

Abstract
Two lineages of Salmonella enterica serovar Typhimurium (S. Typhimurium) of multi-locus sequence type ST313 have been linked with the emergence of invasive Salmonella disease across sub-Saharan Africa. The expansion of these lineages has a temporal association with the HIV pandemic and antibiotic usage. We analysed the whole genome sequence of 129 ST313 isolates representative of the two lineages and found evidence of lineage-specific genome degradation, with some similarities to that observed in S. Typhi. Individual ST313 S. Typhimurium isolates exhibit a distinct metabolic signature and modified enteropathogenesis in both a murine and cattle model of colitis, compared to S. Typhimurium outside of the ST313 lineages. These data define phenotypes that distinguish ST313 isolates from other S. Typhimurium and may represent adaptation to a distinct pathogenesis and lifestyle linked to an-immuno-compromised human population.
Author Summary
Salmonella enterica is a diverse species, isolates of which can colonise or infect many different animals, including humans and can cause different disease syndromes. S. enterica can be sub-typed using serology into serovars. Isolates from some serovars, known as generalists, can infect multiple hosts (e.g. S. Typhimurium) and usually cause gastroenteritis. However, other serovars exhibit host adaptation or even restriction. Host-adapted serovars such as S. Dublin show preference for a particular host but can also infect other hosts, while host-restricted serovars are capable of infecting only a single host (e.g. S. Typhi in humans) and frequently cause febrile systemic disease (typhoid). In this study, we use genotypic and phenotypic methods to investigate clinical isolates representative of populations of two recently emerged S. Typhimurium lineages of type ST313 associated with invasive disease in sub-Saharan Africa. Our results identify potential characteristics in these isolates that may be associated with adaptation to invasive disease in humans with a compromised immunity.
Introduction
Salmonella enterica isolates can infect a range of animals and humans, causing a spectrum of disease syndromes ranging from gastroenteritis through to typhoid and an asymptomatic carrier state [1]. From a clinical perspective S. enterica serovars have been classically assigned to two broad groups, typhoidal or non-typhoidal Salmonella (NTS). Typhoidal Salmonella include the human restricted S. enterica serovar Typhi (S. Typhi), the cause of the systemic disease typhoid fever, which is strictly transmitted within the human population independently of a zoonotic reservoir. NTS, on the other hand, are predominantly associated with self-limiting gastroenteritis, largely originating from zoonotic reservoirs with human-to-human transmission regarded as being relatively rare [2].
Invasive NTS (iNTS) disease in sub-Saharan Africa does not fit well into the classical view of salmonellosis. NTS has emerged as a significant cause of invasive human disease, exceeding S. Typhi in many parts of the region as the leading cause of invasive salmonellosis. Humans can be predisposed to this disease by immune suppression or co-infections, which include severe malaria in children and HIV in adults [3,4]. Invasive NTS clinical syndrome is somewhat dissimilar to both typhoid fever and gastroenteritis, and includes non-specific fever and only sporadic or limited diarrhea [5]. High case fatalities have been reported in children and adults in the absence of adequate treatment [6–9].
We recently reported that the emergence of iNTS disease within the sub-Saharan region has been associated with the emergence of two closely-related, multi-antibiotic resistant lineages of S. Typhimurium that belong to multilocus sequence type (MLST) ST313 [10]. Phylogenetic analysis indicated that these ST313 lineages emerged independently in recent decades, in close temporal association with the HIV pandemic [10]. As no obvious zoonotic source of ST313 S. Typhimurium has been identified, it has been postulated that these lineages may be undergoing host adaptation to humans and may be transmitted, at least in part, directly from human-to-human [11]. Additionally, emergence of the lineages was concomitant with acquisition of multidrug resistance (MDR) including chloramphenicol in one lineage.
Genome sequencing of a representative ST313 isolate, D23580 from Malawi, identified distinct genetic signatures not present in other sequenced non-ST313 S. Typhimurium [5]. For example, the genome of D23580 exhibited considerable genome degradation with some similarity to that observed in S. Typhi [5]. Genome degradation, in the form of the accumulation of so-called pseudogenes, is a signature of some host restricted pathogens including Bordetella pertussis [12,13], S. Typhi [14,15], S. Paratyphi [15,16] and S. Gallinarum [16]. Here, a population-based approach was used to assess how genome degradation emerged within the ST313 lineages. In addition, we used a range of approaches to phenotype representatives of the ST313 in an effort to link the genotypic differences to metabolic and virulence-associated phenotypic differences.
Results
Genome degradation is evident across the S. Typhimurium ST313 lineages
ST313 isolates fall into two closely related phylogenetic lineages that are distinct from other S. Typhimurium (Fig. 1A, S1 Fig, S1 Table) [10]. Previous genome sequence analysis of one ST313 isolate, D23580 from lineage II, revealed both gene acquisition (e.g. novel phage elements) and genome degradation (e.g. deletions and pseudogenes) in comparison to S. Typhimurium in other lineages [5]. To ascertain genome variation within the overall ST313 population, we analysed whole genome sequences of 129 ST313 isolates using the ST19 S. Typhimurium SL1344 genome as a reference.
Fig 1. Characterisation and distribution of SNPs in ST313 lineage I and lineage II.
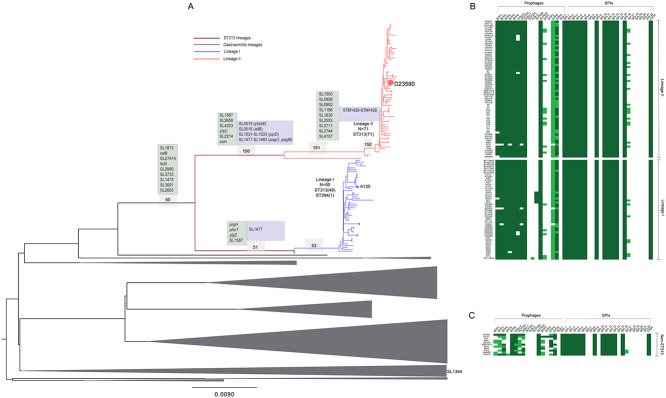
A. Unrooted maximum likelihood tree showing relationships between lineage I (blue), lineage II (red) and gastroenteritis-associated isolates (grey polygons). Size of polygons represents numbers of taxa in the clade. Representative isolates from each group are highlighted within each clade. Scale bar indicates substitutions per variable site. Numbers in light grey boxes indicate parental branches leading to the respective ST313 lineages as shown in Table 1. Text boxes indicate selected degraded genes (pseudogenes—light green and deletions—light blue occurring on the respective branches). Re-used with permission from Okoro et al., Nat Genet. 2012. 44(11): 1215–21. doi: 10.1038/ng.2423. List of degraded complement for strain D23580 is adapted from Kingsley et al., 2009. B. Distribution of prophage elements and Salmonella pathogenicity islands in ST313 lineage I and II. Top left panel represents concatenated phage sequences from strains D23580 (‘BTP’), SL1344 (‘SL’) and DT104 (‘DT’). Top right panel represents concatenated sequences of coding and non-coding sequences of SPI-1 to SPI-22 and CSS4 island. Sequence reads mapping to the complete feature length is represented as a heatmap. Green colour indicates >90% (high) coverage; light green indicates >30% but <90% coverage; and white indicates <30% (low) coverage. Isolate order in lineages I and II on left hand panel although not to scale, is according to phylogenetic positioning in 1A. C Distribution of phages and SPIs in selected non-ST313 isolates.
