

Abstract
Background
Comparative genomics provides insights into the diversification of bacterial species. Bacterial speciation usually takes place with lasting homologous recombination, which not only acts as a cohering force between diverging lineages but brings advantageous alleles favored by natural selection, and results in ecologically distinct species, e.g., frequent host shift in Xanthomonas pathogenic to various plants.
Results
Using whole-genome sequences, we examined the genetic divergence in Xanthomonas campestris that infected Brassicaceae, and X. citri, pathogenic to a wider host range. Genetic differentiation between two incipient races of X. citri pv. mangiferaeindicae was attributable to a DNA fragment introduced by phages. In contrast to most portions of the genome that had nearly equivalent levels of genetic divergence between subspecies as a result of the accumulation of point mutations, 10% of the core genome involving with homologous recombination contributed to the diversification in Xanthomonas, as revealed by the correlation between homologous recombination and genomic divergence. Interestingly, 179 genes were under positive selection; 98 (54.7%) of these genes were involved in homologous recombination, indicating that foreign genetic fragments may have caused the adaptive diversification, especially in lineages with nutritional transitions. Homologous recombination may have provided genetic materials for the natural selection, and host shifts likely triggered ecological adaptation in Xanthomonas. To a certain extent, we observed positive selection nevertheless contributed to ecological divergence beyond host shifting.
Conclusion
Altogether, mediated with lasting gene flow, species formation in Xanthomonas was likely governed by natural selection that played a key role in helping the deviating populations to explore novel niches (hosts) or respond to environmental cues, subsequently triggering species diversification.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1369-8) contains supplementary material, which is available to authorized users.
Keywords: Xanthomonas, Adaptive diversification, Host shift, Parapatric speciation, Homologous recombination, Comparative genomics
Background
Diversification of prokaryotes attracts much attention from ecologists because it plays a critical role in ecosystem equilibrium and dynamics. Speciation of bacteria is distinct from eukaryotes, especially given the predominantly asexual reproduction [1] and the rare occurrence of geographic barriers. Bacterial speciation is often triggered by adaptive divergence [2,3], while homologous recombination, which leads to gene flow cohering diverging populations, simultaneously occurs, a model approximating parapatric or sympatric speciation [4,5]. Accordingly, diversifying selection plays a key role in differentiating sister species. On the other hand, favorable genes could be brought by homologous recombination enable the recipient to explore a new niche [6,7], an example of adaptive processes in bacterial diversification under gene flow [8-11].
Many of the bacteria in the genus Xanthomonas, of the γ-subdivision of Proteobacteria, cause plant diseases, e.g., bacterial spots and blights in leaves and fruits [12]. These plant pathogens often display a high degree of host specificity [13], e.g., X. citri pv. citri exclusively infecting citrus, with various genetic mechanisms associated with the host specificity [14]. Host shifts occurred in pv. mangiferaeindicae that causes bacterial black spot in mango, and in pv. vesicatoria that attacks pepper and tomato, displaying a wide phylogenetic range of hosts in X. citri [15,16]. In contrast, X. campestris pathovars predominantly infect crucifers [17]. It has been known that favorable traits enable deviating populations to explore novel niches in an ecosystem [18]. During habitat shifts such as host transformation, evolutionary footprints of adaptation were often reserved in the genomes [19]. Lu et al. found that six pathogenicity-related gene clusters were associated with the genomic divergences in Xanthomonas [20]. In this study, via a comparative genomic analysis, we comprehensively investigated genomic divergence and adaptation in Xanthomonas, and the contributions of host shifts and homologous recombination to the adaptive diversification between species.
To date, whole genomes of 8 Xanthomonas taxa, including X. campestris pv. campestris (3 strains) [21-23], X. campestris pv. raphani [17], X. citri pv. vesicatoria [24], X. citri pv. citrumelo [25], X. citri pv. citri, X. albilineans [26], X. oryzae pv. oryzae (3 strains) [27-29], and X. oryzicola [30], have been sequenced with conventional shotgun sequencing. The size of their genomes varies from 4.9 to 5.4 Mb, with a high GC content of 63.3–69.7% in the chromosome [12]. Phylogenomic analysis revealed a close affinity among X. citri pv. mangiferaeindicae, X. citri pv. citri, and X. citri pv. citrumelo, three taxa that are closely related to X. citri. pv. vesicatoria [25,31]. Recently, a 5.1-Mb genome of X. citri pv. mangiferaeindicae strain LMG 941 from India with 195 contigs was obtained by pyrosequencing [31]. In order to comprehend the genetic divergence among recently diverging strains, we sequenced the genome of BCRC 13182, a local mangiferaeindicae strain from Taiwan. As the two strains LMG 941 and BCRC 13182 of X. citri pv. mangiferaeindicae diverged only recently, how they became genetically differentiated is quite fascinating. In this study, for exploring the nature of adaptive diversification in Xanthomonas, comparative genomic analyses were conducted on two nonsister species complexes of Xanthomonas, i.e., X. citri and X. campestris. The patterns of genomic divergence, homologous recombination, and genes under positive selection were examined to elucidate the ecological interactions among Xanthomonas taxa.
Results
General features of the Xanthomonas citri pv. mangiferaeindicae genome
The genome of X. citri pv. mangiferaeindicae BCRC 13182 (XCM-B) was sequenced, and 4,292,719,080 bases of paired-end data (read length = 60 bp, Q30 percentage = 98.8%) and 1,185,355,594 bases of mate-paired data (read length = 101 bp, Q30 percentage = 77.0%) were retrieved. These sequences were de novo assembled to 221 contigs, and scaffolded into 43 scaffolds, which comprised 5,355,324 bp with a GC content of 64.75% and indicated the sequence coverage is 1022.92. The largest scaffold was 1,286,619 bp long, and the N50 statistic was 549,958 bp, with an average length of 124,542 bp (Table 1). The protein-coding gene prediction, confirmed by BLAST searches against the NCBI database, identified 5,362 coding sequences, 3,837 of which could be categorized into clusters of orthologous groups (COG) (Figure 1). When compared with the draft genome of XCM strain LMG 941 (XCM-L) (Table 1), these two strains shared similar GC contents and 9 rRNA genes in 3 sets of rRNA operon (16S–23S–5S), whereas the average length (124,542 vs. 26,213), N50 statistic (549,958 vs.67,371), number of protein-coding sequences (5,362 vs. 4,521), and number of tRNA genes (55 vs. 51) were all greater in XCM-B, suggesting higher genome completeness in the genome that we sequenced and assembled. In addition, we aligned 43 scaffolds of XCM-B against those of XCM-L. Among the 195 contigs of XCM-L, 126 could be mapped onto the 18 scaffolds of XCM-B, whereas none of the XCM-L contigs were mapped onto XCM-B with more than one scaffold (Additional file 1: Table S1). Nevertheless, aligning both sequences helped verify the conserved genomes between the two strains. The genomes of two XCM strains shared 4,074 protein-coding genes, with only 203 variables, reaching an average protein sequence identity of 99.89%.
Table 1.
Genome features of X. citri pv. mangiferaeindicae strains BCRC 13182 (XCM-B) and LMG 941 (XCM-L)
| Chromosome feature | XCM-B | XCM- L |
|---|---|---|
| General feature of the genomes | ||
| Scaffold numbers | 43 | 195 |
| N50 size (bp) | 549,958 | 67,371 |
| Average length (bp) | 124,542 | 26,213 |
| Genome size (bp) | 5,355,324 | 5,111,537 |
| G + C content (%) | 64.75 | 64.85 |
| Predicted CDSs | ||
| Protein-coding genes | 5,362 | 4,521 |
| With assigned to COGs | 3,837 | NA |
| Shared protein-coding genes | 4074 | |
| Identical amino acid sequences | 3871 | |
| Sequences with indels | 83 | |
| Noncoding genes | ||
| rRNA | 9 | 9 |
| rRNA operons | 3 | 3 |
| tRNA | 55 | 51 |
NA, not available.
