

Abstract
AphidBase is a centralized bioinformatic resource that was developed to facilitate community annotation of the pea aphid genome by the International Aphid Genomics Consortium (IAGC). The AphidBase Information System designed to organize and distribute genomic data and annotations for a large international community was constructed using open source software tools from the Generic Model Organism Database (GMOD). The system includes Apollo and GBrowse utilities as well as a wiki, blast search capabilities and a full text search engine. AphidBase strongly supported community cooperation and coordination in the curation of gene models during community annotation of the pea aphid genome. AphidBase can be accessed at http://www.aphidbase.com.
Introduction
High quality genome sequence is a prerequisite for whole genome analyses but further, robust and complete annotations are essential for a genome to be fully utilized by the scientific community. Genome annotation involves mapping features such as protein coding genes and their multiple mRNAs, pseudogenes, transposons, repeats, non-coding RNAs, SNPs as well as regions of similarity to other genomes onto the genomic scaffolds. Many of these features can be automatically predicted by sophisticated software packages based on sequence or structure comparisons.
The identification of protein-coding genes is widely considered to be critical to understanding the biology of an organism (Stein, 2001). Gene prediction programs identify protein-coding genes using either ab initio prediction, evidence-based prediction, or a combination of the two methods. Evidence-based prediction programs such as Augustus, Fgenesh++ or the NCBI RefSeq pipeline are generally considered most reliable because they are better able to characterize untranslated regions (UTRs) and alternative splicing especially when cDNA sequences representing full-length mRNAs or large numbers of ESTs are available (Brent, 2008). However, in many cases gene predictions require evaluation by specialist biocurators of a gene family or a pathway.
One of the most challenging aspects of dispersed community annotation is the need to maintain consistent data formats, and to minimize the potential for duplicated annotation made simultaneously by two different annotators (Elsik et al., 2006). This requires annotation tools, standardized methods, oversight by expert curators and a combination of social infrastructure, tool development, training and feedback (Howe et al., 2008). The main mistake during manual annotation from previous genome projects was allowing the submission of incomplete data and data inconsistent with itself. This resulted in annotated genes with missing co-ordinates, protein and mRNA sequences that did not match, and a number of other issues that pollute the databases with incorrect information. To remedy this problem, VectorBase asked submitters to supply data in a spreadsheet format including gene prediction and gene symbol descriptions (Lawson et al., 2009), while BeeBase developed procedures for handling community-annotated gene models, that included mapping, checking for errors and redundancy, assigning identifiers, and incorporating them into the database (Elsik et al., 2006). Here we adopted Apollo (Lewis et al., 2002), a software specialized in the editing of annotation. Apollo provides a graphical, straightforward and controlled approach for manual curation.
The role of the biocurators is not only to inspect and correct automatically predicted gene structures and proteins, but also to add value by connecting information from different sources in a coherent and accessible way (Howe et al., 2008; Elsik et al. 2006). Assembling and curating the datasets generated during annotation of a genome is a labour-intensive and relatively slow process (Wilming et al., 2008) but annotation can be spread over a large number of people to accelerate the process. The efforts of a strong, organized, motivated and voluntary community with many researchers specializing in a variety of gene families of interest, can greatly improve the annotation of a genome sequence; these criteria were met by the International Aphid Genomics Consortium (IAGC), whose goal is to develop genomic resources for aphids. The IAGC recently supervised the sequencing, assembly and analysis of the first aphid genome, that of the pea aphid Acyrthosiphon pisum (IAGC, 2009). Members of the IAGC represent a large community of aphid specialists all over the world collaborating on the analysis of the pea aphid genome. The annotation datasets generated by the IAGC needed an official, centralized repository providing worldwide access, that is now provided by AphidBase.
AphidBase, formerly a web application for the analysis of Aphids ESTs (Gauthier et al., 2007), has been upgraded to a comprehensive genome information resource dedicated to aphids. Incorporating the best features of other eukaryotic model organism databases – such as WormBase (Rogers et al., 2008), Flybase (Wilson et al. 2008) and VectorBase (Lawson et al., 2009), AphidBase provides detailed information about the aphid and its scientific community, includes a genome browser for visualizing genome annotation, and robust search capabilities. AphidBase is also a central node for communication between the aphid community with links to a collaborative wiki and a specialized database on the metabolic networks of aphids and their symbionts (ApicyCyc, http://pbil.univ-lyon1.fr:2555/ACYPI/) and a comprehensive phylogenomic database for the pea aphid (PhylomeDB, Huerta-Cepas et al., 2008).
