

Abstract
Kynurenic acid (KYNA) is a tryptophan metabolite that acts in the brain as an endogenous antagonist at multiple receptors, including glutamate and α7 nicotinic acetylcholine receptors. Increased levels of KYNA have been demonstrated in the brain of patients with a range of neurocognitive disorders, including schizophrenia, and are hypothesized to contribute to cognitive symptoms. Reducing KYNA levels by administering inhibitors of enzymes of the kynurenine pathway, particularly kynurenine aminotransferase II (KAT II), has been proposed as a treatment for such cognitive impairments. Here we report that administration of a systemically available KAT II inhibitor, PF-04859989, restores glutamate release events (“transients”) evoked by pressure ejections of nicotine into the prefrontal cortex of rats exhibiting elevated KYNA levels. Nicotine-evoked glutamatergic transients can be reliably evoked and recorded after repeated pressure ejections of nicotine over 4–5 h. Systemic administration of L-kynurenine (100 mg/kg; i.p.) significantly increased frontal cortical KYNA levels and greatly attenuated the amplitude of nicotine-evoked glutamatergic transients. Systemic administration of PF-04859989 30 min prior to administration of L-kynurenine, but not when administered 30 min after L-kynurenine, restored glutamatergic transients recorded up to 75 min after the administration of the KAT II inhibitor. Furthermore, the KAT II inhibitor significantly reversed L-kynurenine-induced elevations of brain KYNA levels. The KAT II inhibitor did not affect nicotine-evoked glutamatergic transients in rats not pre-treated with L-kynurenine. Because PF-04859989 restores evoked glutamate signaling it therefore is a promising therapeutic compound for benefiting the cognitive symptoms of schizophrenia and other disorders associated with elevated brain KYNA levels.
Keywords: Kynurenic acid L-Kynurenine, KAT II, PF-04859989, Schizophrenia, Cognition
1. Introduction
Kynurenic acid (KYNA) is a tryptophan metabolite and a component of the kynurenine pathway that is released from astrocytes and acts primarily as a noncompetitive antagonist at the strychnine-insensitive glycine recognition site of the NMDA receptor. Additionally, KYNA blocks the α7 nicotinic acetylcholine receptor (nAChR) at similarly low micromolar concentrations (Parsons et al., 1997; Hilmas et al., 2001; Albuquerque and Schwarcz, 2013). Elevated brain levels of KYNA have been documented in the brain and cerebrospinal fluid of patients with schizophrenia and other brain disorders and is hypothesized to contribute to cognitive impairments, primarily by interfering with glutamate neurotransmission (e.g., Erhardt et al., 2007; Linderholm et al., 2012; Nilsson et al., 2005; Schwarcz et al., 2001, 2012). The pathological mechanisms leading to elevated levels of KYNA in the brain are not clear but are likely associated with central and peripheral neuro-inflammatory processes (e.g., Potvin et al., 2008).
The detrimental consequences of elevated KYNA levels on glutamatergic neurotransmission and cognitive and behavioral processes have been widely demonstrated in animals with elevated brain KYNA levels, produced by administration of the precursor, L-kynurenine (KYN; Akagbosu et al., 2010; Alexander et al., 2013; Chess et al., 2007, 2009; Trecartin and Bucci, 2011; Konradsson-Geuken et al., 2010). These findings have suggested that drugs that reduce brain KYNA levels may benefit cognitive function in a wide range of patient groups (e.g., Wonodi and Schwarcz, 2010). Specifically, inhibition of the main enzyme of KYNA production, kynurenine aminotransferase II (KAT II), has been considered a primary test of this hypothesis. Unfortunately, systemically available KAT II inhibitors have remained rare and thus tests of this hypothesis in animals have largely remained restricted to assessing the effects of intracerebral or intracerebroventricular administration of such compounds (e.g., Pellicciari et al., 2006; Amori et al., 2009).
PF-04859989 is a potent, systemically available, brain-penetrant KATII inhibitor (Dounay et al., 2013). We designed experiments to assess the efficacy of this compound when systemically administered, to restore glutamatergic activity in the brains of animals exhibiting elevated KYNA levels. The focus on nicotine-evoked glutamate release as the main dependent measure in these experiments was based on two sets of findings and hypotheses. First, the experimental paradigm used for these experiments was derived from our prior studies on the regulation of glutamate release from thalamic inputs in the prelimbic prefrontal cortex. We previously demonstrated that brief glutamate release events, or “transients”, are evoked by pressure ejections of nicotine (Parikh et al., 2008, 2010; see also Lambe et al., 2003). In attentional task-performing animals, acetylcholine (ACh) released from basal fore-brain projections stimulates nicotinic acetylcholine receptors (nAChRs) in the cortex, expressed in part by thalamic glutamatergic inputs, to modulate circuitry that mediates the detection of cues in attentional contexts (Guillem et al., 2011; Sarter et al., 2009; Hasselmo and Sarter, 2011; Howe et al., 2013). Second, elevated KYNA levels were hypothesized to attenuate nicotine-evoked glutamatergic transients by blocking the effects of nicotine at the α7 nAChR that may also be expressed by glutamatergic thalamic afferents (Rousseau et al., 2005; Livingstone et al., 2010; Dickinson et al., 2008; Konradsson-Geuken et al., 2010), and/or by blocking ionotropic glutamate receptors that are part of additional circuitry linking stimulation of multiple nAChRs with glutamate release (see also Albuquerque and Schwarcz, 2013). The present experiments were not designed to isolate these mechanisms. Rather, the results from six experiments (for overview see Fig. 1) collectively indicated that elevated KYNA levels greatly suppress nicotine-evoked glutamatergic transients and that PF-04859989, when administered prior to KYN, effectively and lastingly restored nicotine-evoked glutamatergic signaling.
Fig. 1.