We first identified the synonymous and non-synonymous single nucleotide polymorphisms (SNPs) in the S. Typhimurium ST313 lineages compared to the reference (Fig. 1A, Table 1). The dN/dS ratios for the parental branch of ST313, and that of each lineage since divergence from the last common ancestor, were similar (0.41 ± 0.13 s.d and 0.35 ± 0.007 s.d, respectively, Table 1). Thus, the dN/dS values were smaller than one but still elevated and similar to those expected for recently evolved lineages where time has been too short for purifying selection to act to a significant level [17,18]. The relatively high proportion of non-synonymous SNPs in the two lineages may also represent segregating polymorphisms rather than fixed mutations. Many of these SNPs, both synonymous and non-synonymous, were in metabolic genes and genes involved in degradation of small molecules in both lineages, when compared with SL1344 (Table 1, Fig. 2A). A proportion of the SNPs were also found in genes with no assigned function.
Table 1. Genetic variation detected in identified human invasive S. Typhimurium lineages.
| Variation type | Node | SNPS (Total) | Synonymous | Non-synonymous | Nonsense | Intergenic | Homoplasic | dN/dS | Mean±s.d |
|---|---|---|---|---|---|---|---|---|---|
| Conserved Variation | |||||||||
| Lineage I & II | 50 | 126 | 51 (40.5%) | 51 (40.5%) | 4 (3.2%) | 20 (15.9%) | 22(17.5%) | 0.32 | |
| Lineage I | 51 | 12 | 3 (25.0%) | 4 (33.3%) | 0 (0.0%) | 5 (41.7%) | 5(41.7%) | 0.43 | |
| 53 | 215 | 74 (34.4%) | 99 (46.0%) | 3 (1.4%) | 39 (18.1%) | 12 (5.6%) | 0.43 | ||
| Lineage II | 150 | 127 | 36 (28.3%) | 72 (56.7%) | 2 (1.6%) | 17 (13.4%) | 5 (3.9%) | 0.65 | |
| 151 | 88 | 32 (36.4%) | 37 (42.0%) | 1 (1.1%) | 18 (20.5%) | 2 (2.3%) | 0.37 | ||
| 152 | 54 | 7 (13.0%) | 6 (11.1%) | 0 (0.0%) | 41 (75.9%) | 3 (5.6%) | 0.28 | 0.41±0.13 | |
| Most recent variation | |||||||||
| Lineage I | 335 | 106 (31.6%) | 144 (43.0%) | 13 (3.9%) | 72 (21.5%) | 35 (10.4%) | 0.35 | ||
| Lineage II | 316 | 84 (26.6%) | 159 (50.3%) | 8 (2.5%) | 64 (20.3%) | 28 (8.9%) | 0.34 | 0.35 ±0.007 | |
The frequencies of mutation are in two groups. Percentages give relative frequency of SNP classes within each group and the two ST313 lineages. Last two columns give the mean dN/dS for lineage and standard deviation (s.d.) from the mean.
Fig 2. Functional characterisation of SNPs, pseudogenes and small indels in ST313 populations.

A. Functional characterisation of non-synonymous (black bars) and synonymous (grey bars) of SNPs in lineages I and II. x-axis shows functional categories used for SNP characterisations; y-axis shows proportion of mutated genes (%) to the no of genes belonging to the functional categories. B. Functional characterisation of degraded gene complement (pseudogenes and deletions) in lineage I (blue bars) and lineage II (red bars). x-axis represents % proportion of pseudogenes in each functional group. Asterisks indicate significantly over-represented group with p < 0.05 using a two-way ANOVA with post-tests performed using the Bonferroni method.
To further characterise the acquisition or loss of genetic material by the ST313 lineages we analysed the whole genome sequence of the additional sequenced isolates. There was little variation in the arrangement of genes within the major virulence-associated Salmonella pathogenicity islands (SPIs), including SPIs -1 to -6, -9, -11 to -14, and -16 (Fig. 1B). The previously described prophage elements BTP1, BTP3, BTP4 and BTP6 [5] were present in all isolates and putative deletion events have led to the loss of the phage remnants SLP281 and Fels2 compared to S. Typhimurium SL1344 and other non-ST313 isolates included in the analyses. Sequences with similarity to the phage SLP289 were found in a subset of lineage I isolates from Uganda and Kenya but were absent from the rest of the ST313 population. Whole or partial sequences of the S. Typhimurium DT104-associated prophage 5 (Fig. 1B) were present in both ST313 lineages, although there was no obvious pattern to the distribution of particular rearrangements of this phage within the ST313 tree. Thus, these data catalogue the major insertions and potential deletions that have occurred since the divergence of ST313 lineages from the last common ancestor.
Pseudogene formation common to both ST313 lineages
The ST313 sequences were next analysed for evidence of pseudogene formation arising from nonsense SNPs and frame-shift mutation caused by insertions or deletions (<20bp) impacting on all ST313 S. Typhimurium used in our analyses. Ten pseudogenes were present in all ST313 isolates but intact in SL1344, including the genes ttdA, ratB and SL1567 (Fig. 1A, S3 Table). ttdA encodes L(+)-tartrate dehydratase, involved in glyoxylate and dicarboxylate metabolism [19]. The gene ttdA is also a pseudogene in S. Typhi and S. Paratyphi A. The ratB gene which encodes an outer membrane protein implicated in intestinal persistence in a murine model, is also a pseudogene in S. Typhi, S. Paratyphi A, S. Paratyphi B and the fowl-restricted S. Gallinarum [5,20,21]. Other pseudogenes found in all sequenced ST313 include SL1567, a membrane associated protein with different independently acquired nonsense SNPs in lineage I and II, SL2747A, a putative exported protein which may be involved in phospholipid biosynthesis and a transposase, SL1873 (S3 Table). Five pseudogenes were a result of frame-shift mutations (Fig. 1A, S3 Table). These affected the genes SL2990, SL3733, SL1475, SL13051, SL2653, which are predicted to be involved in transcriptional regulation, metabolism and transport and annotated as possessing conserved hypothetical functions, respectively. Most of these genomic signatures represent degradation that occurred before divergence from the last common ancestor of both ST313 lineages.