Figure 1.
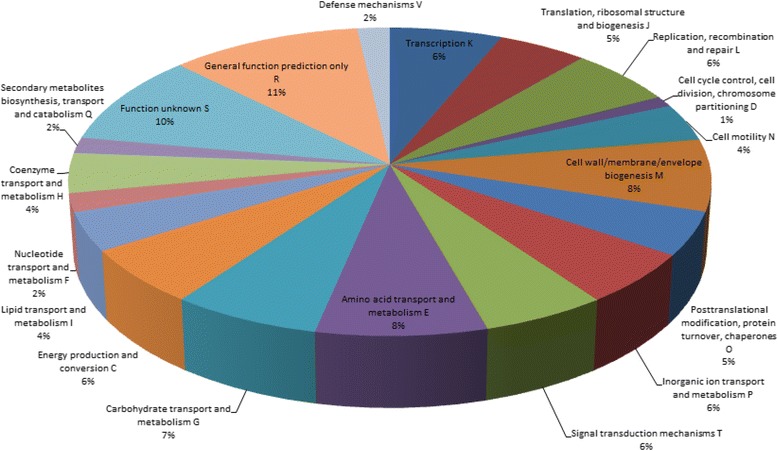
Functional categories of annotated genes in the genome of Xanthomonas citri pv. mangiferaeindicae BCRC 13182. Colors on the pie chart represent predicted clusters of orthologous groups (COG).
Phylogenetic relationships and homologous recombination among Xanthomonas species
As XCM-B that infects mango was once classified as a member of X. campestris [32], we tested the robustness of the classification of 5 X. citri taxa (XCM-B, XCM-L, X. citri pv. citri [XCC], X. citri pv. citrumelo [XCCM], X. citri pv. vesicatoria [XCV]) and 4 X. campestris taxa (X. campestris pv. campestris ATCC33913, 8004, B100 [XCCA, XCC8, XCCB], X. campestris pv. raphani [XCR]) (Additional file 2: Table S2). Based on a concatenated alignment of 2851 orthologous core genes shared by the 9 taxa, the best maximum likelihood (ML) tree identified two major clades corresponding to species. In the citri-clade, 2 XCM strains were clustered with XCC, while XCCM was sister to XCV; within the campestris-clade, XCCA and XCC8 were closely related, both being affined to XCCB and XCR (Figure 2).
Figure 2.
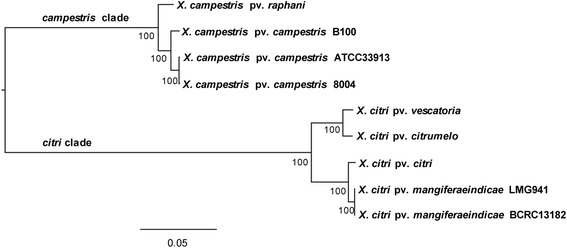
Molecular phylogeny of 9 Xanthomonas taxa based on 2851 core orthologous genes. Numbers at the nodes represent bootstrap values.
Recombination can cohere the taxa within a species, but dissociates sister taxa when it occurs between long-split species. Here, we combined the alignment-based programs geneconv and PhiPack to detect genes affected by recombination between or within species. In total, 283 core genes (9.9% of the genome) were detected with genetic recombination, which included 83 genes with cross-overs occurring between X. citri and X. campestris, 172 genes only detected within species, while 28 have likely derived from other distant species (Additional file 3: Table S3). Single gene trees were generated with the neighbor-joining method. For 2786 of the 2851 trees (97.7%), the topologies agreed with the division between citri and campestris, suggesting a long split between the 2 species.
Genome-wide variations and recombination events
Genetic divergences were not homogeneously distributed along the chromosome. Some genomic regions displayed higher divergences than the others, as so-called “genomic islands of divergence” [33]. Here, we used synonymous substitution rates (Ks), a near-neutral indicator of genetic divergence, to assess the divergence along the genome. In the comparison between 2 XCM strains based on the orthologous genes between XCM and XCC, only one major peak was observed in the putative prophage region, while the remaining regions displayed more or less uniform levels of genomic variation (Additional file 4: Figure S1). We further investigated the genic structure of the prophage region. Of the genes shared between the two strains of XCM, 53 remained intact, while 18 genes had become eliminated (see Additional file 5: Figure S2A). In the comparison between XCM-B and XCC, 33 genes were lost in XCM-B (Additional file 5: Figure S2B). All these results supported rapid evolution with dramatic gain or loss of genes in the prophage region. Furthermore, to examine the associations between host shifting and genomic divergence, pairwise comparisons were conducted in pairs of XCM vs. XCC (shifting between citrus and mango) and XCCM vs. XCV (between citrus and pepper) using 2851 core orthologous genes. Two major Ks peaks of 3.20–3.25 M bp and 4.20–4.35 M bp were shared by the host-shifting pairs, while only a few recombination events were detected in these regions (Figure 3A). It is evident that numerous divergence peaks overlap with peaks of the density of recombination events, implying a correlation between homologous recombination and genetic divergence (Figure 3A). As shown by Spearman’s rank correlation test, both comparisons were significant in the correlation between Ks values and the number of genes with recombination (Spearman’s rho = 0.29–0.35, P < 0.001). In addition, comparisons between closely related strains, i.e., XCM-B vs. XCM-L and XCCA vs. XCC8, were also performed. Similar to the comparisons of host-shifting pairs, significant positive correlations were also observed in the strain pairs without host-shifting (Spearman’s rho = 0.21–0.30, both P < 0.001, Figure 3B). At the gene level, the genes with recombination showed higher Ks than those without recombination based on pairwise comparisons (Mann–Whitney U test, P < 0.001) (Additional file 6: Figure S3), implying that these genes may facilitate the diversification among Xanthomonas strains. Taken together, these results suggest that homologous recombination largely affect the pattern of genomic divergence between Xanthomonas species.
Figure 3.
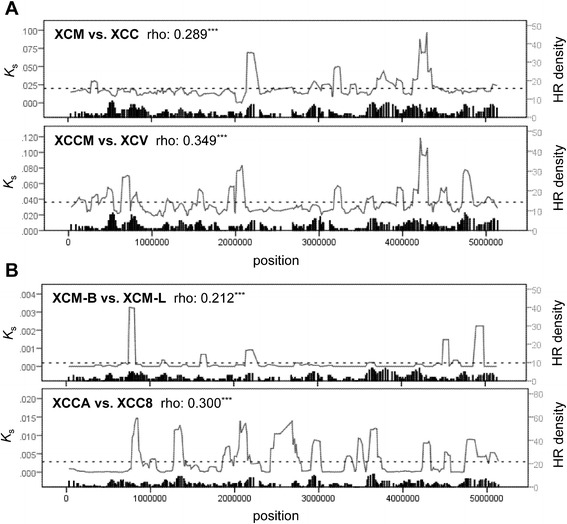
Genome-wide distributions of K s divergence and the density of homologous recombination (HR) events. Ks mean values and the density of homologous recombination events were plotted with a 50-gene window along the X. citri pv. citri (XCC) genome. The asterisks on rho value denote significant Spearman’s correlation (***, P < 0.001). Dashed lines represent the mean of K s values. A. Two host-shifting pairs: XCC vs. X. citri pv. mangiferaeindicae BCRC 13182 (XCM-B), between citrus and mango; X. citri pv. citrumelo (XCCM) vs. X. citri pv. vescicatoria (XCV), between citrus and mango. B. Two closely-related pairs: XCM-B vs. X. citri pv. mangiferaeindicae LMG 941 (XCM-L); X. campestris pv. campestris ATCC 33913 (XCCA) vs. X. campestris pv. campestris 8004 (XCC8).