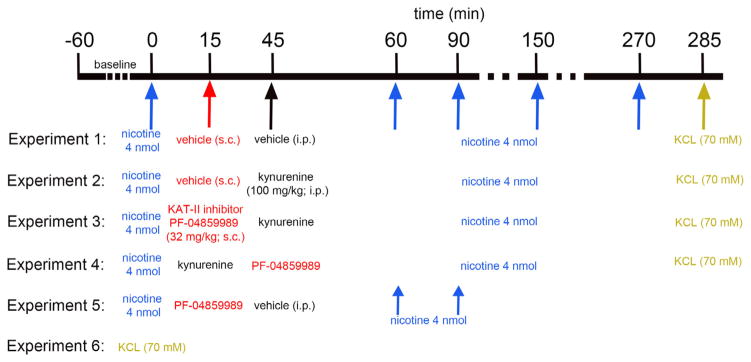
General experimental design used to determine the effects of systemic administration of KYN, in the presence or absence of the KAT II inhibitor PF-04859989 (PF), on nicotine-evoked glutamate release. Amperometric recordings of glutamatergic activity were initiated after implantation of glutamate-sensitive microelectrodes in the mPFC. Experiments began after 60 min of baseline recording. Nicotine (4 nmol) was pressure-ejected into the recording area and evoked glutamatergic transients were measured. Recordings at t = 0 were treated as baselines. In Experiment 1, (saline/saline group; n = 6), saline was administered 15 min and 45 min after the baseline nicotine pressure ejection. To assess the effects of KYN on nicotine-evoked glutamatergic transients, Experiment 2 (saline/KYN group; n = 6) received systemic administration of saline at 15 min and KYN at 45 min after baseline nicotine ejection. To determine whether the systemic KAT II inhibitor PF is effective in reversing the effects of KYN on nicotine-evoked glutamate release, PF was systemically administered 30 min before (Experiment 3) or after (Experiment 4) KYN (n = 5 each). In all 4 experiments, a series of four nicotine ejections followed 15 min (T60), 45 min (T90), 105 min (T150), and 225 min (T270) after the second systemic administration of saline/drug. To determine the integrity of glutamatergic terminals and the viability of the sensor following multiple pressure ejections over 270 min, KCl (70 mM) was pressure ejected 15 min (T285) after the last nicotine infusion in all the animals and compared with the effects of KCl given at t = 0 (Experiment 6). Experiment 5 tested the effects of PF alone on nicotine-evoked glutamatergic transients.
2. Materials and methods
2.1. Subjects
The electrochemical recording studies were conducted at the University of Michigan and the microdialysis studies at Pfizer in Boston. For the electrochemical recordings, adult male Wistar rats (Harlan, Indianapolis, IN), weighing 300–480 g at the beginning of the experiments, were used. Animals were housed in facilities accredited by the American Association of Accreditation of Laboratory Animal Care. Food and water were available ad libitum (Rodent chow; Harlan Teklad, Madison, WI). Procedures were carried out in accordance with protocols approved by the Committee on the Use and Care of Animals at the University of Michigan. Male Sprague-Dawley rats were used for the microdialysis studies and obtained from Charles River Laboratories (Raleigh, NC) with BAS microdialysis guide cannulas (BASi, West Lafayette, IN, catalog# MD-2251) implanted into the prelimbic cortex. Upon arrival, the animals were single housed and allowed to acclimate for at least five days prior to use. These rats were maintained on a 12-h light/dark cycle and allowed free access to food and water. The microdialysis studies were conducted in accordance with animal use protocols approved by the Pfizer Institutional Care and Use Committee and the National Institutes of Health Guidelines.
2.2. Drugs and chemicals
Glutamate oxidase (GO; EC 1.4.3.11) was obtained from Seikagaku America (East Falmouth, MA). Bovine serum albumin (BSA), glutaraldehyde, ascorbic acid (AA), dopamine (DA), L-glutamic acid, L-kynurenine (KYN), potassium chloride (KCl) and nicotine were procured from Sigma (St. Louis, MO). PF-04859989 (PF) was provided by Pfizer Inc. (Groton, CT). Meta-phenylene diamine (m-PD) was obtained from Fluka Biochemika (Buchs, Switzerland). HPLC grade water (Fisher Scientific, Davis, CA) was used to prepare all solutions unless otherwise specified. PF was prepared fresh on each experimental day in 0.9% NaCl, pH ~7.0. KYN was initially dissolved in 2 N sodium hydroxide (NaOH), made up to a final volume of 25 mg/mL with 0.1 M 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer and finally brought to a neutral pH with 1 N hydrochloric acid (Chess et al., 2009).
2.3. Preparation and calibration of glutamate-sensitive microelectrodes
Real-time glutamate release was measured using ceramic-based microelectrodes equipped with four 15 × 333 μm Platinum (Pt) recording sites arranged in side-by-side pairs (Quanteon, Nicholasville, KY). Glutamate-sensitive microelectrodes were prepared by coating GO onto the recording sites as described previously (Hascup et al., 2008; Parikh et al., 2008, 2010). Briefly, GO was cross-linked with the BSA-glutaraldehyde mixture and immobilized onto the bottom pair of recording sites. The remaining upper two recording sites were coated only with the BSA-glutaraldehyde solution, which served to monitor current generated by electrochemically active interferents and was used for self-referencing. GO coated microelectrodes were air-dried for at least 48 h before use. On the day of the experiment, m-PD was electro-polymerized onto the recording sites to enhance selectivity for glutamate by preventing access of potential electroactive interferents including AA and catecholamines. m-PD electroplating was performed by applying a constant voltage of 0.5 V to the microelectrode against a Ag/AgCl reference electrode (Bioanalytical Systems, West Lafayette, IN) in a beaker containing a solution of 5 mM m-PD and 10 μM AA (in 0.05 M PBS), bubbled with nitrogen gas and maintained at 37 °C for 60 min. AA and nitrogen gas served to protect m-PD from oxidation. Calibrations were performed using fixed potential amperometry by applying a constant voltage of 0.7 V against a Ag/AgCl reference electrode in a beaker containing a stirred solution of 0.05 M PBS (40 mL) maintained at 37 °C using a FAST-16 electrochemical system (Quanteon). Amperometric currents were digitized at a frequency of 1 Hz. After a baseline current is achieved, aliquots of stock solutions of AA (20 mM), glutamate (20 mM), and DA (2 mM) were added to the calibration beaker such that the final concentrations were 250 μM AA, 20, 40, 60, and 80 μM glutamate and 2 μM DA. The slope (sensitivity), limit of detection (LOD), and linearity (R2) for glutamate, as well as selectivity ratio for AA, was calculated for individual recording sites.