Further lineage-specific degradation across the ST313 lineage
In addition to shared genome degradation, lineage-specific nonsense SNPs, frame-shift mutations and small deletions are also present in genes of isolates from ST313 lineages I or II. These represent degradation that occurred after divergence of the two ST313 lineages. Three additional candidate pseudogenes were found in all lineage I isolates. These were in prfH, a peptide chain release factor, phnT, a probable ATP-binding component of 2-aminoethylphosphonate transporter and ybjZ a putative ABC transporter (Fig. 1A, S3 Table); Lineage I isolates also harbour a 700 bp partial deletion within a putative phage gene SL1477. In lineage II, three candidate pseudogenes were a consequence of nonsense SNPs while there was an insertion within SL2214, a putative phage protein in an O-antigen modification locus. The lineage II candidate pseudogenes arising from nonsense SNPs include the gene encoding a conserved hypothetical protein, SL2659 and the membrane proteins yhjU and SL4223. The sseI gene, which encodes a type III effector, is inactivated by an IS200 element in all lineage II but not lineage I isolates in our collection. Genes associated with allantoin metabolism or transport e.g. allB, gcl, glp and ybbW are likely pseudogenes in lineage II. Interestingly genes associated with allantoin metabolism are also inactivated in S. Typhi, S. Paratyphi A and S. Gallinarum. All lineage II isolates possess a partial deletion of the pipD gene, encoding a SPI-5 associated protein implicated in persistence in murine macrophages and fluid secretion in bovine models [22,23] (S2 Table). The phoP/phoQ regulated genes, pagO and pagM harbour deletions in all lineage II isolates (Fig. 1A, S2 Table). It is important to note that a number of phoP/phoQ-regulated genes are associated with virulence [24] and pagO has been previously linked to virulence in porcine models [25]. Additionally, a 4.2kb region encoding plasmid stability proteins was also deleted in all lineage II isolates (S2 Table). There is a statistically significant over-representation of surface/membrane associated and exported proteins inactivated in both lineages (p value <0.05) (Fig. 2B).
S. Typhimurium ST313 isolates exhibit a distinct metabolic profile
A systematic analysis of 576 metabolic activities was performed using Biolog phenotype microarrays (PM)[26] on three representatives each of the two ST313 lineages and four S. Typhimurium ST19 isolates including SL1344 that acted as experimental controls and comparators (S1 Table). A principal component analysis (PCA) (Fig. 3A) and a hierarchical clustering of Biolog signal values (Fig. 3B) were employed to assess the data sets (S1 Text, S2 Fig). The results from both analyses support the conclusion that ST313 isolates share similar metabolic capacity distinct from ST19 S. Typhimurium isolates. For example, analyses of cellular respiration over 48 hours of incubation showed that ST313 isolates exploit particular carbon sources such as meso-tartaric acid (meso-tartrate) and tricarballylic acid more readily than S. Typhimurium ST19 isolates included in the experiment (Table 2). Conversely, the ST19 isolates, which includes SL1344, utilised carbon sources such as L-tartaric acid and dihydroxyacetone (Table 2). The differing metabolism of L-tartaric acid and meso-tartaric acid by ST313 and ST19 corroborates the observation that ttdA, encoding the stereo-specific enzyme tartrate dehydratase, is a pseudogene in ST313 isolates [27,28] [29].
Fig 3. Metabolic profile of ST313 lineage I and lineage II isolates.
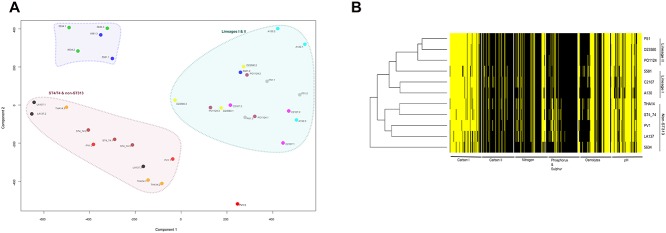
A. Principal component analyses of experiments on isolates and replicates used for 576 metabolic and physiological tests. The axes represent the two principal components (components 1 and 2) that explain the largest amounts of variation observed. Large coloured polygons represent identified clusters as labelled. Replicates of isolates sharing the same polygon colour are labelled with the name of isolates with number suffixes indicating replicate numbers. B. Representation of active (yellow) and non-active (black) wells. Varying shades of grey represent ambiguous (possibly active) results.
Table 2. Summary of metabolites with significantly different impact on respiration.
| Test metabolite / comparison | P-adjusted | Signal value difference |
|---|---|---|
| 48 hours: SL1344 vs. lineages 1 & II | ||
| Dihdroxy Acetone | 3.24E-02 | 123.71 |
| L-Tartaric Acid | 1.49E-03 | 153.96 |
| Tricarballylic Acid | 6.10E-04 | -198.57 |
| M-Tartaric acid | 4.19E-08 | -262.43 |
| 48 hours: lineage 1 vs lineage II | ||
| No significant results | ||
| 15 hours: SL1344 vs. lineage I | ||
| Guanosine- 2'—monophosphate | 1.90E-05 | 104.27 |
| pH 4.5 + L-Histidine | 8.00E-02 | 110.03 |
| D-Glucosaminic acid | 9.63E-03 | 113.88 |
| Cytidine—2', 3'—cyclic monophosphate | 3.98E-11 | 115.94 |
| Guanosine- 3'—monophosphate | 1.16E-05 | 115.97 |
| pH 4.5 + L-Aspartic acid | 4.07E-02 | 126.93 |
| Cytidine—3'—monophosphate | 6.65E-06 | 129.25 |
| Uridine-3'-monophosphate | 6.11E-06 | 132.29 |
| Uridine- 2',3' cyclic monophosphate | 1.37E-11 | 134.18 |
| Cytidine-2'- monophosphate | 1.90E-05 | 138.46 |
| Tricarballylic acid | 3.32E-02 | -135.86 |
| M-Tartaric acid | 5.90E-02 | -85.28 |
| 15 hours: SL1344 vs. lineage II | ||
| Oxalomalic acid | 8.02E-12 | 185.83 |
| Cytidine- 2'—monophosphate | 1.12E-07 | 148.82 |
| Uridine-3-monophosphate | 3.41E-07 | 136.92 |
| Uridine-2-rsquo;3'-cyclic monophosphate | 2.83E-12 | 136.14 |
| Cytidine- 3'-monophosphate | 1.92E-08 | 135.73 |
| D-Glucosamic acid | 8.18E-07 | 129.02 |
| Guanosine-5'-monophosphate | 9.46E-12 | 124.43 |
| Cytidine-2-3'-cyclic monophosphate | 2.83E-12 | 116.93 |
| alpha-methyl-D-Galactoside | 1.20E-05 | 116.19 |
| Bromo Succinic acid | 6.93E-04 | 115.53 |
| Guanosine-3'—monophosphate | 1.85E-06 | 108.37 |
| M-tartaric acid | 3.21E-03 | -71.64 |
| Tricarballylic acid | 6.87E-03 | -121.87 |
| I5 hours: lineages I & II vs. non-ST313 | ||
| Uridine- 2-3'-cyclic monophosphate | 5.15E-18 | -140.74 |
| Cytidine-3'-monophosphate | 1.05E-14 | -138.78 |
| Cytidine-2'-monophosphate | 1.06E-12 | -134.05 |
| D-Glucosaminic acid | 7.35E-10 | -132.12 |
| Cytidine- 2-3'- cyclic monophosphate | 7.63E-16 | -124.80 |
| Melibionic Acid | 3.03E-03 | -109.58 |
| Guanosine- 3'-monophosphate | 1.09E-08 | -101.13 |
Negative signal value difference represent metabolites that were utilised more by isolates in lineages I and II; positive signal values represent metabolites utilized more by SL1344 or other non-ST313 isolates. Significance is reported at signal values ≥ 100 and p-adjusted values of ≤ 0.05. Results similar to the 48 hour profiles with adjusted p-values of <0.05 were also reported for the 15 hour profiles.