Genes under positive selection
For identifying positively selected genes within each Xanthomonas lineage, codeml analyses were conducted with the branch-site model on all branches. In total, 179 genes (6.3% of the core genome) were detected under positive selection. Of these positively selected genes, only 3 were shared between X. citri and X. campestris, while 125 were exclusively in X. citri, and 51 were only in X. campestris (see Additional file 7: Table S4), implying different diversification scenarios.. Among the tree branches of the citri-group, along the branches of XCV and XCM, with host shifts, 25 and 18 positively selected genes were detected, respectively (Figure 4A). As for the remaining branches without host-shift events, the numbers of positively selected genes are as follows: 1 in XCM-L, 6 in XCM-B, 15 in XCCM, and 25 in XCC. Likewise, in the campestris group, 6 positively selected genes occurred in the XCCA lineage, followed by 6 in XCC8, 12 in XCCB, and 15 in XCR. Interestingly, the lineages in citri-group coupled with the host-shift events possessed more genes under positive selection (P < 0.001, g-test). We further tested the association between homologous recombination and positive selection. In total, 98 (57%) out of 184 positively selected genes were identified with recombination, with 73 in the citri-group, and 26 in the campestris-group, all significantly deviating from a random distribution (P < < 0.001, Fisher’s exact test) (Table 2). Intriguingly, the majority of the genes under positive selection did not display tree topologies deviating from the species tree, as shown by 98.4% (126/128) in the citri-group and 96.3% (52/54) in the campestris-group agreeing with the species tree (both P > > 0.05, Fisher’s exact test) (Table 3).
Figure 4.
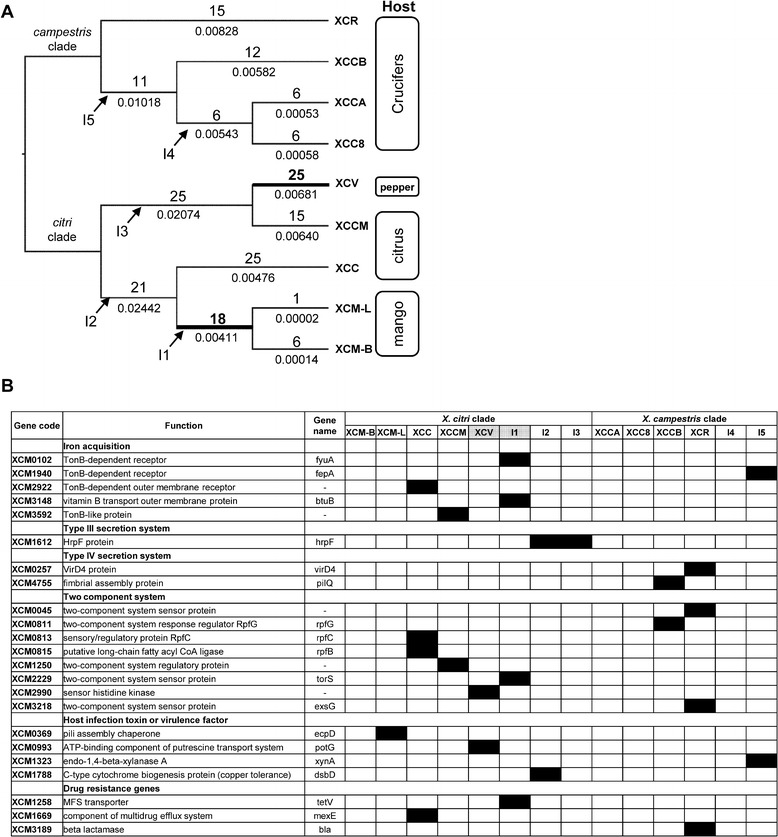
Summary of positively selected genes. (A) Distribution of positively selected genes in the phylogenetic cladogram. The numbers above and under the branches denote the number of positively selected genes and branch lengths, respectively. Thick lines and bold numbers denote the occurrences of host shift events. Internal branches are labeled from I1 to I5 with arrows. (B) Positively selected genes related to plant pathogenicity. Putative functions were based on the gene annotations of X. citri pv. citri. Genes under positive selection are filled in black at the corresponding branch, which is denoted with taxon name (terminal branch) or codes I1–I5 (internal branch). Host-shift branches were labeled in grey.
Table 2.
Distributions of the genes with recombination and the genes under positive selection in Xanthomonas
| Positively selected | Non-positively selected | Fisher’s exact test | ||
|---|---|---|---|---|
| X. citri | ||||
| With recombination | 73 | 210 | P < 0.0001 | |
| Without recombination | 55 | 2513 | ||
| X. campestris | ||||
| With recombination | 26 | 257 | P < 0.0001 | |
| Without recombination | 28 | 2540 | ||
| Total | ||||
| With recombination | 98 | 185 | P < 0.0001 | |
| Without recombination | 81 | 2487 |
Table 3.
Distributions of positively selected genes and tree topologies in Xanthomonas
| Separation of species groups | ||||
|---|---|---|---|---|
| Agree | Disagree | Fisher’s exact test | ||
| X. citri | ||||
| Positively selected | 126 | 2 | P = 1 | |
| Non-positively selected | 2660 | 63 | ||
| X. campestris | ||||
| Positively selected | 52 | 2 | P = 0.3503 | |
| Non-positively selected | 2734 | 63 | ||
| Total | ||||
| Positively selected | 175 | 4 | P = 1 | |
| Non-positively selected | 2611 | 61 | ||
Of the numerous genes putatively under the positive selection, 23 loci were likely to be associated with the plant pathogenicity [34-37] (Figure 4B). For example, 5 genes were involved in the iron acquisition, especially under the environment with the low availability of free iron from the host [38]. Of these genes for protein secretion systems that help the transportation of virulence factor from bacteria to host, 4 loci were exclusively favored in the citri lineage that with 1 genes of the type III, and 2 gene of the type IV [36,37]. Besides, 8 genes related to the two component system of bacteria, which acts as a sensor to environmental cues and an activator of pathogenic genes; 3 genes were associated with the antibiotic resistance, and 4 other genes were related to the various functions with the plant pathogenicity. For the positively selected genes that might have ecological impacts, we listed them in Table 4 and fully discussed in the next section.
Table 4.
Ecological effects of positively selected genes
| Positively selected genes | Selected lineage | Related to host shift | Related to homologous recombination | Putative ecological effects |
|---|---|---|---|---|
| RpfB, RpfC (XCM0813, XCM0815) | X. citri pv. citri | Yes | Yes | Altering pathogenicity to favor exploration of different host |
| DsbD (XCM1788) | I2 branch | No | Yes | Increasing copper tolerance in escaping from copper-containing bactericides |
| PotG (XCM0993) | X. citri pv. vesicatoria | Yes | Yes | Reducing AMF colonization of host plant and increasing infectivity |
| BtuB (XCM3148) | I1 branch (X. citri pv. mangiferaeindicae) | Yes | No | Increasing iron uptake with host-shifting from citrus to mango |
| HrpF (XCM1612) | I2, I3 branches | Yes | Yes | Controlling invasive routes and host specificity of X. citri |
| EcpD (XCM0369) | XCM-L | No | No | Helping pili assembly and associating with virulence |
| XynA (XCM1323) | I5 branch | No | No | Breaking down xylan and causing virulence |
Discussion
Speciation processes in Xanthomonas
Bacterial diversity, which often results from species diversification through ecological interactions, greatly influences the ecosystem health. Xanthomonas species, which can cause serious diseases and economic loss of crops, are an excellent group for examining speciation models. The genomes of 8 taxa that belong to X. citri and X. campestris have been completely sequenced, providing a large data set as a sound foundation for investigating the extent of genetic divergence between species. Their host specificity makes the group even more useful as a system for examining adaptation over the host shifts because the host range of the group as a whole is quite broad. In this study, two strains of X. citri pv. mangiferaeindicae from Taiwan (XCM-B) and India (XCM-L) and two strains of X. campestris (XCCA and XCC8) displayed slight genetic differentiation, providing a window for investigating incipient genetic divergence. As expected, given a shorter time for isolation, strains had lower levels of genetic divergence than that of species/pathovars. It is noticeable that prophage-introduced DNA fragments were detected in X. citri pv. mangiferaeindicae and pv. citri [39], an event that exclusively occurred in X. citri. Several salient features were observed in these inserted DNA fragments. First, long DNA fragments of 28,787 bp were lost in genomes of the two pathovars (Additional file 5: Figure S2). Second, a high proportion of genes (18 of 71 between strains of mangiferaeindicae, and 33 of 71 between pathovars) became eliminated along with the divergence time. Third, higher nucleotide substitution rates were detected in these inserted DNAs than in the host bacterial genome. These facts indicate that the foreign genes introduced by the phage tended to lose functions and were likely to be removed from the genome eventually (e.g., gene XCM0436), although gene residues may still remain [40].