Results
Manual curation
A subset of 10,248 genes predicted by Gnomon were strongly supported by biological evidence and have been inserted in RefSeq, the NCBI database Reference Sequences database (Pruitt et al., 2009). The high quality of these RefSeq predictions allowed their inclusion in the first Acyrthosiphon pisum reference set (Acyr 1.0). When no RefSeq gene was available, Glean (Elsik et al., 2007), a tool that integrates gene predictions from distinct softwares (listed in Table 1) was used to create consensus gene models. The first official reference gene set of Acyrthosiphon pisum, is composed of 34,603 automatically predicted genes, corresponding to 34,821 transcripts and proteins (IAGC, 2009). Following assembly of this official gene set, the IAGC commenced manual curation for the appraisal of this set.
Table 1. The content of AphidBase.
| Category | Software | Results | Comments |
|---|---|---|---|
| Protein coding gene predictions | Acypi 1.0 (reference annotation) | 34,603 genes | This first reference incorporates set is a subset of the Gnomon genes predictions strongly supported by biological evidence and inserted in RefSeq (Pruitt et al., 2009). This subset has been enriched with Glean predictions that do not overlap with RefSeq genes |
| Gnomon | 37,994 genes | Gnomon (http://www.ncbi.nlm.nih.gov/projects/genome/guide/gnomon.shtml), is the NCBI gene prediction program. | |
| Glean | 36,606 genes | Glean (Elsik et al., 2007) is a software that computes consensus gene predictions. We input Augustus, Fgenesh++, Gnomon, GeneID Genscan SNAP and Maker (Cantarel et al. 2008) gene models. | |
| Augustus | 34,677 genes | Augustus (Stanke et al., 2006) predictions incorporate extrinsic evidence with sequence intrinsic evidence. Extrinsic evidence was taken from GMAP (Wu & Watanabe 2005) alignments of Acyrthosiphon pisum ESTs and alignments of proteins from 3 other insect species (Nasonia vitripennis, Tribolium castaneum and Daphnia). | |
| GeneID | 55,644 genes | GeneID (Parra et al. 2000) was applied to the A.. pisum genomic sequences masked against Repbase Repeat database invertebrate division (Jurka et al., 2005). | |
| Fgenesh++ | 26,773 genes | The fgenesh++ or fgenesh pipeline (http://www.softberry.com) is a combination of two rounds of the ab initio algorithm fgenesh and two rounds of fgenesh+ which takes into account homologous protein alignments. | |
| Maker | 22,738 genes | MAKER aligns ESTs and proteins to a genome, produces ab initio gene predictions, and automatically synthesizes these data into gene annotations (Cantarel et al. 2008). | |
| Genscan | 32,322 genes | Genscan is an ab initio gene prediction software (Burge & Karlin, 1997). | |
| Non coding RNA predictions | miRNA finder | 189 miRNAs | miRNA were identified by coupling a computational approach using sequence similarity with known miRNA genes and training and structure recognition algorithms to biological validation by high-throughput sequencing (Legeai F, Rizk G, Walsh T, Edwards O, Gordon K, Lavenier D, Leterme N, Méreau A, Nicolas J, Tagu D, Jaubert-Possamai S. microRNAs of the insect crop pest Acyrthosiphon pisum, in preparation). |
| tRNAscan-SE | 348 tRNAs | tRNAscan-SE (Lowe & Eddy, 1997) is a widely used software for tRNA identification. | |
| Similarities to A. pisum transcripts | EST mapping | 235,621 alignments | The ESTs were extracted from the NCBI nucleotide database using Entrez and aligned using sim4 (Florea et al., 1998). |
| Unigenes mapping | 27,427 alignments | The unigenes were assembled using the tgicl software (http://compbio.dfci.harvard.edu/tgi/software/) and the resulting unigenes were mapped on the genome using sim4 (Florea et al. 1998). | |
| Similarities to transcripts from other aphid species | tblastn or tblastx | 30,840 alignments | Putative coding sequences from EST of other aphid species (Toxoptera citricida, Myzus persicae, Aphis gossypii, Rhopalosipum padi) were predicted by Frame D (Schiex et al., 2003) and used with tblasx to directly compare to the genome sequence |
| Similarities to protein databanks | Blastx vs Flybase | 27,900 alignments | Blastx against Flybase, Drosophila melanogaster release 5.6 |
| Blastx vs Beebase | 14,480 alignments | Blastx against Beebase Apis mellifera protein database release 1 | |
| Blastx vs Uniprot | 194,290 alignments | Blastx against Uniprot trembl and swissprot release 14.2 |
Manual annotation of the pea aphid genome was completed by a group of 96 people from 10 countries self-organized into 27 annotation groups of 1 - 30 individuals (Table 2). Forty-nine members of this group of expert biologists appraised the automatic annotations and in doing so corrected genes boundaries, found new genes and increased the information content of gene models by specifying functional characteristics, or simply by delivering comments or evaluations. To facilitate the process of manual curation, Apollo was set up in AphidBase. The Apollo genome editor is a Java application for browsing and annotating genomic sequences. It offers many functionalities facilitating the correction of gene structures and allowing users to probe, manipulate and alter the interpretation of gene models. Within Apollo, annotations can be created, deleted, merged, split, classified and commented on. For example, one can easily locate and correct incorrect splice sites or start/stop codons, classify a gene as a pseudogene, and even create a new alternatively spliced RNA. Using AphidBase's Apollo configuration, a curator validates or modifies a reference annotation or creates new annotations by a simple drag and drop of any form of gene evidence (predictions) from one panel to the other. Apollo then automatically generates a unique identifier. As a result, the curators are not directly modifying the reference set but rather append a new annotation layer. Finally, at each release, curated genes automatically replace their previous referenced versions. This process does not require reviewing or double-checking; however, author names are attached to each annotated gene in order to facilitate collaborative work.