S. Typhimurium ST313 isolates exhibit reduced enteropathogenicity
To determine if ST313 isolates are virulent in a mouse systemic infection model we orally inoculated genetically susceptible mice (NRAMP1-, C57bl/6) with representative ST313 isolates from lineage I and II. The resulting data showed that the tested ST313 isolates are indeed able to colonise systemic sites in this model (S3 Fig). We therefore investigated the ability of representative isolates of ST313 (A130 from lineage I and D23580 from lineage II) to induce an inflammatory response in the caecum of orally inoculated streptomycin pre-treated C57bl/6 mice, compared with SL1344 (Fig. 4). No significant difference in Salmonella colonisation of the caecum was evident at 48 hours post-inoculation (S4 Fig). SL1344 induced pronounced inflammation characterised by marked oedema in the submucosa with moderate to marked cellular inflammatory infiltrates in the submucosa and mucosa, with numerous crypt abscesses and erosive changes in the surface epithelium (Fig. 4C & 4D). However, these pathological signatures were less common in mice infected with A130 (Fig. 4E) or D23580 (Fig. 4F), although there was some evidence of mild to moderate submucosal oedema and mild inflammatory cell infiltration into the submucosa and mucosa. The epithelial surface changes and crypt abscesses were also much less prominent. SL1344ΔorgA, a SPI-1 defective derivative induced similar levels of inflammatory cell infiltration into the mucosa and submucosa of the caeca to A130 and D23580 (Fig. 4B). Uninfected caecum exhibited no noticeable oedema or neutrophil infiltration (Fig. 4A). The histopathological scores of the replicate experiments summarised in Fig. 4G & 4H illustrate these observations. In further experiments, groups of streptomycin pretreated 129P2/olaHsd mice were independently inoculated with the same S. Typhimurium isolates as in the previous experiment in C57bl/6 mice. Similar differences in intestinal pathology in the caecum were observed 48 hours post-inoculation (Fig. 4H).
Fig 4. Histopathological analysis of Salmonella induced caecal inflammation of streptomycin-pre-treated mice.

A single representative caecal wall image from each group is shown. A. naïve uninfected mouse. B. Infection with SL1344ΔorgA (SPI-1 mutant derivative). C & D. Infection with SL1344. E. Infection with A130 (lineage 1); F. Infection with D23580 (lineage II). Images A, C, D, E & F were taken at x100 magnification of original size (image B is x50 magnification). Abbreviations: l, intestinal lumen; sm, submucosa; oe, submucosal oedema; pmn (polymorphonuclear leukocytes) infiltrate; sc, surface changes; ca, crypt abcesses. Histopathology score of inflammatory changes in the caecum of streptomycin pretreated C57bl/6 mice (G) or 129P2/olaHsd mice (H) with ST313 isolates or strain SL1344, two or three days post inoculation. The scores of five inflammatory markers according to the key are indicated.
The overall virulence profiles observed in the streptomycin-treated mouse model of colitis were also evident in a bovine ligated ileal loop model. In these studies, ligated segments of the mid-ileum of two calves were infected in triplicate with representative invasive ST313 isolates from the two lineages (lineage I—A130 & 5597; lineage II- D23580 & 5579) and compared to bovine virulent ST19 S. Typhimurium strains ST4/74, DT104 and IR715 (S1 Table) and internal negative controls. Secretory and inflammatory responses in this model are strongly influenced by SPI-1 (prgH mutation; Fig. 5), as previously described [30]. In pair-wise t-tests, a significant difference in fluid accumulation was detected 12 hours post-inoculation between ST19 and ST313 isolates in almost all cases (Fig. 5). Mean values for the secretory response to the three ST19 isolates also differed significantly from the mean value for the four ST313 isolates (p = 0.02). Recruitment of 111Indium oxinate-labelled polymorphonuclear leukocytes (PMN) relative to the negative control (PMN influx) also differed significantly for a number of ST19 and ST313 isolates in pair-wise combinations (Fig. 5). Though the difference in mean values for PMN influx for all ST19 vs. ST313 was marginally not significant (p = 0.065), the difference was significant for PMN recruitment to the luminal contents by ST19 vs. ST313 (p = 0.04).
Fig 5. Secretory and inflammatory responses induced by ST19 (grey bars) and ST313 S. Typhimurium strains (maroon bars) in bovine ligated ileal loops.

A. Mean fluid accumulation normalised to loop length [volume (mL)/length (cm)]. B. Influx of 111In-labelled PMN for test strains normalized to loop length and negative control loops, as described in Methods. Values represent the mean ± SEM of triplicate determinations in two independent calves. Right panel of A and B shows summary of paired T-test results. Asterisks show statistically significant pairwise comparisons. C. Influx of 111Indium oxinate-labelled polymorphonuclear leukocytes (PMN) induced by ST313 isolates relative to the negative control.
Discussion
Here, we have identified lineage-specific signatures and phenotypic changes that differentiate ST313 from other S. Typhimurium, including isolates associated with gastroenteritis. These findings extend on previous analyses of D23580, a lineage II ST313 isolate to the broader ST313 population [5]. We identified and highlighted lineage specific gene acquisition and loss events, some common to both ST313 lineages and others restricted to either lineage I or II. Among the collective changes that have accompanied the emergence of ST313 are a relatively high proportion of genomic changes found in metabolic genes (Fig. 2A). This is worthy of note since altered metabolic capacity has previously been associated with adaptation of Salmonella serotypes to extra-intestinal niches [31]. The high numbers of SNPs within this class of gene could also be indicative of evolutionary pressure acting on the ST313 isolates. Examples of genes within this group include ttdA, that are also found either deleted or are pseudogenes in host-restricted or host-adapted serovars such as S. Typhi, S. Paratyphi A, S. Paratyphi B and S. Gallinarum. This ability to utilise less common carbon sources such as meso-tartrate and tricarballylic acid by the ST313 isolates may positively influence their fitness in a new ecological niche.
Degradation of aerobic metabolic genes in the isolates of the two lineages may suggest a preferential loss or reduction of aerobic metabolic capacity in ST313. This observation could be indicative of a heightened ability for anaerobic metabolism following internalisation within macrophages, as anaerobic respiration and metabolism takes precedence over aerobic metabolism within this niche. These metabolic activities impact the interaction of the pathogen with the host in the intracellular niche and have implications for intracellular compartmentalisation within tissues such as the bone marrow, as previously reported [32,33].
Further evidence for the clonality of the two epidemic lineages in sub-Saharan Africa is emphasized by the predominantly conserved pattern of the known S. Typhimurium phages and genomic islands. Although we present details of common SNPs in ncRNAs found in intergenic regions of the genomes in ST313 (S1 Text), the impact of these SNPs on regulation and subsequently on metabolism or virulence-associated phenotypes are difficult to predict and will be the subject of future investigation.