2.4. Amperometric recordings of nicotine-evoked glutamatergic transients in vivo
Animals were anesthetized with urethane (1.25–1.5 g/kg, i.p.) and placed on a stereotaxic frame (David Kopf Instruments, Tujunga, CA). As is the case for most anesthetics, urethane anesthesia likely affects neurotransmission (but see Sceniak and MacIver, 2006). The acute insertion of a microelectrode/micropipette assembly and the measurement of electrochemical currents evoked by pressure ejections of drugs into the vicinity of recordings sites necessitated experimentation in anesthetized rats. Animal body temperature was maintained at 37 °C during the entire experimental session by using a water-heated circulating isothermal pad. Single-barrel glass capillaries (1.0 × 0.58 mm, 6 inches; A-M Systems, Everett, WA) were pulled using a micropipette puller (model 51210; Stoelting, Wood Dale, IL) and then bumped against a glass slide to attain an inner diameter of ~15 μm. Micropipettes were attached to the microelectrode with the tip centered between the two pairs of recording sites, ~70 μm from the surface of the electrode. The micropipette was loaded with one of the test solutions (see below, Experimental design) before microelectrode implantation. The microelectrode/micropipette assembly was slowly lowered into the right mPFC (AP: +3.0 mm from bregma; ML: 0.7 mm; ventral: −3.0 mm from dura). Glutamatergic transients were measured in the thalamic input layers (layers III and V) of the prelimbic region; nAChR agonist-evoked glutamatergic transients recorded in this region has previously been shown to reflect release from mediodorsal thalamic afferents (Parikh et al., 2008; Hasselmo and Sarter, 2011). An Ag/AgCl reference electrode prepared from a miniature silver wire (200 μm diameter) was implanted at a site remote from the recording area and a fixed potential of +0.7 V was held between the reference and recording electrodes. Amperometric recordings were made at 1 Hz, and data was digitized using the FAST-16 system (Quanteon LLC, Nicholasville, KY). Experiments began after the stabilization of baseline current (60 min). Drug solutions were pressure ejected from the micropipettes using polytetrafluoroethylene tubing connected to a pressure ejection system (Picospritzer III; Parker Hannifin, Fairfield, NJ), and ejection volumes were monitored using a stereomicroscope equipped with a reticule.
2.5. Experimental design for electrochemical recordings
Fig. 1 provides a summary of the experiments conducted to determine the effects of systemic administration of KYN (precursor of KYNA) and the KATII inhibitor PF on nicotine-evoked glutamatergic transients. The concentration of nicotine (4 nmol) was adopted based on dose–response data described in our prior study and was the lowest dose that generated maximum transient amplitudes (Parikh et al., 2008). First, we recorded nicotine-evoked glutamatergic transients in the presence of systemic vehicles for KYN (i.p.) and PF (s.c.). The effects of pressure ejections of nicotine were recorded prior to the systemic administration of drugs (15 and 45 min after the first pressure ejection of nicotine), and 4 more times between 60 and 270 min following the first test of nicotine (see Fig. 1).
Experiment 2 determined the effects of KYN (100 mg/kg; i.p.) on nicotine-evoked glutamatergic transients. The dose for KYN was adopted from studies showing robust impairments on measures of learning and memory in studies on rats (Chess et al., 2007, 2009). The 3rd and main experiment assessed the effects of PF (32 mg/kg) administered 30 min prior to KYN administration. This dose for PF was selected prior to the in vivo measurement of brain KYNA levels (see Results) and was based on data indicating 80% inhibition of basal brain KYNA levels, plateauing about 1 h after administration and lasting for over 4 h (W. Horner, Pfizer, personnel communication). As will be described in Results, this dose of PF nearly completely attenuated the KYN-induced increase in brain KYNA levels. The 4th experiment tested the hypothesis that the KATII inhibitor does not restore nicotine-evoked transients if administered 30 min after KYN. Experiment 5 assessed the effects of PF on nicotine-evoked transients in animals that did not receive an injection of KYN. At the end of experiments 1–4 (Fig. 1), potassium (70 mM; 200 nL) -evoked glutamatergic transients were recorded to test sensor viability and the integrity of the recording area. To determine potential effects of time and prior pressure ejections on KCL-evoked transients, such transients were obtained in a separate group of rats that did not receive any other treatment (KCl effects measured at t = 0; Experiment 6). Finally, we also tested whether pressure ejections of vehicle alone (0.9% saline; 200 nL) evoked glutamatergic transients (not listed in Fig. 1).
2.6. Glutamate signal analyses
Currents recorded via GO-coated sites were corrected by those recorded on non-GO-coated recordings sites if relatively high background noise levels or artifacts resulted from pressure ejection occurred. Because m-PD-coated electrodes are completely protected against currents produced by dopamine (Parikh et al., 2008), currents recorded from non-GO-coated sites simply were subtracted from currents recorded via GO-coated sites. Glutamatergic signals were analyzed with respect to peak amplitudes and signal decay rate, defined as the time required for the signal to decline by 50% from peak amplitude (t50).
2.7. Histological methods
After completion of amperometric recording sessions, animals were transcardially perfused with 100 mL of ice-cold 0.1 M PBS followed by 300 mL of 4% paraformaldehyde in 0.1 M PBS, pH 7.4. The brains were removed and postfixed overnight at 4 °C and stored in 30% sucrose in 0.1 M PBS for 72 h. Coronal sections (50 μm) were cut using a freezing microtome (CM 2000R; Leica, Chantilly, VA) and stored in cryoprotectant solution (15% glucose, 30% ethylene glycol, and 0.04% sodium azide in 0.05 M PBS, pH 7.4) at 20 °C until further processing. Serial sections were Nissl-stained to verify the placement of microelectrodes.
2.8. Statistical analyses of electrochemical recordings
Statistical analyses were conducted using SPSS/PC+ (V19.0; IBM SPSS Statistics for Windows, Armonk, NY: IBM Corp). Mixed model repeated-measures ANOVA was used to analyze the effect of time (5 levels) on nicotine-evoked glutamatergic transients (Experiment 1). Post hoc multiple comparisons were conducted using the Bonferroni test. One-way ANOVAs were used to compare baseline nicotine-evoked as well as KCl-evoked glutamate signal amplitudes and decay rates between the 4 experiments and groups of animals. Across the 4 conditions, repeated-measures mixed-factor ANOVAs were used to analyze the effect of time (4 levels) on the percentage change in glutamate signal amplitude and on decay rates from baseline. Post hoc multiple comparisons were conducted using the least significant difference (LSD) test. Alpha was set at 0.05.