Table 2. Twenty-seven annotation groups, self-organized by members of the International Aphid Genomics Consortium via an interactive web form.
| Group | No. Members |
|---|---|
| Bacterial genes in the aphid genome | 31 |
| Chromatin remodelling | 3 |
| Circadian rhythms | 7 |
| Cuticular proteins | 5 |
| Development | 16 |
| DNA methyltransferase | 3 |
| Germ line determination | 9 |
| Ion channels | 2 |
| Juvenile Hormone related | 6 |
| Meiosis and Mitosis | 5 |
| Metabolism | 18 |
| microRNAs | 5 |
| Mitochondrial genes in the aphid genome | 4 |
| Neuropeptides | 3 |
| Odorant Binding Proteins | 6 |
| Olfactory Gustatory Receptors | 3 |
| Peritrophic membrane components | 2 |
| Pesticide resistance | 8 |
| Proteases and Carbohydrases | 12 |
| Ribosomal proteins | 5 |
| Sex Determination | 6 |
| Small noncoding RNA machinery | 4 |
| Stress, Immunity and Defense | 17 |
| Telomerase | 1 |
| Transport | 8 |
| Transposable Elements | 8 |
| Virus Transmission, Transcytosis | 9 |
Despite development of the Apollo bioinformatic environment, annotating genes remains a laborious process that requires rigor, to this end the IAGC developed recommended practices and standardized procedures that were published by IAGC on The Aphid Genomics Collaboration Wiki (https://dgc.cgb.indiana.edu/display/aphid/Annotation+Guidelines, https://dgc.cgb.indiana.edu/display/aphid/Manual+annotation+using+Apollo).
Because, curators are naming genes manually, and because names are most useful when they are descriptive, particular attention was made to clarify aspects of nomenclature. Two types of nomenclature are associated with a given Acyrthosiphon pisum gene, the gene symbol (a symbol or abbreviation) and the full gene name (gene description). When possible, and if the orthology is clear, the drosophila or human gene names or descriptions have been used by the curator because they are controlled by the Flybase and HUGO consortiums, respectively.
Nine months after the beginning of the manual curation process, 2,010 genes had been manually annotated. Among these manually annotated genes, 1,536 genes were tagged as “finished”, i.e. their current structures were considered correct according to the available biological data and current knowledge. While most of these genes correspond to a RefSeq prediction, 50 (3.3%) genes were not present in the first reference set (Table 3). Within annotation groups, curators predominantly investigated genes with at least some biological evidence and similarity with known proteins. The fact that generation of the RefSeq set requires biological evidence explains the over-representation of genes having a RefSeq source in the curated set. Only 19% of RefSeq genes, compared to 80% of the Glean predicted genes, were hand-corrected by annotators. This difference in the frequency of hand-correction reflects a lower confidence level on predictions when no biological evidence was available. In summary, about 28% of the predictions needed correction; a rate similar to that associated with the best methods used on the human genome (Guigó et al. 2006).
Table 3. Curated gene statistics according to their origin in the reference prediction set.
