxis and signal relay in Dictyostelium. Mol Biol Cell. 2005;16:4572–4583. doi: 10.1091/mbc.E05-04-0342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the Rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 15.Konijn TM, van Haastert PJ. Measurement of chemotaxis in Dictyostelium. Methods Cell Biol. 1987;28:283–298. doi: 10.1016/s0091-679x(08)61652-0. [DOI] [PubMed] [Google Scholar]