Surface proteins are often antigenic in nature and are part of the first line of contact with the host immune system. Changes in surface proteins can thus impact on the host response to colonisation and invasion by these pathovariants. The so-called ‘stealth’ methods employed by host-adapted serovars to evade host gut inflammatory responses leading to increased invasive capability have been well documented [34]. Thus, potential inactivation of genes such as pagO [23,25], pipD [22], ratB [20] and sseI [5] in ST313 isolates is interesting in this regard (Table 3). These proteins are all exported or membrane surface-associated proteins implicated in the establishment of gastrointestinal infection or long-term systemic infections in animal models. Such defects have been observed in the differential virulence profiles observed in pigs infected with S. Typhimurium, which cause non-fatal but acute enteritis, and S. Choleraesuis, which is host-adapted and frequently causes a severe systemic disease in pigs. In a porcine ligated ileal loop model, S. Typhmiurium elicits a profound inflammatory response, which subsequently controls and confines the pathogen to the intestinal mucosa. Conversely, the host-adapted S. Choleraesuis replicated slowly and elicited weaker pro-inflammatory responses both of which may facilitate avoidance of the host immune response by stealth [35]. Although we do not show a direct causal link, it is also possible that the inactivation of these virulence-associated proteins has led to the reduction in the enteropathogenic potential of isolates in the lineages I and II (Fig. 4 & 5, respectively).
Table 3. Gene accession numbers/IDs.
| Gene | Accession number | |
|---|---|---|
| Uniprot | EMBL | |
| allB | E1W930 | CBW16615 |
| gcl | E1W925 | CBW16610 |
| gip | E1W926 | CBW16611 |
| orgAa | E1WAB5 | CBW18948 |
| orgAb | E1WAB4 | CBW18947 |
| pagM | E1WGA4 | CBW17891 |
| pagO | E1WGA0 | CBW17887 |
| phnT | E1W8T8 | CBW16522 |
| phoP | E1WFA1 | CBW17265 |
| phoQ | E1WFA0 | CBW17264 |
| pipD | E1W7C5 | CBW17129 |
| prgH | E1WAB9 | CBW18952 |
| ratB | CBW18577 | |
| SL3051 | E1WHL5 | CBW19150 |
| SL1475 | E1WBM7 | CBW17570 |
| SL1567 | E1WBW8 | CBW17662 |
| SL1873 | E1WGH9 | CBW17967 |
| SL2214 | E1WC80 | CBW18310 |
| SL2653 | E1WJG7 | CBW18755 |
| SL2659 | E1WJH3 | CBW18761 |
| SL2990 | E1WAQ5 | CBW19089 |
| SL3733 | E1WDJ3 | CBW19825 |
| SL4223 | E1WEX6 | CBW20309 |
| sse1 | E1WCC7 | CBW18358 |
| ttdA | E1WID8 | CBW19423 |
| ybbw | E1W929 | CBW16614 |
| ybjZ | E1W6X5 | CBW16977 |
| yhjU | E1WD54 | CBW19683 |
SL1344 gene names when not given are abbreviated to SLxxxx
S. Typhimurium ST313 is frequently associated with iNTS disease in sub-Saharan Africa. However, the extent to which this genotype is also associated with gastroenteritis in this region is poorly understood. iNTS disease syndrome is distinct from typhoid fever and gastroenteritis and thus lack an established animal model of infection. Mice with a defective Nramp1 gene are also susceptible to invasive NTS disease, so we used this infection model to determine if ST313 isolates differed in their ability to colonise systemic sites organs of the reticuloendothelial system and the gall bladder compared with the non-ST313 SL1344. All ST313 exhibited some virulence in the mice and colonised to a similar level to that observed for SL1344 (S3 Fig). This is consistent with reports that ST313 isolates can establish systemic infections in different models of infection [36]. We also evaluated the ability of ST313 isolates (A130, D23580) and ST19 (SL1344) to invade eukaryotic cells growing in vitro was using Hep2 cells. Although, all three isolates showed evidence of invasion, internalisation and replication in epithelial cells over a time course of 24 hours, A130 and D23580 consistently showed lower invasion compared to SL1344 in this in-vitro model (p < 0.001 at 24 hours) (S5 Fig, SI Text).
We have detailed the shared genomic and phenotypic variation that may contribute to the adaptation of these new pathovariants to the novel niche provided by immunocompromised humans, identifying several changes that are consistent with those found in host-adapted lineages of S. enterica. The high proportion of metabolic genes implicated in the degraded gene component in lineages I and II of ST313 is a signature that is an emerging narrative among invasive pathogens in enterobacteriaceae including Salmonella [16], Shigella [37], Yersinia [38] and E. coli [39]. Our results thus suggest adaptation within a particular human population in ST313. However, the possibility of asymptomatic carriers or environmental reservoirs being integral components of iNTS transmission networks also exists. Elucidating these networks and defining the relationship between zoonotic, environmental and human isolates remains the subject of much needed on-going research.
Methods
Bacterial isolates and culture conditions
Bacterial isolates used in this study have been described in Okoro, et al., 2012[40]. See S1 Table. All bacteria were grown on Luria-Bertani (LB) medium; single colonies were incubated in LB Broth overnight at 37°C. Descriptions of specific growth conditions for experiments are given in the corresponding segments below.
Calculating dN/dS of identified lineages
dN/dS was calculated using the formula adapted from Holt et al., 2008 (N/n)/(S/s), where N = sum of nonsynonymous SNPs, n = nonsynonymous sites in non-repetitive protein-coding sequences, S = sum of synonymous SNPs, s = synonymous sites in non-repetitive protein-coding sequences[14,41]
Functional characterisation of SNP categories
To investigate the origin of SNPs reported on the tree, SNPs were reconstructed back to the phylogenetic tree using parsimony and optimised by both ACCTRAN (accelerated transformation) and DELTRAN (delayed transformation)[42]. Both methods gave comparable results and so the results from the DELTRAN optimisation are presented here. The DELTRAN method allocates or maps SNP origins along the phylogenetic branches as close to the tips as possible [43]. This enabled frameshift mutations and premature stop codons that reduced the length of CDSs relative to their annotation in the reference genome to be detected. SNP positions, type and quality were manually confirmed by checking reads against the reference sequence and visualised using BamView[44].
Detection of distribution of insertion sequences, phages and pathogenicity islands by mapping
Paired-end sequence reads of each isolates were mapped to the multi-fasta sequence features of either insertion sequences, phages or pathogenicity islands using the Burrows-Wheeler Aligner software BWA[45], with minimum base call quality of 50, minimum mapping quality of 30, and minimum read depth of 4. Isolates from each of identified lineages were analyzed separately, by lineage. A cut-off value of < 30% of reads mapped to the length of the feature was selected as an indication of absence and > 70% as presence of the region of interest in an isolate. A heat map of the analysis based on the selected cut-off values was generated.