On the other hand, homologous recombination had started to contribute noticeably to the global diverging patterns among Xanthomonas strains at the incipient stage, as shown by the positive and significant correlation with genomic divergence (Figure 3). Homologous recombination is well documented in prokaryotes with foreign genes introgressed into the recipient species [41]. Recombination often occurs between closely related bacteria, resulting in convergence of two bacterial populations; recombination with divergent bacteria brings variations into the recipient population, facilitating the differentiation among incipient species [42,43].
Furthermore, 1 and 6 genes under positive selection were detected in the lineages leading to XCM-L and XCM-B, respectively. Also, of the 7 genes involved in diversifying selection of pv. mangiferaeindicae, 3 genes were involved in homologous recombination. Similarly, 6 of 9 genes under positive selection were detected with recombination for XCCA and XCC8. At early stages of speciation or diversification, the interplay between natural selection and homologous recombination clearly played a key role in differentiating incipient species/races.
Homologous recombination in Xanthomonas
In this study, we found frequent genetic recombination in Xanthomonas, with about 10% of the core genome mediated with homologous recombination, whereas only 2% of the core genes displayed phylogenies deviating from the species tree. This gap to the expectations simply came from intraspecific crossovers, which constitute the most recombination events (about 88%) with short DNA fragments. Apparently, those genes displaying trees inconsistent with the species tree represented footprints of gene flow between species. This result suggests that the divergence of Xanthomonas tends to follow parapatric speciation, in which gene flow between species can be long lasting and nonstop [41]. This is a pattern well documented in the Archaea and Bacteria [1,7]. It is intriguing that most of the foreign genes were favored by natural selection (Table 2), especially those associated with plant infection (Figure 4B), reflecting a fact that homologous recombination may create advantageous new alleles for a so-called “hopeful monster” [44] in bacteria. Altogether, the topology of the putative species tree in Xanthomonas reflects a scenario of deep divergence between species, mediated by recurrent gene flow at the same time [16].
In this study, 98 out of 283 genes with recombination were under positive selection (Table 2). Via homologous recombination, genes were able to be interchanged between strains, and advantageous alleles that helped bacterial colonization would be favored by natural selection [3,45]. Of these genes, it is noticeable that RpfB (XCM0815) and RpfC (XCM0813) of the Rpf gene cluster, which regulates the virulence of Xanthomonas [20,46,47], were positively selected in X. citri pv. citri lineage (Table 4). Functionally, RpfB encodes a fatty acyl-CoA ligase, which catalyzes the synthesis of an important signal molecule that regulates the expression of virulence genes [48,49]. RpfB signaling is perceived by the two component system of RpfC and RpfG [50], and the mutations of RpfC were found to reduce the pathogenicity of X. citri pv. citri [14]. Accordingly, existence of favorable RpfB alleles suggests that homologous recombination may have helped the adaptation of Xanthomonas. Besides, DsbD (XCM1788) has been reported to be associated with copper tolerance [51]. Copper compounds are frequently used as bactericides in controlling the leaf infection of Xanthomonas species, including X. citri [52,53]. A previous experiment showed that a DsbD knockout mutant was highly sensitive to environmental copper [54]. An effective DsbD allele was capable of reducing the impacts of the copper-containing bactericides, thus enabling the bacterium to escape from the agricultural control.
Host shifts likely triggering genetic adaptation
Like the findings of previous studies, compared with the vast portions of the genome that are usually shaped by purifying selection, adaptive genes only constitute a relatively smaller proportion of the chromosome instead [55]. Furthermore, in this study, genes under positive selection (only 6.3% of the core genome) were unequally distributed along lineages, with significantly more genes located at lineages leading to nodes coupled with host shifts (Figure 4A). For example, at the lineages from X. citri pv. citri leading to X. citri pv. mangiferaeindicae and X. citri pv. vescatoria with hosts shifts among citrus, mango and pepper, respectively, 18 and 25 positively selected genes were detected, revealing faster accumulation of positively selected genes than other lineages without host shifts. However, the lineages of X. campestris pv. raphani and pv. campestris, both infecting the Brassicaceae without host shifting, did not show facilitated positive selection. This contrasting pattern suggested that favorable genes may have helped the bacteria in exploring new niches. The tight association between positively selected genes and host shifts further suggests that Xanthomonas likely followed ecological speciation, which describes species arising from ecological diversification [3].
We found that some positively selected genes were associated with the host shifting events. It is notable that the PotG gene (XCM0993), that encodes ATPase involved in putrescine transport, was shaped by positive selection in the X. citri pv. vesicatoria lineage. Putrescine is a ubiquitous polyamine that enhances the interaction between plants and arbuscular mycorrhizal fungi (AMF) [56], which in turn have been shown to activate plant defense against infectious pathogens such as Xanthomonas [57]. Alternatively, the removal of exogenous putrescine from the soil may impede host colonization by AMF; thus, an effective PotG gene that decreases AMF colonization of host plant will be favored by natural selection, subsequently helping X. citri pv. vesicatoria to explore a new niche (host). In addition, BtuB gene (XCM3148) was positively selected in X. citri pv. mangiferaeindicae lineage. BtuB is responsible for the iron uptake, while iron dependent superoxide dismutases are vital in inhibiting the reactive oxygen species (ROS) responses in host cells, thus increasing the infection rates [46]. Interestingly, previous studies revealed that the iron contents in mango leaves might be hundreds times lower than that in citrus leaves (0.27 vs. 41.7 mg kg-1 dry weight) [58,59]. The sharp difference suggested that an effective iron transporter was needed when the host shifted from citrus to mango.
In addition to the genes related to the host shifting, an Hrp regulon gene XCM1612 that encodes components for the type III secretion system (T3SS) [60] was detected as loci under positive selection in the X. citri. The pathogenicity of Xanthomonas mainly depends on the T3SS, which is highly conserved among plant and animal pathogenic bacteria [61,62]. Curiously, the amino acid sequences of the major subunit of Hrp pili are hypervariable in different subspecies of bacterial pathogens [63,64]. The rapidly evolving Hrp pili provide evidence for strong positive selection in Xanthomonas spp. and diversifying selection in Pseudomonas syringae [65,66]. The acquisition of the Hrp gene cluster has been found to be associated with the adaptation and plant pathogenicity in Xanthomonas [20]. Thus, we hypothesized that the positive selection on HrpF gene may be responsible for the invasion routes and the host specificity of Xanthomonas. Furthermore, two genes involved in type IV secretion system were detected with positive selections exclusively in X. campestris, seemingly agreeing that type IV secretion system may not be involved in the infection of X. citri on citrus [67]. Moreover, EcpD (XCM0369) that encodes an adhesion protein to help pili assembly was positively selected in XCM-L. EcpD has been shown to facilitate the polymerization of EcpA to form pilus in Escherichia coli and involve in host cell recognition or biofilm formation [68]. Xanthomonas species only possessed EcpD while lacks other members of Ecp operon. We therefore hypothesized that EcpD might participate the assembly of the other pili in association with the virulence in Xanthomonas. On the other hand, a xylanase gene XynA (XCM1323) was positively selected at the branch leading to X. campestris pv. campestris (Figure 4B, Table 4). Xylan is a major component in the cell walls of land plants and exists in all plant tissues [69]. Previous studies showed xylanases are responsible for the virulence of X. citri pv. vesicatoria and X. oryzae pv. oryzae [70,71]. Two gene clusters xcs and xps of the Type II secretion system (T2SS) have been shown to control the secretion of xylanases in XCV and are associated with the virulence [70]. Nevertheless, no member of these two gene clusters was detected under positive selection in this study (Additional file 3: Table S3).