Appraised genes are those examined by a biocurators. Corrected genes are those for which biocurators made changes to the automated gene model. Ratio of corrected genes to curated genes is shown in parentheses. Merged genes includes annotated genes that joining 2 or 3 Glean or RefSeq predictions. Resulting number of annotated genes is the final sets of improved genes.
| RefSeq Models | Glean Models | Total | |
|---|---|---|---|
| Number of appraised genes | 1,286 | 218 | 1,504 |
| Number of corrected genes | 250 (19.4%) | 175 (80.3%) | 425 (28.2%) |
| Number of merged genes | 10* | 26** | 36*** |
| Resulting number of annotated genes | 1 281 | 205 | 1486 |
1 RefSeq predictions was merged with another RefSeq prediction
9 Glean predictions was merged with another RefSeq prediction
8 Glean predictions were merged with RefSeq predictions
AphidBase
AphidBase (http://www.aphidbase.com) is an information system set up to safely centralize, manage, mine, disseminate and promulgate data generated by the IAGC. This Information System is based on GMOD (http://www.gmod.org), the Generic Model Organism Database Project, a largely open source project aimed at developing a complete set of software packages for creating and administering the genome database of a model organism. Among others, components of the GMOD project include a genome browser and editor (GBrowse (Stein et al., 2002) and Apollo (Lewis et al., 2002), a robust database scheme Chado (Mungall et al., 2007), as well as biological ontology tools, and a set of standard operating procedures. Implementation of GMOD, a system that is widely used in the bioinformatics community and thus, well supported and documented, gave us the opportunity to simply set up integrated but flexible solutions to meet the majority of our needs for data storage, controlled vocabulary, visualization, and exploration.
A Gbrowse genome browser directly connected to the Chado database offers a large number of configurable tracks that are listed in Table 1. Each Gbrowse detailed feature contains links to other sources of information. For example, each gene is directly connected to the following: (1) its NCBI Entrez page allowing the gathering of functional information and a link to BLink, the NCBI Blast results visualizer tool; (2) its phylogenetic tree established by PhylomeDB (Huerta-Cepas et al., 2008) and (3) AcypiCyc, a metabolism BioCyc database for Acyrthosiphon pisum (Vellozo et al. manuscript in preparation).
AphidBase also provides a configurable Blast search page permitting comparison to A. pisum sequence databanks (reads, scaffolds, official gene and protein sets, predictions and cDNAs). When possible, in order to facilitate web navigation, a reported hit is linked to its genome location in Gbrowse or to its detailed page resources (e.g. NCBI Entrez page or FlyBase gene report). In parallel, a full text search engine monitored by Lucene (http://lucene.apache.org/) allows a rapid keyword search among the gene annotations or the description of their homologous proteins. Finally, AphidBase is a web portal used to centralize indispensable news and documentation about the IAGC, and includes a download area for large sequences and annotations retrievals.
Community organization
In order to facilitate manual annotation and communication among annotators from multiple labs, organizations and even countries, we made use of a range of collaboration tools including an email listserve, teleconferencing, interactive webforms, an annotation workshop, and a collaboration wiki.
The aphidgenomics electronic mailing list (Thomas, 1986), established in 2003, has provided a forum to raise and discuss annotation issues, describe and discuss aphid biology and coordinate writing of the main genome paper. All messages exchanged on the list are automatically archived online and accessible to any listmember at any time via web browsers.
A wiki is a web social software that facilitates online communication (Stein, 2008). The IAGC Collaboration Wiki (https://dgc.cgb.indiana.edu/display/aphid/Introduction) served three purposes. First, it provided an information center where all sorts of information to assist annotators, including annotation guidelines, nomenclature instructions, training resources and important announcements from the IAGC Steering Committee were disseminated to the community. For example, presentation materials used in the annotation workshop were made downloadable from the IAGC Collaboration Wiki to disseminate important guidelines to annotators including those who were not able to attend the annotation workshop. Second, the wiki played a role complementary to the electronic mailing list in that a discussion that would normally run over many back and forth email exchanges could be summarized on a single wiki page. Third, the IAGC Collaboration Wiki served an on-line workspace, where any member could contribute to the community in a decentralized way. Within the wiki, each annotation group has own collaboration site, allowing multiple members of the group to edit the page simultaneously ensuring that the information is current and accurate. Finally, the IAGC Collaboration Wiki is equipped with access control, allowing for restricted access to specific parts of the wiki, thereby facilitating the sharing of prepublication data and free discussions amongst collaborators.
Regular, usually weekly, teleconferences have facilitated progress on all phases of the project. Prior to the workshop weekly calls were restricted to members of the workshop organization committee, representatives from Baylor College of Medicine and NCBI, and a core set of IAGC members. During this first phase, weekly calls were focused on workshop planning and infrastructure development. Following our July 2008 workshop weekly teleconference calls shifted their focus to the biology coming out of our annotation efforts. Each week, two annotation groups were assigned to presenting their results using a slide presentation they had disseminated to the community via the email listserve. Finally, during the writing phase of the project the weekly teleconferences have proved invaluable in facilitating discussion of our publication plans, preparation of the manuscript including the figures and tables and in generally maintaining cohesion and keeping the community on task. Despite the logistical challenges of staging weekly teleconferences in a community that is spread across the globe, there is little doubt that these calls have been a valuable and essential part of our community-based annotation of the pea aphid genome.