2.9. Brain KYNA concentrations following the administration of KYN
On the day prior to the collection of dialysates, rats were lightly anesthetized with isoflurane and a 4.0 mm BAS microdialysis probe (BASi, West Lafayette, IN; catalog # MD-2204) was inserted into the prelimbic cortex (AP +3.2 mm; ML +0.7 mm (left hemisphere); DV 5.3 mm below dura). The dialysis probe was perfused overnight at 0.3 μL/min with artificial cerebrospinal fluid (aCSF; 147 mM NaCl, 1.3 mM CaCl2, 2.7 mM KCl, 1.0 mM MgCl2). At approximately 7:00 AM on the day of testing, the flow was increased to 2 μL/min. Following a 2-hr stabilization period, dialysis samples were collected at 15-min intervals using a refrigerated fraction collector. After collection of 6 baseline samples, vehicle or PF, at the dose used for the electrochemical recording (32 mg/kg, s.c.; see above for justification of dose) was administered. KYN (100 mg/kg; i.p.) was administered to all rats 30 min following the administration of PF or the vehicle for PF. Dialysis samples were collected for an additional 2.5 h. Collections were stored frozen at −20 °C for off-line determination of KYNA levels.
2.9.1. Determination of KYNA levels in collections
A 20-μL aliquot of each dialysis sample was analyzed for KYNA content by HPLC in conjunction with fluorescence detection. KYNA was separated over a C18 column (150 × 3 mm, 3 μm particle sized; Thermo Scientific, catalog # 28103-153030) using a mobile phase consisting of 50 mM sodium acetate, 250 mM zinc acetate and 3% acetonitrile, pH 6.2. The flow rate was set at 0.35 ml/min and the column was maintained at room temperature. KYNA was detected with a fluorescence detector (excitation = 344 nm / emission = 398 nm).
Chromatography data were collected and kynurenic acid levels quantified by comparison to known standards using EZChrom Elite software (Agilent Technologies). KYNA levels were normalized to baseline levels for each animal. Average baseline levels were calculated using the first six microdialysis samples collected prior to treatment. Time course data were then expressed as the fold-change from baseline calculated as the ratio of the response at each time point to the average baseline level. Statistical analyses of fold changes were conducted using repeated measures ANOVA on the effects of time (baseline levels averaged over pre-PF administration time points (−75 – 0 min) versus post-KYN administration time points (60 – 180 min) and condition (vehicle versus PF), followed by post hoc comparisons between individual data pairs using t-tests).
3. Results
The present experiments employed glutamate oxidase-coated microelectrodes to measure nicotine-evoked glutamatergic transients in the medial prefrontal cortex (mPFC) of animals pre-treated with KYN and to determine the efficacy of PF-48599889 (PF), a kynurenine aminotransferase II (KAT II) inhibitor, in restoring KYN-attenuated glutamatergic transients. The design for the main experiments is illustrated in Fig. 1.
3.1. Electrode calibration
Table 1 summarizes the electrochemical properties, determined in vitro and prior to implantation, of glutamate-sensitive micro-electrodes used in all six experiments.
Table 1.
In vitro calibration of glutamate-sensitive microelectrodes.
| Sensitivity (pA/μM) | LOD (nM) | R2 | Selectivity |
|---|---|---|---|
| 10.52 ± 0.95 | 571.32 ± 1116.19 | 0.991 ± 0.002 | 97.20 ± 12.78 |
Data are based on 30 glutamate-sensitive microelectrodes (mean ± SEM); LOD, limit of detection; R2 depicts the linearity of the response of the microelectrode to increasing concentrations of glutamate; Selectivity refers to the ratio of the electrode response to glutamate relative to ascorbic acid.
3.2. Overall analyses of effects of conditions (omnibus ANOVA)
Further below, we will compare the amplitudes of glutamatergic transients observed in Experiments 2–4 (Fig. 1) with the baseline effects of nicotine obtained from Experiment 1, as well as conduct comparisons between data from individual experiments. To statistically justify these comparisons we first conducted omnibus ANOVAS over the data from all 4 experiments and repeated nicotine pressure ejections.
Pressure-ejections of vehicle alone produced small transients, peaking at 4.39 ± 0.56 μM glutamate (M; SEM). As infusions of saline in previous experiments did not evoke such transients (Konradsson-Geuken et al., 2009), we speculate that the vehicle transients produced in this study is a result of the pipette placement and reflect pressure ejection artifact. It is important to note that the nicotine-evoked glutamatergic responses presented in this study are substantially larger. The initial pressure ejections of nicotine evoked glutamatergic transients that peaked at 10.92 ± 2.90 μM and with an average decay rate (t50) of 4.0 ± 0.63 s. First, we confirmed that the effects of the very first pressure ejection of nicotine, prior to the systemic administration of KYN and PF (at t = 0 in Fig. 1), did not differ between the 4 experiments. The amplitudes and decay rates of these transients were consistent with those observed in our previous studies (Parikh et al., 2008, 2010) and they did not differ between experiments 1–4 (amplitude: F(3,18) = 0.08, p > 0.05; decay rate: F(3,18) = 0.33, p > 0.05). This finding allowed us to express the amplitudes and decay rates of transients, that were recorded after the systemic administration of KYN and PF, as percent changes from baseline and use this data for statistical analyses.
A repeated measures ANOVA indicated main effects of Experiment (Experiments 1–4; F(3,18) = 13.38, p < 0.001) and the effects of repeated pressure ejections of nicotine (or Time; F(3,54) = 14.74, p < 0.001), as well as a significant interaction between the effects of these two factors (F(9,54) = 7.24, p < 0.001) on the amplitudes of glutamatergic transients. Transient decay rates were not affected by Experiment or Time (main effects and interactions: all p > 0.05) and, thus, were not further analyzed. Multiple pairwise comparisons of the main effect of Experiment on transient amplitudes indicated that administration of KYN alone, as well as of KYN followed by PF, attenuated nicotine-evoked glutamate release when compared with the data from Experiment 1 (both p < 0.001). In contrast, transient amplitudes evoked by nicotine in Experiment 3 (PF administered prior to KYN) did not differ from the control condition (p > 0.05) and were significantly larger than those seen following KYN alone or PF given after KYN (Experiments 2 and 4; both p < 0.004). These effects and the time course across repeatedly evoked pressure ejections (see Fig. 1) will be detailed in the individual analyses below.