Biolog experiments
Culture and inoculum preparation were preformed according to a modified manufactures’ protocol (see S1 Text). A total of 576 assays were performed for each isolate, with each isolate represented by three biological replicates. Bacteria were incubated for 15–48 hours at 37°C and bacterial respiration on each assayed metabolite was measured by colorimetric redox assay. The metabolic activity and kinetics data files of each strain over time were exported from the OmniLog phenotype MicroArray (PM) program suite. Further analysis proceeded as described previously in R[46]. Signal values were calculated as in Homann et al., 2005 [47]. Log signal values displayed a clear bimodal distribution corresponding to non-respiring (background dye reduction) and respiring modes. Normal distributions were fitted to each mode, and strains were defined as respiring on a particular substrate if all 3 replicates were at least 4 times more likely to originate from the respiring distribution. Significant differences in respiration rates between isolates were assessed using a moderated t-test with the LIMMA R package [48]. P-values were corrected using the Benjamini and Hochberg method [49] to control for the false discovery rate. Results presented here are for respiration for up to 15 hours and 48 hours. Results with adjusted p-values of <0.05 and signal value differences (positive or negative) greater than or equal to 100 at 48 hours were selected as significant. Results similar to the 48 hour profiles with adjusted p-values of <0.05 were also reported for the 15 hour profiles.
Determination of common phenotypes and metabolic pathway analyses
The functions of metabolites significantly utilized to a greater or lesser degree by the invasive isolates commonly relative to SL1344 were identified for each cluster, and the list of the associated metabolites generated and analysed with Pathway Tools [50] to put them in a wider context and predict the metabolic pathways that were involved.
Ethics statement
All mouse experiments were conducted in compliance with the Animals (Scientific Procedures) Act 1986 under Home Office project licence 80/2596 with the consent and approval of the Ethical Review Committee of the Wellcome Trust Sanger Institute, UK. Mice were sacrificed by cervical dislocation at the end of the experiment. Calf ligated ileal loop experiments were conducted in compliance with the Animals (Scientific Procedures) Act 1986 under Home Office project licence 30/2485 with the consent and approval of the Ethical Review Committee of the Institute for Animal Health, UK. General anaesthesia was induced by intravenous administration of propofol and maintained by inhalation of isoflourane in oxygen for the duration of the study. Calves were given an overdose of intravenous sodium pentobarbitone at the end of the study.
Streptomycin pre-treated mouse model of colitis
S. Typhimurium isolates were grown in LB agar supplemented with appropriate antibiotic selection and incubated overnight at 37°C. Single colonies were used to inoculate LB broth and incubated overnight at 37°C. Approximately 1x107 were inoculated into each mouse. Two experiments were conducted and a total of 5 and 3 mice per isolate were used for the infections in first and second experiment, respectively. Specific pathogen-free SPF female mice C57BL/6 (groups of five) or female 129P2/olaHsd mice, (groups of three), 6–8 weeks old, were treated by oral gavage with 0.2 mL of 100 mg/mL streptomycin by oral gavage. At 20 hours after streptomycin treatment, mice were infected with 1x106 (C57BL/6 mice) or 1x107 (129P2/olaHsd mice) of S. Typhimurium in 0.2 ml of PBS pH 7.4 or treated with sterile PBS (control) by oral gavage. At 48 hours (C57BL/6) and 72 hours (129P2/olaHsd), mice were culled and two caecal tissue samples taken for enumeration of viable S. Typhimurium or were fixed with formalin for subsequent wax embedding, sectioning and tissue staining with haematoxylin/eosin (H/E) staining. Enumeration of bacteria was conducted by plating serial dilutions of caecal tissue homogenates on LB agar containing the appropriate antibiotics. Colonies were counted after overnight incubation at 37°C. The H/E stained caeca were histopathologically assessed and scored using a 4-point scale of 0, 1, 2, or 3, for five markers of vascular and cellular inflammation by using a modification of methods described in Kim, J.J. et al., 2012 [51] as follows; mucosal inflammatory cellular infiltration predominantly by neutrophils (PMNs), the presence of crypt abscesses (neutrophils within the lumen of the crypts in the mucosa), erosive and reactive changes to the epithelial surface of the mucosa, the amount of submucosal oedema assessed by the increase in thickness of the submucosa and the level of submucosal inflammatory cellular infiltration predominantly by neutrophils.
Bovine ligated ileal loop experiments
Salmonella-induced secretory and inflammatory responses in calves were quantified essentially as described previously [30]. Briefly, two 4-week-old Friesian bull calves were placed under terminal general anaesthesia, a laparotomy performed and the mid-ileum flushed with sterile PBS. In each calf, twenty-seven 6 cm loops with 1 cm spacers were constructed by ligation of the gut with surgical silk. Representative invasive ST313 isolates from the two lineages (lineage I—A130 & 5597; lineage II- D23580 &5579) and bovine virulent ST19 strains SL1344, DT104 and IR715. IR715 is a nalidixic acid-resistant derivative of strain ATCC 14028 [52]. Triplicate loops in each calf were inoculated in a semi-randomized order with c. 1x109 CFU of the indicated S. Typhimurium strains grown to mid-logarithmic phase in LB broth at 37°C. Three loops in each calf were inoculated with an equivalent volume of sterile LB broth as a negative control. After inoculation the mid-ileum was returned to the abdominal cavity for 12 h then the animals given an overdose of pentobarbitone sodium. At post mortem examination, loops were excised and the volume of fluid accumulated recorded and normalized to loop length [volume (mL)/length (cm)]. To quantify inflammation, c. 80 mL of jugular blood was collected at the start of the experiment and PMN isolated and labelled with 111Indium oxinate as described[30] then injected into the donor calf within 1 h of loop inoculation. Gamma-radioactivity associated with the mucosa and contents of each loop was normalized to loop length (counts per minute/cm), then the mean PMN influx for each set of triplicate loops determined by dividing the mean value for test strains by the value for the negative control. Values shown are the mean ± standard error of the mean (SEM) from two independent animals.
Supporting Information
(DOC)
Maximum likelihood tree inferred from concatenated SNPS in the core genes obtained using the Panseq program[53]. Scale bar indicates substitutions per variable site. Nodes with 100% bootstrap support are indicated by asterisks.
(TIF)
The data illustrates the extent of variability within replicates and the clustering of isolates based on overall metabolic potential on tested metabolites and physiological conditions.
(TIF)
Geometric mean of recovered bacteria (cfu/ml) from mice infected with ST313 isolates (x-axis). Dotted lines running parallel to x-axis indicate numbers (geometric mean) recovered bacteria from SL1344 infections (grey vertical lines).
(TIF)
(TIF)
Asterisks indicate statistically significant differences at p < 0.001 using a two-way ANOVA with post-tests performed using the Bonferroni method.
(TIF)
* ST4/74 is the parent of SL1344 and differs from it by just 8 SNPs. ST4/74 is the strain used Biolog and bovine experiments. For consistency, ST4/74 is referred to as SL1344 in the relevant sections within the text. **IR715 is a nalidixic acid-resistant derivative of strain ATCC 14028.
(XLS)
Boundaries of deleted loci are indicated in the first two columns. SL1344 gene names when not given are abbreviated to SLxxxx.
(XLS)
Single asterisks indicate pseudogenes common to all isolates within a particular lineage. Lineages without asterisks indicate branches leading to terminal nodes. Gene categories were identified using Artemis-based gene classification scheme. SL1344 gene names when not given are abbreviated to SLxxxx.