Possibly the most important collaborative tool used to facilitate community annotation of the pea aphid genome was a two-day annotation workshop. When planning the workshop we set out with the goal of equipping the novice annotator with the skills and tools they would need to annotate their genes of interest. Talks almost exclusively focused on how to use our annotation tools, such as GBrowse, Apollo and the wiki but also included a “mini-conference”, where a handful of people who had made progress on annotation prior to the workshop were invited to communicate their research in short talks. In addition to the Apollo and GBrowse lectures, we had hands-on workshop sessions that small groups of up to 10 people could sign-up for to learn to these tools. One of the most important aspects of the workshop was the scheduled meeting of annotation groups. These annotation group meetings provided an opportunity for many members of the community to meet their collaborators for the first time. Most annotation groups during this time discussed their gene lists and set in place firm goals for their annotation and individual task assignments. A final and important point of the workshop was that it promoted social connection among members of the IAGC. Despite the progress of various online communication tools discussed above, face-to-face communication plays an important role in building an active and organized scientific community.
Future evolution of AphidBase
Annotation of a complete genome provides an opportunity to unite the strengths of a diverse community and yet the success of such a project depends critically on a genome information system, such as AphidBase. The current challenge for AphidBase is to implement and/or develop tools to remain functional and accessible as new aphid data accumulates so as to enable the IAGC to make rapid pure and applied scientific advances with these data.
Although the gene curation process is ongoing, we already noticed that only almost 20% of the inspected genes with cDNA coverage or protein similarities have been manually refined, while about 80 % of the genes from the Glean reference set (i.e. the ab initio gene models), required manual curation to improve their automatically predicted structures. In conclusion, a single and automated annotation of genome is not acceptable when only a few transcription evidence is available and when the well annotated genome of a closely related species is lacking. Hopefully, new and cheaper technologies are producing more and more sequence reads of either cDNAs (RNA-Seq) or whole genomes. Taking into account new future complete genomes and libraries of millions of cDNA sequences will improve the annotation quality, but demand computer and informatic platforms able to deal with such large amounts of data. In this context, new automatic procedures are now able to incorporate the product of massive scales cDNA sequencing projects to correct gene models or to predict more genes or splice variants (Wang et al., 2008, Denoeud et al., 2008. The AphidBase strength will lie in its ability of frequently upgrading gene models by using these strategies combined with the effort made by its scientific community for appraising gene models with regard of new evidences, the re-annotation process impacting a manually curated genes implying it supervision by experts.
Thereafter, the future of AphidBase will be strongly affected by the quantity of its inherent biological data. Pursuing this ambition, AphidBase is working to improve and automatically update gene annotations by adding functional tips such as a gene belonging to a protein family, its known domains, its classification under a Gene Ontology term, or even when it is possible inference of its protein structure. Moreover, AphidBase is expanding annotated features and will soon integrate for example, transposable elements predicted by the Repet pipeline (Quesneville et al., 2005), putative SNPs derived from the comparisons of ESTs, microsatellites, or new non coding RNAs.
The large acceptance of AphidBase will also depend on its panel of given functionalities and tools. For example, one of the outstanding features of the pea aphid genome discovered during the community annotation process is a very high level of gene duplications (IAGC, 2009). So easy navigation between paralogous genes and tools for graphically comparing their surroundings such as Synview (Wang et al., 2006) or Gbrowse_Syn (http://gmod.org/wiki/GBrowse_syn) appear to be the key means for increasing the knowledge of the evolution of the aphid. In addition, Biomart (Smedley et al., 2009) would be a convincing and efficient solution to help AphidBase users to perform advanced and complex queries on biological data sources, regardless their geographical location through a single web interface. Finally, we are now implementing functional web pages about gene, transcript, peptide or ontology terms, which will summarize available information and expertise at a glance.
Finally, AphidBase will also be strongly affected by the quality of its data; in other words in the level of human curation involved in the procedure including expertise or literature references. Consequently, implementing a wiki for gathering functional annotation appears to be a good solution due to its easiness and availability, wide scope and flexibility (Salzberg 2007; Mons et al., 2008). However, wikis are still lacking of integration with database such as Chado, or any other data warehouse system.