Conclusions
In this study, we sequenced a genome of X. citri pv. mangiferaeindicae and conducted genomic analyses of 9 taxa of X. citri and campestris. Between the 2 strains of XCM, only the prophage region displayed sharp differentiation, while gradually losing the constituting genes. In addition, we found homologous recombination frequently occurring in the Xanthomonas genomes, which likely represented footprints of gene flow between species, thus most likely suggesting parapatric speciation. It is noticeable that facilitated accumulations of positively selected genes occurred along the lineages with host shifts. Interestingly, most of the favored genes were acquired from homologous recombination. Taken together, the genes with recombination enabled Xanthomonas species to explore novel niches and respond to environmental stresses, subsequently resulting in adaptive diversification in this pathogenic genus.
Methods
Whole sequencing, assembly, and annotation of Xanthomonas citri pv. mangiferaeindicae
Bacterial strain BCRC 13182 (NCHUPP Xma1) from Taiwan of X. citri pv. mangiferaeindicae was purchased from the Food Industry Research and Development Institute and sequenced in this study. The whole genome was sequenced using a high-throughput sequencing technique with Illumina GA IIx and HiSeq sequencers [72]. Paired-end sequence reads of 60-bp were obtained, with an average distance of 150 bp between pair reads. Mate-paired sequence reads of 100 bp were obtained with an average distance of 3000 bp between pair reads. All raw sequences were stored in NCBI SRA database (SRP049288). A total of 110,671,234 and 15,543,685 high-quality sequences of 60 and 100 bp, respectively, were assembled into contigs using the de novo assembler ABySS version 1.2.7 [73]. After contigs shorter than 200 bp were removed, 221 contigs were generated. Scaffolding was performed with these contigs using SSPACE v2.0 [74], and 43 scaffolds were built based on the distance information between paired reads.
The draft genome was analyzed using an integrated annotation pipeline glued by the perl programming language. Glimmer version 3.02 [75] was used to predict the protein-coding regions, which were annotated with BLAST [76] against the nonredundant protein database (http://ncbi.nlm.nih.gov) and the Clusters of Orthologous Groups (COG) database [77] with a cutoff of E-value < 1 × 10-5. All protein-coding regions were manually curated with the EMBOSS analysis package according to the BLAST results [77]. We used tRNAscan-SE and RNAmmer version 1.2 to predict the prokaryotic transfer RNA genes and the ribosomal RNA genes [78,79].
Comparative genomics analysis and orthologous protein identification
For comparative analyses, we downloaded 8 bacteria genomes from the NCBI GenBank (http://www.ncbi.nlm.nih.gov/genbank/) in addition to the newly assembled genomes of X. citri pv. mangiferaeindicae BCRC 13182, including X. citri pv. citri 306, X. citri pv. citrumelo F1, X. citri pv. vesicatoria 85–10, X. campestris pv. campestris ATCC 33913, X. campestris pv. campestris 8004, X. campestris pv. campestris B100, X. campestris pv. raphani 756C, and a draft genome of X. citri pv. mangiferaeindicae LMG 941 (Additional file 1: Table S1). Orthologous genes in these genomes were identified using bidirectional best hits (BBH) in a BLAST search [80] based on the criteria set at E-value < 1 × 10-5, identity > 60%, and a threshold of 70% of orthologous alignment length. The core genome with 2,851 orthologous genes and common proteins shared by Xanthomonas species were recognized. In total, 43 scaffolds of the X. citri pv. mangiferaeindicae strain BCRC 13182 were aligned against the strain LMG 941, which has 195 contigs. We used BLAST for pairwise nucleotide alignment of the two draft genomes.
Phylogeny of Xanthomonas
For aligning nucleotide sequences of protein-encoding genes without interrupting codons, the protein sequences were first aligned by ClustalW2 [81], and the corresponding nucleotide sequences were aligned accordingly using PAL2NAL [82]. A maximum-likelihood (ML) phylogeny based on the concatenated 2851 genes was generated with the program RAxML [83]. We used the GTRGAMMA substitution model suggested by jModelTest 2.1.4 [84,85]. Confidence in the internal nodes of the ML tree was tested with 1000 rapid bootstrap replicates. For each gene phylogeny, we used ClustalW2 to generate a neighbor-joining tree with default settings.
Detection of genetic recombination
Putative genetic recombination events occurring in Xanthomonas genomes were examined by using the software geneconv [86] and PhiPack [87]. First, the alignments of the 2851 orthologous genes were tested with geneconv, and a recombination event can be recognized when a Bonferroni corrected KA p-value is less than 0.05. Notably, the geneconv program is able to distinguish a recombination occurring within or between species groups. In addition, to reduce the probability of detecting false positive when merely using a single method, we used the PhiPack program, which performs three different methods (Neighbor similarity score, MaxChi, and Phi) for identifying genetic recombination, to confirm the results of geneconv. Using the PhiPack program, recombination was identified for p-values lower than 0.05. Taken together, a gene with recombination was recognized when recombination was detected with geneconv and at least two additional methods implemented in PhiPack.
Divergence estimation
Numbers of substitutions per synonymous site (Ks) and per nonsynonymous site (Ka) were calculated using codeml program implemented in the PAML package with the codon-based model [88]. All core orthologous genes were mapped onto the XCC genome to perform window-sliding analyses. The genome-wide comparisons of Ks values and the number of genes with recombination were made with a window size of 50 genes and an overlap of 10 genes. Spearman’s rho coefficient was used to examine the correlations between Ks values and the number of the genes with recombination. In addition, the significant of difference of Ks distributions between genes with and without recombination were evaluated with Mann–Whitney U test. Both tests were performed by using SPSS Statistics 17.0.
Detection of genes under positive selection
For detecting genes under positive selection, we applied the codeml program from the PAML package to all core genes. The multiple alignments were fit to the F3X4 model of codon frequencies, and the branch-site alternative model (model = 2, NSsites = 2) was adopted [89]. Three independent simulations were performed with initial omega (Ka/Ks) values of 0.5, 1, and 1.5, respectively, while the null model was fixed with the omega value at 1. Likelihood ratio tests were used to assess the significance between the test and null models, and the P values were adjusted with a false discovery rate (FDR) of 0.01 for multiple testing corrections. We performed codeml analyses on each branch of individual gene trees. For above three tests, branches detected with significance were deemed to be affected by positive selection. Assuming the number of positively selected genes was proportional to the branch length, we used a chi-square test to examine the homogeneity in the distribution of positively selected genes between branches with and without host shifts. Fisher’s exact tests were also used to assess the differences of the number of positively selected genes between outlier and background genes, as well as between the genes with and without homologous recombination.
Availability of supporting data
Alignments and phylogenetic trees of the core orthologous genes were deposited in TreeBASE (http://treebase.org/treebase-web/home.html).