3.3. Experiment 1: nicotine-evoked glutamatergic transients and effects of repeated administration
The analysis of effects of a total of 5 successive pressure ejections of nicotine, distributed over 4.5 h (see Fig. 1) indicated a significant decrease in the peak amplitude of transients (F(4,20) = 4.41, p < 0.05), with post hoc comparisons indicating that the 5th and last pressure ejection produced lower amplitudes when compared with the initial 2 pressure ejections of nicotine (p < 0.05; Fig. 2a). Decay rates remained unaffected by repeated pressure ejections (F(4,20) = 0.61, p > 0.05; Fig. 2b). The nature of the decrease in amplitude of nicotine-evoked glutamatergic transients, observed following lasting anesthesia (>5 h) and after 4 prior pressure ejections will be addressed in Discussion and in the context of the amplitudes of transients evoked by potassium at the end of all experiments. Importantly, even at 270 min after the first pressure ejections, evoked glutamatergic transients remained viable and thus available to determine the duration of effects of KYN in interaction with pre- or post-treatment of PF.
Fig. 2.
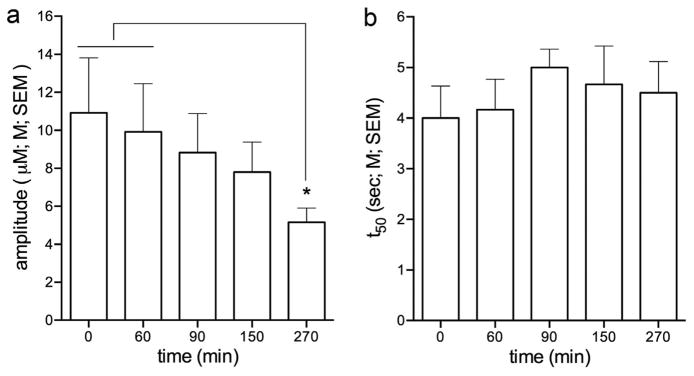
Effect of repeated pressure ejections of nicotine on the amplitudes and decay rates of glutamatergic transients (Experiment 1). a The first administration of nicotine (4 nmol) into mPFC evoked glutamatergic transients with a peak amplitude of 10.92 ± 2.90 μM. Except for the amplitudes evoked by the 5th and last pressure ejection of nicotine, at t = 270 min, amplitudes remained stable over time (see Results for ANOVA). b The average decay rate (t50) of nicotine-evoked glutamate signals at T0 was 4.0 ± 0.63 s and there were no effects of repeated pressure ejections of nicotine on this measure. The amplitudes and decay rates of glutamatergic transients evoked by nicotine at t = 0 did not differ between Experiments 1–4 (see Results; post hoc comparisons indicated in figures: *,**,***: p < 0.05, 0.01, 0.001).
3.4. Experiments 2 and 4: KYN preloading suppresses glutamatergic transients
Compared with transients recorded in the control condition (Experiment 1), administration of KYN (100 mg/kg; i.p.) attenuated the amplitudes of nicotine-evoked glutamatergic transients. Furthermore, the administration of PF 30 min after KYN preloading did not restore KYN-suppressed transients (Fig. 3a and b).
Fig. 3.

Effects of PF administered following KYN (Experiments 2 and 4). a Compared with the amplitudes of glutamatergic transients evoked by nicotine in animals treated with vehicle, systemic KYN administration significantly attenuated amplitudes (see Results for ANOVAs). Post hoc comparisons indicated significant attenuation of the amplitudes of glutamatergic transients for all time points except for the last time point (270 min) when amplitudes in vehicle-treated rats declined and thereby limited the demonstration of an additional dampening by KYN. Administration of the KAT II inhibitor PF after KYN was ineffective. b Representative traces depicting the effect of KYN and PF on nicotine-evoked glutamatergic transients recorded at t = 90 min.
Compared with the results from Experiment 1, systemic KYN, whether followed by PF or the vehicle for PF, attenuated glutamate amplitudes at all time points (p < 0.001) except for the 5th and last (at 270 min) when relatively low baseline amplitudes prevented the detection of significant effects of KYN (p > 0.05). The amplitude reductions caused by KYN, and KYN followed by PF, did not differ significantly (all p > 0.05).
3.5. Experiment 3: administration of PF prior to KYN preloading restores glutamatergic transients
In this experiment, the KATII inhibitor was administered 30 min prior to KYN. For the first two tests of nicotine-evoked glutamate release, at 15 and 45 min after KYN injections, the amplitudes of nicotine-evoked glutamatergic transients were significantly greater than those recorded after KYN administration alone (p < 0.01 and 0.001, respectively) and were statistically similar to the amplitudes observed in the absence of KYN (Fig. 4a and b). The restoring effect of PF was no longer observed 105 min following KYN (or 135 min after administering PF). At this point, transient amplitudes following PF-KYN were indistinguishable from those recorded following the administration of KYN alone.
Fig. 4.
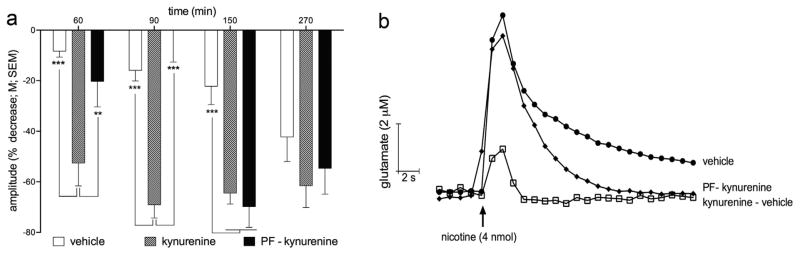
Effects of PF administered prior to KYN (Experiment 3). a Compared with the amplitudes of glutamatergic transients evoked by nicotine in animals treated with KYN alone, PF administered prior to KYN attenuated the effects of KYN at 15 and 45 min after KYN administration. PF was ineffective at rescuing glutamatergic transients at late time points (105 and 225 min after KYN, or 135 and 255 min after the administration of PF). b Representative traces depicting the effect of KYN and PF, given prior to KYN, on nicotine-evoked glutamatergic transients recorded at t = 90 min.
3.6. Nicotine-evoked transients following the administration of PF alone
Experiment 5 addressed the question of whether the administration of the KATII inhibitor alone, that is, in the absence of KYN preloading, affects glutamate transients. Prior to the administration of PF, the amplitudes of nicotine-evoked transients (13.63 ± 1.90 μM) were similar to those seen following the first pressure-ejections of nicotine in Experiments 1–4 (t = 0 in Fig. 1; F(4,21) = 0.39, p > 0.05). Pressure ejections of nicotine at 45 and 75 min following the administration of PF yielded glutamate transient amplitudes that likewise did not differ from these baseline transients F(2,6) = 2.91, p > 0.05).