Materials and Methods
Aphidbase
Aphidbase is a Chado database v0.5. Various softwares were used and several bioinformatics groups were engaged in annotation of the pea aphid genome sequence (IAGC, 2009). BioPerl (http://www.bioperl.org) was used to parse and transform all data files into the standard GFF3 format (http://www.sequenceontology.org/gff3.shtml) required by the Chado database loader. As a result Gbrowse directly connected to the database offers a large number of configurable tracks (Table 1).
Apollo
Apollo is connected to a duplicate of the public AphidBase Chado database (Figure 1), enabling users to directly load and save their modifications and editions to this database.
Figure 1. Data flow of AphidBase.
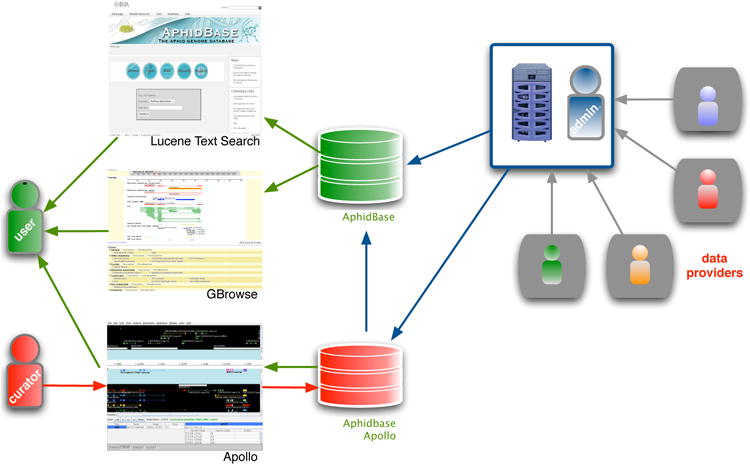
Two databases are fed in parallel with data computed by the administrator or submitted by providers. Regular users are accessing data stored in the databases using different front-ends. Authorized curators are inserting and modifying their annotations through Apollo, saving changes to their gene models directly in the specialized database. “Finished” annotations are frequently exported to the public AphidBase database.
Both databases are fed synchronously, in such a manner that experts or users get the same information either while browsing through Gbrowse or while editing through Apollo. The single difference between the duplicates is that the Apollo dedicated AphidBase copy contains current manual annotation data. All curated genes marked as « finished » in the Apollo “Annotation Editor” dialog box are routinely released into the public GBrowse AphidBase database.
For reasons of safety and traceability, the AphidBase administrators assigned usernames and passwords to authorized curators. Thus, only authorized curators can modify or comment on genome annotations in the Apollo copy of AphidBase.
Aphidbase's Apollo database can be started with Java WebStart, allowing the application to be started directly from the Aphidbase web site. Furthermore, before launching the application, Java WebStart automatically looks for an update via the internet, and downloads it if necessary.
Blast
Aphidbase offers a web blast search (version 2.2.15) that allows the parameterized comparison of nucleic and protein sequences against various databanks (transcripts and protein predictions, reads and scaffolds and ESTs).
Lucene full text search
The AphidBase full text search is based on the Apache Lucene indexation of flat files of the description of RefSeq predictions and the Uniprot proteins aligned on the genome, extracted from the Chado database. It has been generated with the help of the Lucene Java API encapsulated into an Apache Tomcat server.
Community Organization
The aphidgenomics electronic mailing list is managed using GNU Mailman, an open source mailing list management software written Python (http://www.list.org/). The aphidgenomics server is hosted in Department of Ecology and Evolution at Princeton University (http://www.eco.princeton.edu/mailman/listinfo/aphidgenomics). The IAGC Wiki is run on Confluence, an enterprise wiki engine (Atlassian, Sydney, Australia, http://www.atlassian.com/software/confluence/) and hosted at Indiana University.
Acknowledgments
The authors would like to acknowledge Terence Murphy (NCBI) for his advice and support, the International Aphid Genomics Consortium and in particular the all the curators involved in gene model improvement. We thank Ed Lee, Olivier Arnaiz, Scott Cain, Dave Clements and the rest of the GMOD team, Stéphanie Sibide-Bocs, Joëlle Amselem and Michaël Alaux from ANR GnpAnnot. The assistance of IT staff of AphidGenomics at Princeton University, and of Phillip Steinbachs for the establishment of the IAGC Wiki hosted by The Center for Genomics and Bioinformatics, Indiana University were fully appreciated. AphidBase is partly funded by ANR Génoplante “GnpAnnot”. Pea Aphid Genome Annotation Workshop I was supported by an American Genetic Association Special Event Award and an NRI, CSREES, USDA award 2007-04628 to ACCW.