Additional files
Comparisons of assembled scaffolds and lengths between strains BCRC 13182 and LMG 941 of X. citri pv. mangiferaeindicae. (XCM).
Information on 9 Xanthomonas species.
Summary of orthologous genes among 9 Xanthomonas taxa.
Distribution of mean of K s of Xanthomonas citri pv. mangiferaeindicae BCRC 13182 (XCM-B) vs. Xanthomonas citri pv. mangiferaeindicae LMG941 (XCM-L).
Schematic presentation of the putative prophage region among two Xanthomonas citri pv. mangiferaeindicae (XCM) strains and Xanthomonas citri pv. citri (XCC).
Comparisons of K s values between the genes with and without recombination.
List of positively selected genes in Xanthomonas.
Footnotes
Chao-Li Huang, Pei-Hua Pu, Hao-Jen Huang, Huang-Mo Sung, Hung-Jiun Liaw and Yi-Min Chen contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
PHP designed and conducted molecular experiments. HMS and YTC assembled the genome and completed gene annotations. CLH, PHP, HJH, HJL, YMC, CMC, MBH, and YTC conducted the genetic analysis. TWP and CCH computed the genomic divergence. CLH, YTC, and TYC drafted the manuscript. NO and TG improved the writing. CCH and TYC supervised the work. All authors read and approved the final manuscript.
Contributor Information
Chao-Li Huang, Email: clhuang65535@gmail.com.
Pei-Hua Pu, Email: pph941027@yahoo.com.tw.
Hao-Jen Huang, Email: haojen@mail.ncku.edu.tw.
Huang-Mo Sung, Email: hmsung@mail.ncku.edu.tw.
Hung-Jiun Liaw, Email: liawh@mail.ncku.edu.tw.
Yi-Min Chen, Email: cohen@mail.ncku.edu.tw.
Chien-Ming Chen, Email: d98570003@mail.ntou.edu.tw.
Ming-Ban Huang, Email: h2o41040272@gmail.com.
Naoki Osada, Email: nosada@nig.ac.jp.
Takashi Gojobori, Email: tgojobor@nig.ac.jp.
Tun-Wen Pai, Email: twp@mail.ntou.edu.tw.
Yu-Tin Chen, Email: wytchen@gmail.com.
Chi-Chuan Hwang, Email: chchwang@mail.ncku.edu.tw.
Tzen-Yuh Chiang, Email: tychiang@mail.ncku.edu.tw.
References
- 1.Cadillo-Quiroz H, Didelot X, Held NL, Herrera A, Darling A, Reno ML, et al. Patterns of gene flow define species of thermophilic Archaea. PLoS Biol. 2012;10:e1001265. doi: 10.1371/journal.pbio.1001265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kopac S, Wang Z, Wiedenbeck J, Sherry J, Wu M, Cohan FM. Genomic heterogeneity and ecological speciation within one subspecies of Bacillus subtilis. Appl Environ Microbiol. 2014;80:4842–53. doi: 10.1128/AEM.00576-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vos M. A species concept for bacteria based on adaptive divergence. Trends Microbiol. 2011;19:1–7. doi: 10.1016/j.tim.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 4.Oakley BB, Carbonero F, van der Gast CJ, Hawkins RJ, Purdy KJ. Evolutionary divergence and biogeography of sympatric niche-differentiated bacterial populations. ISME J. 2010;4:488–97. doi: 10.1038/ismej.2009.146. [DOI] [PubMed] [Google Scholar]
- 5.Sikorski J, Nevo E. Adaptation and incipient sympatric speciation of Bacillus simplex under microclimatic contrast at “Evolution Canyons” I and II, Israel. Proc Natl Acad Sci U S A. 2005;102:15924–9. doi: 10.1073/pnas.0507944102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohan FM, Koeppel AF. The origins of ecological diversity in prokaryotes. Curr Biol. 2008;18:R1024–34. doi: 10.1016/j.cub.2008.09.014. [DOI] [PubMed] [Google Scholar]
- 7.Vos M, Didelot X. A comparison of homologous recombination rates in bacteria and archaea. ISME J. 2008;3:199–208. doi: 10.1038/ismej.2008.93. [DOI] [PubMed] [Google Scholar]
- 8.Luo C, Walk ST, Gordon DM, Feldgarden M, Tiedje JM, Konstantinidis KT. Genome sequencing of environmental Escherichia coli expands understanding of the ecology and speciation of the model bacterial species. Proc Natl Acad Sci U S A. 2011;108:7200–5. doi: 10.1073/pnas.1015622108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohan FM. Genetic exchange and evolutionary divergence in prokaryotes. Trends Ecol Evol. 1994;9:175–80. doi: 10.1016/0169-5347(94)90081-7. [DOI] [PubMed] [Google Scholar]
- 10.Nogueira T, Rankin DJ, Touchon M, Taddei F, Brown SP, Rocha EP. Horizontal gene transfer of the secretome drives the evolution of bacterial cooperation and virulence. Curr Biol. 2009;19:1683–91. doi: 10.1016/j.cub.2009.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wiedenbeck J, Cohan FM. Origins of bacterial diversity through horizontal genetic transfer and adaptation to new ecological niches. FEMS Microbiol Rev. 2011;35:957–76. doi: 10.1111/j.1574-6976.2011.00292.x. [DOI] [PubMed] [Google Scholar]
- 12.Vauterin L, Hoste B, Kersters K, Swings J. Reclassification of xanthomonas. Int J Syst Bacteriol. 1995;45:472–89. doi: 10.1099/00207713-45-3-472. [DOI] [Google Scholar]
- 13.Dye D, Bradbury J, Goto M, Hayward A, Lelliott R, Schroth MN. International standards for naming pathovars of phytopathogenic bacteria and a list of pathovar names and pathotype strains. Rev Plant Pathol. 1980;59:153–68. [Google Scholar]
- 14.Siciliano F, Torres P, Sendín L, Bermejo C, Filippone P, Vellice G, et al. Analysis of the molecular basis of Xanthomonas axonopodis pv. citri pathogenesis in Citrus limon. Electron J Biotechnol. 2006;9:200–4. doi: 10.2225/vol9-issue3-fulltext-20. [DOI] [Google Scholar]
- 15.Gagnevin L, Pruvost O. Epidemiology and control of mango bacterial black spot. Plant Dis. 2001;85:928–35. doi: 10.1094/PDIS.2001.85.9.928. [DOI] [PubMed] [Google Scholar]
- 16.Mhedbi-Hajri N, Hajri A, Boureau T, Darrasse A, Durand K, Brin C, et al. Evolutionary history of the plant pathogenic bacterium Xanthomonas axonopodis. PLoS One. 2013;8:e58474. doi: 10.1371/journal.pone.0058474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bogdanove AJ, Koebnik R, Lu H, Furutani A, Angiuoli SV, Patil PB, et al. Two new complete genome sequences offer insight into host and tissue specificity of plant pathogenic Xanthomonas spp. J Bacteriol. 2011;193:5450–64. doi: 10.1128/JB.05262-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang C-L, Ho C-W, Chiang Y-C, Shigemoto Y, Hsu T-W, Hwang C-C, et al. Adaptive divergence with gene flow in incipient speciation of Miscanthus floridulus/sinensis complex (Poaceae) Plant J. 2014;80:834–47. doi: 10.1111/tpj.12676. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt H, Greshake B, Feldmeyer B, Hankeln T, Pfenninger M. Genomic basis of ecological niche divergence among cryptic sister species of non-biting midges. BMC Genomics. 2013;14:384. doi: 10.1186/1471-2164-14-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu H, Patil P, Van Sluys M-A, White FF, Ryan RP, Dow JM, et al. Acquisition and evolution of plant pathogenesis–associated gene clusters and candidate determinants of tissue-specificity in Xanthomonas. PLoS One. 2008;3:e3828. doi: 10.1371/journal.pone.0003828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.da Silva AR, Ferro JA, Reinach F, Farah C, Furlan L, Quaggio R, et al. Comparison of the genomes of two Xanthomonas pathogens with differing host specificities. Nature. 2002;417:459–63. doi: 10.1038/417459a. [DOI] [PubMed] [Google Scholar]