(XLS)
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was funded by a Wellcome Trust Grant 098051 (http://www.wellcome.ac.uk). CKO is supported by a Society in Science, Branco Weiss Fellowship, administered by the ETH Zurich (http://www.society-in-science.org). LB is supported by a Research Fellowship from the Alexander von Humboldt Stiftung/Foundation (http://www.humboldt-foundation.de/web/sponsorship.html). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Popoff MY, Bockemuhl J, Gheesling LL (2004) Supplement 2002 (no. 46) to the Kauffmann-White scheme. Research in microbiology 155: 568–570. [DOI] [PubMed] [Google Scholar]
- 2. Langridge GC, Nair S, Wain J (2009) Nontyphoidal Salmonella serovars cause different degrees of invasive disease globally. The Journal of infectious diseases 199: 602–603. 10.1086/596208 [DOI] [PubMed] [Google Scholar]
- 3. Graham SM (2010) Nontyphoidal salmonellosis in Africa. Current opinion in infectious diseases 23: 409–414. 10.1097/QCO.0b013e32833dd25d [DOI] [PubMed] [Google Scholar]
- 4. Berkley JA, Bejon P, Mwangi T, Gwer S, Maitland K, et al. (2009) HIV infection, malnutrition, and invasive bacterial infection among children with severe malaria. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America 49: 336–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kingsley RA, Msefula CL, Thomson NR, Kariuki S, Holt KE, et al. (2009) Epidemic multiple drug resistant Salmonella Typhimurium causing invasive disease in sub-Saharan Africa have a distinct genotype. Genome research 19: 2279–2287. 10.1101/gr.091017.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gordon MA, Banda HT, Gondwe M, Gordon SB, Boeree MJ, et al. (2002) Non-typhoidal salmonella bacteraemia among HIV-infected Malawian adults: high mortality and frequent recrudescence. AIDS (London, England) 16: 1633–1641. [DOI] [PubMed] [Google Scholar]
- 7. Gordon MA, Graham SM, Walsh AL, Wilson L, Phiri A, et al. (2008) Epidemics of invasive Salmonella enterica serovar enteritidis and S. enterica Serovar typhimurium infection associated with multidrug resistance among adults and children in Malawi. Clin Infect Dis 46: 963–969. 10.1086/529146 [DOI] [PubMed] [Google Scholar]
- 8. Gordon MA (2008) Salmonella infections in immunocompromised adults. The Journal of infection 56: 413 10.1016/j.jinf.2008.03.012 [DOI] [PubMed] [Google Scholar]
- 9. Cheesbrough JS, Taxman BC, Green SD, Mewa FI, Numbi A (1997) Clinical definition for invasive Salmonella infection in African children. The Pediatric infectious disease journal 16: 277–283. [DOI] [PubMed] [Google Scholar]
- 10. Okoro CK, Kingsley RA, Connor TR, Harris SR, Parry CM, et al. (2012) Intracontinental spread of human invasive Salmonella Typhimurium pathovariants in sub-Saharan Africa. Nat Genet 44: 1215–1221. 10.1038/ng.2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kariuki S, Revathi G, Kariuki N, Kiiru J, Mwituria J, et al. (2006) Invasive multidrug-resistant non-typhoidal Salmonella infections in Africa: zoonotic or anthroponotic transmission? Journal of medical microbiology 55: 585–591. [DOI] [PubMed] [Google Scholar]
- 12. Parkhill J, Sebaihia M, Preston A, Murphy LD, Thomson N, et al. (2003) Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat Genet 35: 32–40. [DOI] [PubMed] [Google Scholar]
- 13. Parkhill J, Wren BW, Thomson NR, Titball RW, Holden MT, et al. (2001) Genome sequence of Yersinia pestis, the causative agent of plague. Nature 413: 523–527. [DOI] [PubMed] [Google Scholar]
- 14. Holt KE, Parkhill J, Mazzoni CJ, Roumagnac P, Weill FX, et al. (2008) High-throughput sequencing provides insights into genome variation and evolution in Salmonella Typhi. Nature genetics 40: 987–993. 10.1038/ng.195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Holt KE, Thomson NR, Wain J, Langridge GC, Hasan R, et al. (2009) Pseudogene accumulation in the evolutionary histories of Salmonella enterica serovars Paratyphi A and Typhi. BMC genomics 10: 36 10.1186/1471-2164-10-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thomson NR, Clayton DJ, Windhorst D, Vernikos G, Davidson S, et al. (2008) Comparative genome analysis of Salmonella Enteritidis PT4 and Salmonella Gallinarum 287/91 provides insights into evolutionary and host adaptation pathways. Genome research 18: 1624 10.1101/gr.077404.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kryazhimskiy S, Plotkin JB (2008) The population genetics of dN/dS. PLoS genetics 4: e1000304 10.1371/journal.pgen.1000304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rocha EP, Smith JM, Hurst LD, Holden MT, Cooper JE, et al. (2006) Comparisons of dN/dS are time dependent for closely related bacterial genomes. J Theor Biol 239: 226–235. [DOI] [PubMed] [Google Scholar]
- 19. Hurlbert RE, Jakoby WB (1965) Tartaric Acid Metabolism. I. Subunits of L(+)-Tartaric Acid Dehydrase. The Journal of biological chemistry 240: 2772–2777. [PubMed] [Google Scholar]
- 20. Kingsley RA, Humphries AD, Weening EH, De Zoete MR, Winter S, et al. (2003) Molecular and phenotypic analysis of the CS54 island of Salmonella enterica serotype typhimurium: identification of intestinal colonization and persistence determinants. Infection and immunity 71: 629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thomson NR, Clayton DJ, Windhorst D, Vernikos G, Davidson S, et al. (2008) Comparative genome analysis of Salmonella Enteritidis PT4 and Salmonella Gallinarum 287/91 provides insights into evolutionary and host adaptation pathways. Genome Res 18: 1624–1637. 10.1101/gr.077404.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wood MW, Jones MA, Watson PR, Hedges S, Wallis TS, et al. (1998) Identification of a pathogenicity island required for Salmonella enteropathogenicity. Molecular microbiology 29: 883–891. [DOI] [PubMed] [Google Scholar]
- 23. Lawley TD, Chan K, Thompson LJ, Kim CC, Govoni GR, et al. (2006) Genome-wide screen for Salmonella genes required for long-term systemic infection of the mouse. PLoS pathogens 2: e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miller SI, Loomis WP, Alpuche-Aranda C, Behlau I, Hohmann E (1993) The PhoP virulence regulon and live oral Salmonella vaccines. Vaccine 11: 122–125. [DOI] [PubMed] [Google Scholar]
- 25. Carnell SC, Bowen A, Morgan E, Maskell DJ, Wallis TS, et al. (2007) Role in virulence and protective efficacy in pigs of Salmonella enterica serovar Typhimurium secreted components identified by signature-tagged mutagenesis. Microbiology 153: 1940–1952. [DOI] [PubMed] [Google Scholar]
- 26. Bochner BR (2003) New technologies to assess genotype-phenotype relationships. Nature reviewsGenetics 4: 309 [DOI] [PubMed] [Google Scholar]
- 27. Blattner FR, Plunkett G 3rd, Bloch CA, Perna NT, Burland V, et al. (1997) The complete genome sequence of Escherichia coli K-12. Science 277: 1453–1462. [DOI] [PubMed] [Google Scholar]
- 28. Tipton PA, Peisach J (1990) Characterization of the multiple catalytic activities of tartrate dehydrogenase. Biochemistry 29: 1749–1756. [DOI] [PubMed] [Google Scholar]