References
- Brent MR. (2008) Steady progress and recent breakthroughs in the accuracy of automated genome annotation. Nat Rev Genet. 2008 Jan;9(1):62–73. doi: 10.1038/nrg2220. [DOI] [PubMed] [Google Scholar]
- Burge C, Karlin S. Prediction of Complete Gene Structures in Human Genomic DNA. J Mol Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- Cantarel BL, Korf I, Robb SMC, Parra G, Ross E, Moore B, Holt C, Sánchez Alvarado A, Yandell M. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008;18:188–196. doi: 10.1101/gr.6743907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denoeud F, Aury JM, Da Silva C, Noel B, Rogier O, Delledonne M, Morgante M, Valle G, Wincker P, Scarpelli C, Jaillon O, Artiguenave F. (2008) Annotating genomes with massive-scale RNA sequencing. Genome Biol. 2008;9(12):R175. doi: 10.1186/gb-2008-9-12-r175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsik CG, Worley KC, Zhang L, Milshina NV, Jiang H, Reese JT, Childs KL, Venkatraman A, Dickens CM, Weinstock GM, Gibbs RA. Community annotation: Procedures, protocols, and supporting tools. Genome Res. 2006;16:1329–1333. doi: 10.1101/gr.5580606. [DOI] [PubMed] [Google Scholar]
- Elsik CG, Mackey AJ, Reese JT, Milshina NV, Roos DS, Weinstock GM. Creating a honey bee consensus gene set. Genome Biology. 2007;8:R13. doi: 10.1186/gb-2007-8-1-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florea L, Hartzell G, Zhang Z, Rubin GM, Miller W. (1998) A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res. 1998;8:967–74. doi: 10.1101/gr.8.9.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier JP, Legeai F, Zasadzinski A, Rispe C, Tagu D. AphidBase: a database for aphid genomic resources. Bioinformatics. 2007 Mar 15;23(6):783–4. doi: 10.1093/bioinformatics/btl682. [DOI] [PubMed] [Google Scholar]
- Guigó R, Flicek P, Abril JF, Reymond A, Lagarde J, Denoeud F, Antonarakis S, Ashburner M, Bajic VB, Birney E, Castelo R, Eyras E, Ucla C, Gingeras TR, Harrow J, Hubbard T, Lewis SE, Reese MG. (2006) EGASP: the human ENCODE Genome Annotation Assessment Project. Genome Biol. 2006;7(Suppl 1):S2.1–31. doi: 10.1186/gb-2006-7-s1-s2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Nagy A, Reymond A, Alioto T, Patthy L, Antonarakis SE, Guigó R. (2009) Identifying protein-coding genes in genomic sequences. Genome Biology. 2009;10:201. doi: 10.1186/gb-2009-10-1-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe D, Costanzo M, Fey P, Gojobori T, Hannick L, Hide W, Hill DP, Kania R, Schaeffer M, St Pierre S, Twigger S, White O, Rhee SY. The future of biocuration. Nature. 2008;455:47–50. doi: 10.1038/455047a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J, Bueno A, Dopazo J, Gabaldoń T. PhylomeDB: a database for genome-wide collections of gene phylogenies. Nucleic Acids Res. 2008;36:D491–D496. doi: 10.1093/nar/gkm899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Aphid Genomics Consortium. Genome Sequence of the Pea Aphid Acyrthosiphon pisum. PLoS Biology. 2009 doi: 10.1371/journal.pbio.1000313. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogentic and Genome Research. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- Lawson D, Arensburger P, Atkinson P, Besansky NJ, Bruggner RV, Butler R, Campbell KS, Christophides GK, Christley S, Dialynas E, Hammond M, Hill CA, Konopinski N, Lobo NF, MacCallum RM, Madey G, Megy K, Meyer J, Redmond S, Severson DW, Stinson EO, Topalis P, Birney E, Gelbart WM, Kafatos FC, Louis C, Collins FH. VectorBase: a data resource for invertebrate vector genomics. Nucleic Acids Res. 2009;37:D583–D587. doi: 10.1093/nar/gkn857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis SE, Searle SMJ, Harris N, Gibson M, Iyer V, Richter J, Wiel C, Bayraktaroglu L, Birney E, Crosby MA, Kaminker JS, Matthews BB, Prochnik SE, Smith CD, Tupy JL, Rubin GM, Misra S, Mungall CJ, Clamp ME. Apollo: a sequence annotation editor. Genome Biology. 2002;3(12) doi: 10.1186/gb-2002-3-12-research0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mons B, Ashburner M, Chichester C, vanMulligen E, Weeber M, denDunnen J, van Ommen GJ, Musen M, Cockerill M, Hermjakob H, Mons A, Packer A, Pacheco R, Lewis S, Berkeley A, Melton W, Barris N, Wales J, Meijssen G, Moeller E, Roes PJ, Borner K, Bairoch A. Calling on a million minds for community annotation in WikiProteins. Genome Biol. 2008;9(5):R89. doi: 10.1186/gb-2008-9-5-r89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mungall CJ, Emmert DB The FlyBase Consortium. A Chado case study: an ontology-based modular schema for representing genome-associated biological information. BioInformatics. 2007;23:i337–i346. doi: 10.1093/bioinformatics/btm189. [DOI] [PubMed] [Google Scholar]