3.7. Depolarization-evoked glutamatergic transients at the end of experiments
Because of repeated pressure ejections, we were concerned that the low amplitudes observed in response to the final nicotine pressure ejection (at t = 270 min in Fig. 1) reflected a decline in tissue integrity or other effects of repeated pressure ejections. Furthermore, we wished to determine whether the treatments affected the fundamental excitability of glutamatergic terminals, and also verify that sensors maintained their sensitivity following >5 h of implantation into the brain. Potassium pressure ejections evoked transients with amplitudes of 5–6 μM and decay rates of 4– 5 s. KCl-evoked transients did not differ between the Experiments (amplitude: F(3,18) = 0.11, p > 0.05; decay rate: F(3,18) = 0.07, p > 0.05; Fig. 5a and b). Thus, electrodes remained viable and the excitability of the local networks depolarized by potassium was not affected by the prior presence of KYN or PF.
Fig. 5.

Glutamate signals evoked by KCl. Bar charts depicting the average peak amplitudes (a) and decay rates (b) of KCl-evoked glutamatergic transients in Experiments 1–4, and 6 (see Fig. 1). c Representative traces depicting KCl-evoked glutamate signals recorded at t = 285 min for Experiments 1–4 and t = 0 min for Experiment 6. Peak amplitudes and decay rates of KCL-evoked glutamatergic transients did not differ between the Experiments (see Results for ANOVA).
Finally, we compared the KCl-evoked glutamatergic transients in Experiments 1–4 with transients evoked in a separate group of rats (n = 4) that did not receive any other manipulation (KCl administered at t =0; see Experiment 6 in Fig.1). Neither the amplitudes nor the decay rates differed between KCl-evoked transients recorded in Experiments 1–4, and 6 (amplitudes: F(4,21) = 0.26, p > 0.05; decay rate: F(4,21) = 0.23, p > 0.05).
3.8. Brain KYNA levels
Administration of KYN, at the dose used in the electrochemical recording studies, elevated prefrontal KYNA levels approximately 100-fold over baseline values, peaking at 45–90 min after injection of the precursor. A main effect of time (F(9,45) = 12.64, p < 0.001) indicated that even in the presence of PF, KYN-induced KYNA levels were higher than at baseline. Importantly, the analysis of the effects of time and condition (PF versus vehicle) indicated a significant interaction between the two factors (F(9,45) = 5.88, p < 0.001), reflecting significantly higher levels of KYNA in the vehicle condition. Post hoc comparisons indicated significant differences between KYNA levels of the two groups at 60–180 min (all p < 0.05).
4. Discussion
The main result from the present experiments indicates that elevation of brain KYNA levels attenuate nicotine-evoked glutamatergic transients recorded in prefrontal cortex, and that systemic administration of the KAT II inhibitor PF-04859989 restored these transients. This finding suggests that this compound is a promising candidate for testing the hypothesis that lowering KYNA levels benefits the cognitive abilities of a range of patients, including patients with schizophrenia.
Repeated pressure ejections of nicotine reliably evoked glutamatergic transients, except for the 5th and last ejection that was administered at t = 270 min. Because the subsequent test, at t = 285 min, of potassium-evoked glutamatergic transients generated release levels that did not differ from those recorded in animals that did not receive any other manipulation (Experiment 6 in Fig. 1), it is not likely that the diminished amplitude seen after the 5th pressure ejection of nicotine was due to decline in tissue integrity or of the release capacity of glutamatergic terminals. Rather, this late decrease in the amplitudes of glutamatergic transients may have been due to repeated nicotine-induced down-regulation of nAChRs (e.g., Papke et al., 2009).
KYN administration lowered the amplitude of glutamatergic transients by 60–70%. At t = 60 and t = 90, administration of PF-04859989 restored amplitudes to levels statistically similar to nicotine-evoked transients at baseline. PF was no longer effective 135 min following administration. This finding would not be predicted based on the more persistent attenuation of KYN-induced elevations of KYNA observed in our microdialysis experiments (Fig. 6; see also the comparable effects of KYN on KYNA levels in Alexander et al., 2012). Note also that KYNA levels were not fully suppressed in the presence of PF. Thus, the relationships between chronically elevated KYNA levels and KYNA-induced nAChR antagonism remain unclear. KYNA brain levels per se may not be sufficient to predict the status of the multiple receptors antagonized by this compound (see also Albuquerque and Schwarcz, 2013). Persistent with this observation, it has been previously observed that the activity of KAT II accounts on partly for KYNA synthesis (reviewed in Schwarcz et al., 2012).
Fig. 6.

Levels of kynurenic acid (KYNA), measured in the prelimbic cortex of rats, using in vivo microdialysis. Vehicle or PF (32 mg/kg; s.c.) was administered prior to KYN (KYN; 100 mg/kg; i.p.; see arrows). Repeated measures ANOVA and post hoc comparisons indicated that at 60–180 min, KYNA levels were significantly higher in rats given vehicle when compared to the KATII inhibitor (absolute KYNA levels [M ± SEM: baseline: vehicle + KYN: 2.17 ± 0.06 nM; PF + KYN: 1.77 ± 0.16 nM; average 60– 180 min: vehicle + KYN: 164.48 ± 10.09 nM; PF + KYN: 30.53 ± 1.64 nM).
The present experiments were not designed to identify the receptor-based mechanisms via which elevated KYNA levels suppressed nicotine-evoked glutamatergic transients. Although blockade of α7 nAChR represents a plausible hypothesis (references in Introduction), we previously reported that nicotine-evoked glutamatergic transients were unaffected by knockout of this nAChR (Parikh et al., 2010). It is more likely, therefore, that elevated KYNA levels suppressed nicotine-evoked transients by interfering with microcircuitry that includes ionotropic glutamate receptors and that links nicotinergic stimulation of other nAChR subtypes with the regulation of glutamate release from thalamic afferents.