- 29. Chen R, Jeong SS (2000) Functional prediction: identification of protein orthologs and paralogs. Protein science: a publication of the Protein Society 9: 2344–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Watson PR, Galyov EE, Paulin SM, Jones PW, Wallis TS (1998) Mutation of invH, but not stn, reduces Salmonella-induced enteritis in cattle. Infect Immun 66: 1432–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nuccio SP, Baumler AJ (2014) Comparative analysis of Salmonella genomes identifies a metabolic network for escalating growth in the inflamed gut. MBio 5: e00929–00914. 10.1128/mBio.00929-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gordon MA, Kankwatira AM, Mwafulirwa G, Walsh AL, Hopkins MJ, et al. (2010) Invasive non-typhoid salmonellae establish systemic intracellular infection in HIV-infected adults: an emerging disease pathogenesis. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America 50: 953 10.1086/651080 [DOI] [PubMed] [Google Scholar]
- 33. Okoro CK, Kingsley RA, Quail MA, Kankwatira AM, Feasey NA, et al. (2012) High-resolution single nucleotide polymorphism analysis distinguishes recrudescence and reinfection in recurrent invasive nontyphoidal Salmonella typhimurium disease. Clin Infect Dis 54: 955–963. 10.1093/cid/cir1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tsolis RM, Young GM, Solnick JV, Baumler AJ (2008) From bench to bedside: stealth of enteroinvasive pathogens. Nat Rev Microbiol 6: 883–892. 10.1038/nrmicro2012 [DOI] [PubMed] [Google Scholar]
- 35. Paulin SM, Jagannathan A, Campbell J, Wallis TS, Stevens MP (2007) Net replication of Salmonella enterica serovars Typhimurium and Choleraesuis in porcine intestinal mucosa and nodes is associated with their differential virulence. Infect Immun 75: 3950–3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Parsons BN, Humphrey S, Salisbury AM, Mikoleit J, Hinton JC, et al. (2013) Invasive non-typhoidal Salmonella typhimurium ST313 are not host-restricted and have an invasive phenotype in experimentally infected chickens. PLoS Negl Trop Dis 7: e2487 10.1371/journal.pntd.0002487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Maurelli AT, Fernandez RE, Bloch CA, Rode CK, Fasano A (1998) "Black holes" and bacterial pathogenicity: a large genomic deletion that enhances the virulence of Shigella spp. and enteroinvasive Escherichia coli. Proc Natl Acad Sci U S A 95: 3943–3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Reuter S, Connor TR, Barquist L, Walker D, Feltwell T, et al. (2014) Parallel independent evolution of pathogenicity within the genus Yersinia. Proc Natl Acad Sci U S A 111: 6768–6773. 10.1073/pnas.1317161111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Touchon M, Hoede C, Tenaillon O, Barbe V, Baeriswyl S, et al. (2009) Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet 5: e1000344 10.1371/journal.pgen.1000344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Okoro C, K., Kingsley R, A., Connor T, R., Harris S, R., Parry C, M., et al. (2012) Intracontinental spread of human invasive Salmonella Typhimurium pathovariants in sub-Saharan Africa. Nature Genetics advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nei M, Gojobori T (1986) Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol 3: 418–426. [DOI] [PubMed] [Google Scholar]
- 42. Swofford DL, Maddison WP (1992) Parsimony, character-state reconstructions, and evolutionary inferences In: Mayden RL, editor. Systematics, Historical Ecology, and North American Freshwater Fishes. Stanford, CA: Stanford University Press; pp. pp. 187–223. [Google Scholar]
- 43. Agnersson I, Miller JA (2008) Is ACCTRAN better than DELTRAN? Cladistics 24: 1–7. [DOI] [PubMed] [Google Scholar]
- 44. Carver T, Harris SR, Otto TD, Berriman M, Parkhill J, et al. (2012) BamView: visualizing and interpretation of next-generation sequencing read alignments. Briefings in bioinformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Croucher NJ, Mitchell AM, Gould KA, Inverarity D, Barquist L, et al. (2013) Dominant role of nucleotide substitution in the diversification of serotype 3 pneumococci over decades and during a single infection. PLoS Genet 9: e1003868 10.1371/journal.pgen.1003868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Homann OR, Cai H, Becker JM, Lindquist SL (2005) Harnessing natural diversity to probe metabolic pathways. PLoS Genet 1: e80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Smyth GK (2004) Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Statistical applications in genetics and molecular biology 3: Article3 [DOI] [PubMed] [Google Scholar]
- 49. Benjamini Y, Hochberg Y (1995) Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B (Methodological) 57: 289–300. [Google Scholar]
- 50. Karp PD, Paley S, Romero P (2002) The Pathway Tools software. Bioinformatics 18 Suppl 1: S225–232. [DOI] [PubMed] [Google Scholar]
- 51. Kim JJ, Shajib MS, Manocha MM, Khan WI (2012) Investigating intestinal inflammation in DSS-induced model of IBD. Journal of visualized experiments: JoVE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Stanley J, Baquar N, Threlfall EJ (1993) Genotypes and phylogenetic relationships of Salmonella typhimurium are defined by molecular fingerprinting of IS200 and 16S rrn loci. J Gen Microbiol 139 Pt 6: 1133–1140. [DOI] [PubMed] [Google Scholar]
- 53. Laing C, Buchanan C, Taboada EN, Zhang Y, Kropinski A, et al. (2010) Pan-genome sequence analysis using Panseq: an online tool for the rapid analysis of core and accessory genomic regions. BMC Bioinformatics 11: 461 10.1186/1471-2105-11-461 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC)
Maximum likelihood tree inferred from concatenated SNPS in the core genes obtained using the Panseq program[53]. Scale bar indicates substitutions per variable site. Nodes with 100% bootstrap support are indicated by asterisks.
(TIF)
The data illustrates the extent of variability within replicates and the clustering of isolates based on overall metabolic potential on tested metabolites and physiological conditions.
(TIF)
Geometric mean of recovered bacteria (cfu/ml) from mice infected with ST313 isolates (x-axis). Dotted lines running parallel to x-axis indicate numbers (geometric mean) recovered bacteria from SL1344 infections (grey vertical lines).
(TIF)
(TIF)
Asterisks indicate statistically significant differences at p < 0.001 using a two-way ANOVA with post-tests performed using the Bonferroni method.
(TIF)
* ST4/74 is the parent of SL1344 and differs from it by just 8 SNPs. ST4/74 is the strain used Biolog and bovine experiments. For consistency, ST4/74 is referred to as SL1344 in the relevant sections within the text. **IR715 is a nalidixic acid-resistant derivative of strain ATCC 14028.
(XLS)
Boundaries of deleted loci are indicated in the first two columns. SL1344 gene names when not given are abbreviated to SLxxxx.
(XLS)
Single asterisks indicate pseudogenes common to all isolates within a particular lineage. Lineages without asterisks indicate branches leading to terminal nodes. Gene categories were identified using Artemis-based gene classification scheme. SL1344 gene names when not given are abbreviated to SLxxxx.
(XLS)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.