- Parra G, Blanco E, Guigó R. GeneID in Drosophila. Genome Res. 2000;10:511–515. doi: 10.1101/gr.10.4.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Klimke W, Maglott DR. NCBI Reference Sequences: current status, policy and new initiatives. Nucleic Acids Res. 2009;37:D32–D36. doi: 10.1093/nar/gkn721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesneville H, Bergman CM, Andrieu O, Autard D, Nouaud D, Ashburner M, Anxolabehere D. Combined Evidence Annotation of Transposable Elements in Genome Sequences. PLoS Comp Biol. 2005;1(2):e22. doi: 10.1371/journal.pcbi.0010022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers A, Antoshechkin I, Bieri T, Blasiar D, Bastiani C, Canaran P, Chan J, Chen WJ, Davis P, Fernandes J, Fiedler TJ, Han M, Harris TW, Kishore R, Lee R, McKay S, Müller HM, Nakamura C, Ozersky P, Petcherski A, Schindelman G, Schwarz EM, Spooner W, Tuli MA, Van Auken K, Wang D, Wang X, Williams G, Yook K, Durbin R, Stein LD, Spieth J, Sternberg PW. WormBase 2007. Nucleic Acids Res. 2008;36:D612–D617. doi: 10.1093/nar/gkm975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg SL. (2007) Genome re-annotation: a wiki solution? Genome Biol. 2007;8(1):102. doi: 10.1186/gb-2007-8-1-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiex T, Gouzy J, Moisan A, de Oliveira Y. FrameD: a flexible program for quality check and gene prediction in prokaryotic genomes and noisy matured eukaryotic sequences. Nucleic Acids Res. 2003;31:3738–3741. doi: 10.1093/nar/gkg610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedley D, Haider S, Ballester B, Holland R, London D, Thorisson G, Kasprzyk A. BioMart - biological queries made easy. BMC Genomics. 2009 Jan 14;10(1):22. doi: 10.1186/1471-2164-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Schöffmann O, Morgenstern B, Waack S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics. 2006;7:62. doi: 10.1186/1471-2105-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein L. (2001) Genome annotation: from sequence to biology. Nat Rev Genet. 2001 Jul;2(7):493–503. doi: 10.1038/35080529. [DOI] [PubMed] [Google Scholar]
- Stein LD, Mungall C, Shu S, Caudy M, Mangone M, Day A, Nickerson E, Stajich JE, Harris TW, Arva A, Lewis S. (2002) The generic genome browser: a building block for a model organism system database. Genome Res. 2002 Oct;12(10):1599–610. doi: 10.1101/gr.403602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein L. (2008) Towards a cyberinfrastructure for the biological sciences: progress, visions and challenges. Nat Rev Genet. 2008;9(9):678–88. doi: 10.1038/nrg2414. [DOI] [PubMed] [Google Scholar]
- Thomas E. LISTSERV. L-Soft International Inc.; Landover, MD: 1986. [Google Scholar]
- Wang H, Su Y, Mackey AJ, Kraemer ET, Kissinger JC. SynView: a GBrowse-compatible approach to visualizing comparative genome data. Bioinformatics. 2006;22(18):2308–2309. doi: 10.1093/bioinformatics/btl389. [DOI] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. (2008) Alternative isoform regulation in human tissue transcriptomes. Nature. 2008 Nov 27;(7221):456. 470–6. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilming LG, Gilbert JGR, Howe K, Trevanion S, Hubbard T, Harrow JL. The vertebrate genome annotation (Vega) database. Nucleic Acids Res. 2008;36:D753–D760. doi: 10.1093/nar/gkm987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RJ, Goodman JL, Strelets VB The FlyBase Consortium. FlyBase: integration and improvements to query tools. Nucleic Acids Res. 2008;36:D588–D593. doi: 10.1093/nar/gkm930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. BioInformatics. 2005;21(9):1859–1875. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]