It will be important to demonstrate the efficacy of KAT II inhibitors in animal models exhibiting elevated levels of KYNA as a component of a disease process. KYN “preloading” is a convenient experimental approach to demonstrate the impact of increased KYNA levels on neuronal and behavioral functions (references in Introduction). Furthermore, as indicated by the absence of effects of PF in animals not pretreated with KYN, it is not clear whether endogenous KYNA levels in the brain of intact rats are present at levels sufficient to block cholinergic or glutamatergic receptors and to demonstrate efficacy of KATII inhibitors. However, such “preloading” may not fully reproduce important alterations in the kynurenine pathway that are associated with chronic, pathophysiological elevation of KYNA levels. For example, several animal models of schizophrenia have focused on disturbing the maturation of mesolimbic-telencephalic circuitry and demonstrated important post-pubertal impairments modeling aspects of schizophrenia (Lipska et al., 2002; Alexander et al., 2009; Brooks et al., 2012). The status of the kynurenine pathway and brain levels of KYNA in such models appears unknown. If KYNA levels are elevated in such models, they could be employed to test of the beneficial effects of KAT II inhibitors. Such experiments would also provide important information concerning the efficacy of KATII inhibitors when given chronically to animals with elevated brain levels of KYNA.
Acknowledgments
The authors thank Drs. Brian M. Campbell, Christine A. Strick, Kathryn A. Welch, and Aarti Sawant-Basak (all at Pfizer) for their assistance in the preparation of this manuscript.
Footnotes
Financial disclosures
This study was support by a Grant from Pfizer Inc. Martin Sarter has received honoraria from Pfizer Inc. during the past 3 years.
References
- Akagbosu CO, Evans GC, Gulick D, Suckow RF, Bucci DJ. Exposure to kynurenic acid during adolescence produces memory deficits in adulthood. Schizophr Bull. 2010;38:769–778. doi: 10.1093/schbul/sbq151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albuquerque EX, Schwarcz R. Kynurenic acid as an antagonist of α7 nicotinic acetylcholine receptors in the brain: facts and challenges. Biochem Pharmacol. 2013;85:1027–1032. doi: 10.1016/j.bcp.2012.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander KS, Brooks JM, Sarter M, Bruno JP. Disruption of mesolimbic regulation of prefrontal cholinergic transmission in an animal model of schizophrenia and normalization by chronic clozapine treatment. Neuropsychopharmacology. 2009;34:2710–2720. doi: 10.1038/npp.2009.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander KS, Pocivavsek A, Wu -Q, Pershing L, Schwarcz R, Bruno P. Early developmental elevations of brain kynurenic acid impair cognitive flexibility in adults: reversal with galantamine. Neuroscience. 2013;238:19–28. doi: 10.1016/j.neuroscience.2013.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander KS, Wu HQ, Schwarcz R, Bruno JP. Acute elevations of brain kynurenic acid impair cognitive flexibility: normalization by the alpha7 positive modulator galantamine. Psychopharmacology. 2012;220:627–637. doi: 10.1007/s00213-011-2539-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amori L, Wu -Q, Marinozzi M, Pellicciari R, Guidetti P, Schwarcz R. Specific inhibition of kynurenate synthesis enhances extracellular dopamine levels in the rodent striatum. Neuroscience. 2009;159:196–203. doi: 10.1016/j.neuroscience.2008.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks JM, Pershing ML, Thomsen MS, Mikkelsen JD, Sarter M, Bruno JP. Transient inactivation of the neonatal ventral hippocampus impairs attentional set-shifting behavior: reversal with an α7 nicotinic agonist. Neuropsychopharmacology. 2012;37:2476–2486. doi: 10.1038/npp.2012.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chess AC, Simoni K, Alling E, Bucci DJ. Elevations of endogenous kynurenic acid produce spatial working memory deficits. Schizophr Bull. 2007;33:797–804. doi: 10.1093/schbul/sbl033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chess AC, Landers AM, Bucci DJ. L-kynurenine treatment alters contextual fear conditioning and context discrimination but not cue-specific fear conditioning. Behav Brain Res. 2009;201:325–331. doi: 10.1016/j.bbr.2009.03.013. [DOI] [PubMed] [Google Scholar]
- Dickinson JA, Kew JN, Wonnacott S. Presynaptic alpha 7- and beta 2-containing nicotinic acetylcholine receptors modulate excitatory amino acid release from rat prefrontal cortex nerve terminals via distinct cellular mechanisms. Mol Pharmacol. 2008;74:348–359. doi: 10.1124/mol.108.046623. [DOI] [PubMed] [Google Scholar]
- Dounay AB, Anderson M, Bechle BM, Evrard E, Gan X, Kim JY, McAllister LA, Pandit J, Rong S, Salafia MA, Tuttle JB, Zawadzke LE, Verhoest PR. PF-04859989 as a template for structure-based drug design: identification of new pyrazole series of irreversible KAT II inhibitors with improved lipophilic efficiency. Bioorg Med Chem Lett. 2013;23:1961–1966. doi: 10.1016/j.bmcl.2013.02.039. [DOI] [PubMed] [Google Scholar]
- Erhardt S, Schwieler L, Nilsson L, Linderholm K, Engberg G. The kynurenic acid hypothesis of schizophrenia. Physiol Behav. 2007;92:203–209. doi: 10.1016/j.physbeh.2007.05.025. [DOI] [PubMed] [Google Scholar]
- Guillem K, Bloem B, Poorthuis RB, Loos M, Smit AB, Maskos U, Spijker S, Mansvelder HD. Nicotinic acetylcholine receptor β2 subunits in the medial prefrontal cortex control attention. Science. 2011;333:888–891. doi: 10.1126/science.1207079. [DOI] [PubMed] [Google Scholar]