- 22.Qian W, Jia Y, Ren S-X, He Y-Q, Feng J-X, Lu L-F, et al. Comparative and functional genomic analyses of the pathogenicity of phytopathogen Xanthomonas campestris pv. campestris. Genome Res. 2005;15:757–67. doi: 10.1101/gr.3378705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vorhölter F-J, Schneiker S, Goesmann A, Krause L, Bekel T, Kaiser O, et al. The genome of Xanthomonas campestris pv. campestris B100 and its use for the reconstruction of metabolic pathways involved in xanthan biosynthesis. J Biotechnol. 2008;134:33–45. doi: 10.1016/j.jbiotec.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 24.Thieme F, Koebnik R, Bekel T, Berger C, Boch J, Büttner D, et al. Insights into genome plasticity and pathogenicity of the plant pathogenic bacterium Xanthomonas campestris pv. vesicatoria revealed by the complete genome sequence. J Bacteriol. 2005;187:7254–66. doi: 10.1128/JB.187.21.7254-7266.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jalan N, Aritua V, Kumar D, Yu F, Jones JB, Graham JH, et al. Comparative genomic analysis of xanthomonas axonopodis pv. citrumelo f1, which causes citrus bacterial spot disease, and related strains provides insights into virulence and host specificity. J Bacteriol. 2011;193:6342–57. doi: 10.1128/JB.05777-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pieretti I, Royer M, Barbe V, Carrere S, Koebnik R, Cociancich S, et al. The complete genome sequence of Xanthomonas albilineans provides new insights into the reductive genome evolution of the xylem-limited Xanthomonadaceae. BMC Genomics. 2009;10:616. doi: 10.1186/1471-2164-10-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ochiai H, Inoue Y, Takeya M, Sasaki A, Kaku H. Genome sequence of Xanthomonas oryzae pv. oryzae suggests contribution of large numbers of effector genes and insertion sequences to its race diversity. JARQ-Jpn Agr Res Q. 2005;39:275. doi: 10.6090/jarq.39.275. [DOI] [Google Scholar]
- 28.Lee B-M, Park Y-J, Park D-S, Kang H-W, Kim J-G, Song E-S, et al. The genome sequence of Xanthomonas oryzae pathovar oryzae KACC10331, the bacterial blight pathogen of rice. Nucleic Acids Res. 2005;33:577–86. doi: 10.1093/nar/gki206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salzberg SL, Sommer DD, Schatz MC, Phillippy AM, Rabinowicz PD, Tsuge S, et al. Genome sequence and rapid evolution of the rice pathogen Xanthomonas oryzae pv. oryzae PXO99A. BMC Genomics. 2008;9:204. doi: 10.1186/1471-2164-9-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 31.Midha S, Ranjan M, Sharma V, Pinnaka AK, Patil PB. Genome sequence of Xanthomonas citri pv. mangiferaeindicae strain LMG 941. J Bacteriol. 2012;194:3031. doi: 10.1128/JB.00433-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ah-You N, Gagnevin L, Chiroleu F, Jouen E, Neto JR, Pruvost O. Pathological variations within Xanthomonas campestris pv. mangiferaeindicae support its separation into three distinct pathovars that can be distinguished by amplified fragment length polymorphism. Phytopathology. 2007;97:1568–77. doi: 10.1094/PHYTO-97-12-1568. [DOI] [PubMed] [Google Scholar]
- 33.Du P, Yang Y, Wang H, Liu D, Gao G, Chen C. A large scale comparative genomic analysis reveals insertion sites for newly acquired genomic islands in bacterial genomes. BMC Microbiol. 2011;11:135. doi: 10.1186/1471-2180-11-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caza M, Kronstad JW. Shared and distinct mechanisms of iron acquisition by bacterial and fungal pathogens of humans. Front Cell Infect Microbiol. 2013;3:80. doi: 10.3389/fcimb.2013.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gotoh Y, Eguchi Y, Watanabe T, Okamoto S, Doi A, Utsumi R. Two-component signal transduction as potential drug targets in pathogenic bacteria. Curr Opin Microbiol. 2010;13:232–9. doi: 10.1016/j.mib.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 36.Alfano JR, Collmer A. Type III secretion system effector proteins: double agents in bacterial disease and plant defense. Annu Rev Phytopathol. 2004;42:385–414. doi: 10.1146/annurev.phyto.42.040103.110731. [DOI] [PubMed] [Google Scholar]
- 37.Christie PJ, Vogel JP. Bacterial type IV secretion: conjugation systems adapted to deliver effector molecules to host cells. Trends Microbiol. 2000;8:354–60. doi: 10.1016/S0966-842X(00)01792-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown JS, Holden DW. Iron acquisition by gram-positive bacterial pathogens. Microb Infect. 2002;4:1149–56. doi: 10.1016/S1286-4579(02)01640-4. [DOI] [PubMed] [Google Scholar]
- 39.Marraffini LA, Sontheimer EJ. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008;322:1843–5. doi: 10.1126/science.1165771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chiang T-Y, Chiang YC, Pan C-H, Wang W-K, Chen C-N, Hsu T-W, et al. Plant evolutionary genetics and biology of weeds. Nantou, Taiwan: Endemic Species Research Institute; 2004. Comparative genomics of horizontal transfer between chloroplast and nuclear genomes in rice and Arabidopsis; pp. 1–10. [Google Scholar]
- 41.Mallet J. Hybridization, ecological races and the nature of species: empirical evidence for the ease of speciation. Phil Trans R Soc B. 2008;363:2971–86. doi: 10.1098/rstb.2008.0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Didelot X, Maiden MCJ. Impact of recombination on bacterial evolution. Trends Microbiol. 2010;18:315–22. doi: 10.1016/j.tim.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guttman DS, Dykhuizen DE. Clonal divergence in Escherichia coli as a result of recombination, not mutation. Science. 1994;266:1380–3. doi: 10.1126/science.7973728. [DOI] [PubMed] [Google Scholar]
- 44.Goldschmidt R. The material basis of evolution. New Haven, CT: Yale University Press; 1940. [Google Scholar]
- 45.Koeppel A, Perry EB, Sikorski J, Krizanc D, Warner A, Ward DM, et al. Identifying the fundamental units of bacterial diversity: a paradigm shift to incorporate ecology into bacterial systematics. Proc Natl Acad Sci U S A. 2008;105:2504–9. doi: 10.1073/pnas.0712205105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roohani R, Najafabadi MS, Alavi SM, Farrokhi N, Shams-bakhsh M. Transcript analysis of some pathogenicity-related elements in an Iranian A* isolate of Xanthomonas citri subsp. citri. J Crop Prot. 2012;1:337–47. [Google Scholar]
- 47.Büttner D, Bonas U. Regulation and secretion of Xanthomonas virulence factors. FEMS Microbiol Rev. 2010;34:107–33. doi: 10.1111/j.1574-6976.2009.00192.x. [DOI] [PubMed] [Google Scholar]
- 48.Ryan RP, Fouhy Y, Lucey JF, Jiang B-L, He Y-Q, Feng J-X, et al. Cyclic di-GMP signalling in the virulence and environmental adaptation of Xanthomonas campestris. Mol Microbiol. 2007;63:429–42. doi: 10.1111/j.1365-2958.2006.05531.x. [DOI] [PubMed] [Google Scholar]
- 49.Slater H, Alvarez-Morales A, Barber CE, Daniels MJ, Dow JM. A two-component system involving an HD-GYP domain protein links cell–cell signalling to pathogenicity gene expression in Xanthomonas campestris. Mol Microbiol. 2000;38:986–1003. doi: 10.1046/j.1365-2958.2000.02196.x. [DOI] [PubMed] [Google Scholar]
- 50.Ryan RP, Fouhy Y, Lucey JF, Crossman LC, Spiro S, He YW, et al. Cell-cell signaling in Xanthomonas campestris involves an HD-GYP domain protein that functions in cyclic di-GMP turnover. Proc Natl Acad Sci U S A. 2006;103:6712–7. doi: 10.1073/pnas.0600345103. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 51.Gupta SD, Wu HC, Rick PD. A Salmonella typhimurium genetic locus which confers copper tolerance on copper-sensitive mutants of Escherichia coli. J Bacteriol. 1997;179:4977–84. doi: 10.1128/jb.179.16.4977-4984.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Palmieri ACB, Amaral AM, Homem RA, Machado MA. Differential expression of pathogenicity- and virulence-related genes of Xanthomonas axonopodis pv. citri under copper stress. Genet Mol Biol. 2010;33:348–53. doi: 10.1590/S1415-47572010005000030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ritchie D. Copper-containing fungicides/bactericides and their use in management of bacterial spot on peaches. Southeast Reg Newsl. 2004;4:1. [Google Scholar]
- 54.Katzen F, Beckwith J. Role and location of the unusual redox-active cysteines in the hydrophobic domain of the transmembrane electron transporter DsbD. Proc Natl Acad Sci U S A. 2003;100:10471–6. doi: 10.1073/pnas.1334136100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sella G, Petrov DA, Przeworski M, Andolfatto P. Pervasive natural selection in the Drosophila genome? PLoS Genet. 2009;5:e1000495. doi: 10.1371/journal.pgen.1000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu Q-S, Zou Y-N, He X-H. Exogenous putrescine, not spermine or spermidine, enhances root mycorrhizal development and plant growth of trifoliate orange (Poncirus trifoliata) seedlings. Int J Agr Biol. 2010;12:576–80. [Google Scholar]
- 57.Erb M, Lenk C, Degenhardt J, Turlings TC. The underestimated role of roots in defense against leaf attackers. Trends Plant Sci. 2009;14:653–9. doi: 10.1016/j.tplants.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 58.Pestana M, de Varennes A, Goss M, Abadía J, Faria E. Floral analysis as a tool to diagnose iron chlorosis in orange trees. Plant Soil. 2004;259:287–95. doi: 10.1023/B:PLSO.0000020979.83817.b4. [DOI] [Google Scholar]
- 59.Hossain T, Alam Z, Absar N. Changes in different nutrients and enzyme contents in mango leaves infected with Colletotrichum glaeosorioides. Ind Phytopayhol. 1999;52:75–6. [Google Scholar]
- 60.Cornelis GR. The type III secretion injectisome. Nat Rev Microbiol. 2006;4:811–25. doi: 10.1038/nrmicro1526. [DOI] [PubMed] [Google Scholar]
- 61.Cornelis GR, Van Gijsegem F. Assembly and function of type III secretory systems. Annu Rev Microbiol. 2000;54:735–74. doi: 10.1146/annurev.micro.54.1.735. [DOI] [PubMed] [Google Scholar]
- 62.Tampakaki A, Fadouloglou V, Gazi A, Panopoulos N, Kokkinidis M. Conserved features of type III secretion. Cell Microbiol. 2004;6:805–16. doi: 10.1111/j.1462-5822.2004.00432.x. [DOI] [PubMed] [Google Scholar]
- 63.Lee YH, Kolade OO, Nomura K, Arvidson DN, He SY. Use of dominant-negative HrpA mutants to dissect Hrp pilus assembly and type III secretion in Pseudomonas syringae pv. tomato. J Biol Chem. 2005;280:21409–17. doi: 10.1074/jbc.M500972200. [DOI] [PubMed] [Google Scholar]
- 64.Weber E, Koebnik R. Domain structure of HrpE, the Hrp pilus subunit of Xanthomonas campestris pv. vesicatoria. J Bacteriol. 2005;187:6175–86. doi: 10.1128/JB.187.17.6175-6186.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weber E, Koebnik R. Positive selection of the Hrp pilin HrpE of the plant pathogen Xanthomonas. J Bacteriol. 2006;188:1405–10. doi: 10.1128/JB.188.4.1405-1410.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guttman DS, Gropp SJ, Morgan RL, Wang PW. Diversifying selection drives the evolution of the type III secretion system pilus of Pseudomonas syringae. Mol Biol Evol. 2006;23:2342–54. doi: 10.1093/molbev/msl103. [DOI] [PubMed] [Google Scholar]
- 67.Jacob TR, de Laia ML, Moreira LM, Goncalves JF, de Souza Carvalho FM, Ferro MIT, et al. Type IV secretion system is not involved in infection process in citrus. Int J Microbiol. 2014;2014:9. doi: 10.1155/2014/763575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Garnett JA, Martínez-Santos VI, Saldaña Z, Pape T, Hawthorne W, Chan J, et al. Structural insights into the biogenesis and biofilm formation by the Escherichia coli common pilus. Proc Natl Acad Sci U S A. 2012;109:3950–5. doi: 10.1073/pnas.1106733109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wong KKY, Tan LUL, Saddler JN. Multiplicity of beta-1,4-xylanase in microorganisms: functions and applications. Microbiol Rev. 1988;52:305–17. doi: 10.1128/mr.52.3.305-317.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Szczesny R, Jordan M, Schramm C, Schulz S, Cogez V, Bonas U, et al. Functional characterization of the Xcs and Xps type II secretion systems from the plant pathogenic bacterium Xanthomonas campestris pv vesicatoria. New Phytol. 2010;187:983–1002. doi: 10.1111/j.1469-8137.2010.03312.x. [DOI] [PubMed] [Google Scholar]
- 71.Rajeshwari R, Jha G, Sonti RV. Role of an in planta-expressed xylanase of Xanthomonas oryzae pv. oryzae in promoting virulence on rice. Mol Plant-Microbe Interact. 2005;18:830–7. doi: 10.1094/MPMI-18-0830. [DOI] [PubMed] [Google Scholar]
- 72.Bentley DR. Whole-genome re-sequencing. Curr Opin Genet Dev. 2006;16:545–52. doi: 10.1016/j.gde.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 73.Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM, Birol İ. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19:1117–23. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinforma. 2011;27:578–9. doi: 10.1093/bioinformatics/btq683. [DOI] [PubMed] [Google Scholar]
- 75.Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999;27:4636–41. doi: 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 77.Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, et al. The COG database: an updated version includes eukaryotes. BMC Bioinforma. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:0955–64. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lagesen K, Hallin P, Rødland EA, Stærfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–8. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fuchsman CA, Rocap G. Whole-genome reciprocal BLAST analysis reveals that planctomycetes do not share an unusually large number of genes with Eukarya and Archaea. Appl Environ Microbiol. 2006;72:6841–4. doi: 10.1128/AEM.00429-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Larkin M, Blackshields G, Brown N, Chenna R, McGettigan P, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–8. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 82.Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34:W609–12. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinforma. 2006;22:2688–90. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 84.Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 86.Padidam M, Sawyer S, Fauquet CM. Possible emergence of new geminiviruses by frequent recombination. Virol. 1999;265:218–25. doi: 10.1006/viro.1999.0056. [DOI] [PubMed] [Google Scholar]
- 87.Bruen TC, Philippe H, Bryant D. A simple and robust statistical test for detecting the presence of recombination. Genet. 2006;172:2665–81. doi: 10.1534/genetics.105.048975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–91. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 89.Yang Z, Wong WS, Nielsen R. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol. 2005;22:1107–18. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]