- Hascup KN, Hascup ER, Pomerleau F, Huettl P, Gerhardt GA. Second-by-second measures of L-glutamate in the prefrontal cortex and striatum of freely moving mice. J Pharmacol Exp Ther. 2008;324:725–731. doi: 10.1124/jpet.107.131698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselmo ME, Sarter M. Modes and models of forebrain cholinergic neuromodulation of cognition. Neuropsychopharmacology. 2011;36:52–73. doi: 10.1038/npp.2010.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilmas C, Pereira EF, Alkondon M, Rassoulpour A, Schwarcz R, Albuquerque EX. The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: physiopathological implications. J Neurosci. 2001;21:7463–7473. doi: 10.1523/JNEUROSCI.21-19-07463.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe MW, Berry AS, Francois F, Gilmour G, Carp JM, Tricklebank M, Lustig C, Sarter M. Prefrontal cholinergic mechanisms instigating shifts from monitoring for cues to cue-guided performance: converging electrochemical and fMRI evidence from rats and humans. J Neurosci. 2013;33:8742–8752. doi: 10.1523/JNEUROSCI.5809-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konradsson-Geuken A, Gash CR, Alexander K, Pomerleau F, Huettl P, Gerhardt GA, Bruno JP. Second-by-second analysis of alpha 7 nicotine receptor regulation of glutamate release in the prefrontal cortex of awake rats. Synapse. 2009;63:1069–1082. doi: 10.1002/syn.20693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konradsson-Geuken A, Wu HQ, Gash CR, Alexander KS, Campbell A, Sozeri Y, Pellicciari R, Schwarcz R, Bruno JP. Cortical kynurenic acid bi-directionally modulates prefrontal glutamate levels as assessed by micro-dialysis and rapid electrochemistry. Neuroscience. 2010;169:1848–1859. doi: 10.1016/j.neuroscience.2010.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambe EK, Picciotto MR, Aghajanian GK. Nicotine induces glutamate release from thalamocortical terminals in prefrontal cortex. Neuro-psychopharmacology. 2003;28:216–225. doi: 10.1038/sj.npp.1300032. [DOI] [PubMed] [Google Scholar]
- Linderholm R, Skogh E, Olsson K, Dahl -L, Holtze M, Engberg G, Samuelsson M, Erhardt S. Increased levels of kynurenine and kynurenic acid in the CSF of patients with schizophrenia. Schizophr Bull. 2012;38:426–432. doi: 10.1093/schbul/sbq086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipska BK, Halim ND, Segal PN, Weinberger DR. Effects of reversible inactivation of the neonatal ventral hippocampus on behavior in the adult rat. J Neurosci. 2002;22:2835–2842. doi: 10.1523/JNEUROSCI.22-07-02835.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingstone PD, Dickinson JA, Srinivasan J, Kew JN, Wonnacott S. Glutamate-dopamine crosstalk in the rat prefrontal cortex is modulated by alpha7 nicotinic receptors and potentiated by PNU-120596. J Mol Neurosci. 2010;40:172–176. doi: 10.1007/s12031-009-9232-5. [DOI] [PubMed] [Google Scholar]
- Nilsson LK, Linderholm KR, Engberg G, Paulson L, Blennow K, Lindström LH, Nordin C, Karanti A, Persson P, Erhardt S. Elevated levels of kynurenic acid in the cerebrospinal fluid of male patients with schizophrenia. Schizophr Res. 2005;80:315–322. doi: 10.1016/j.schres.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Papke RL, Kem WR, Soti F, Lopez-Hernandez GY, Horenstein NA. Activation and desensitization of nicotinic alpha7-type acetylcholine receptors by benzylidene anabaseines and nicotine. J Pharmacol Exp Ther. 2009;329:791–807. doi: 10.1124/jpet.108.150151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh V, Ji J, Decker MW, Sarter M. Prefrontal beta2 subunit-containing and alpha7 nicotinic acetylcholine receptors differentially control glutamatergic and cholinergic signaling. J Neurosci. 2010;30:3518–3530. doi: 10.1523/JNEUROSCI.5712-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh V, Man K, Decker MW, Sarter M. Glutamatergic contributions to nicotinic acetylcholine receptor agonist-evoked cholinergic transients in the prefrontal cortex. J Neurosci. 2008;28:3769–3780. doi: 10.1523/JNEUROSCI.5251-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons CG, Danysz W, Quack G, Hartmann S, Lorenz B, Wollenburg C, Baran L, Przegalinski E, Kostowski W, Krzascik P, Chizh B, Headley PM. Novel systemically active antagonists of the glycine site of the N-methyl-D-aspartate receptor: electrophysiological, biochemical and behavioral characterization. J Pharmacol Exp Ther. 1997;283:1264–1275. [PubMed] [Google Scholar]
- Pellicciari R, Rizzo R, Costantino G, Marinozzi M, Amori L, Guidetti P, Wu HQ, Schwarcz R. Modulators of the kynurenine pathway of tryptophan metabolism: synthesis and preliminary biological evaluation of (S)-4-(Ethyl-sulfonyl)benzoylalanine, a potent and selective kynurenine aminotransferase II (KAT II) inhibitor. Chem Med Chem. 2006;1:528–531. doi: 10.1002/cmdc.200500095. [DOI] [PubMed] [Google Scholar]
- Potvin S, Stip E, Sepehry AA, Gendron A, Bah R, Kouassi E. Inflammatory cytokine alterations in schizophrenia: a systematic quantitative review. Biol Psychiatry. 2008;63:801–808. doi: 10.1016/j.biopsych.2007.09.024. [DOI] [PubMed] [Google Scholar]
- Rousseau SJ, Jones IW, Pullar IA, Wonnacott S. Presynaptic alpha7 and non-alpha7 nicotinic acetylcholine receptors modulate [3H]d-aspartate release from rat frontal cortex in vitro. Neuropharmacology. 2005;49:59–72. doi: 10.1016/j.neuropharm.2005.01.030. [DOI] [PubMed] [Google Scholar]
- Sarter M, Parikh V, Howe WM. nAChR agonist-induced cognition enhancement: integration of cognitive and neuronal mechanisms. Biochem Pharmacol. 2009;78:658–667. doi: 10.1016/j.bcp.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sceniak MP, MacIver MB. Cellular actions of urethane on rat visual cortical neurons in vitro. J Neurophysiol. 2006;95 (95):3865–3874. doi: 10.1152/jn.01196.2005. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ. Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci. 2012;13:465–477. doi: 10.1038/nrn3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarcz R, Rassoulpour A, Wu HQ, Medoff D, Tamminga CA, Roberts RC. Increased cortical kynurenate content in schizophrenia. Biol Psychiatry. 2001;50:521–530. doi: 10.1016/s0006-3223(01)01078-2. [DOI] [PubMed] [Google Scholar]
- Trecartin KV, Bucci DJ. Administration of kynurenine during adolescence, but not during adulthood, impairs social behavior in rats. Schizophr Res. 2011;133:156–158. doi: 10.1016/j.schres.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wonodi I, Schwarcz R. Cortical kynurenine pathway metabolism: a novel target for cognitive enhancement in schizophrenia. Schizophr Bull. 2010;36:211–218. doi: 10.1093/schbul/sbq002. [DOI] [PMC free article] [PubMed] [Google Scholar]