

Abstract
Multiple sclerosis (MS) is a common neurodegenerative disease that presents after an auto-reactive immune response against constituents of the central nervous system. Demyelination, inflammation, and white matter lesions are all hallmarks of this disease. Clinical research supports the use of mesenchymal stem cells (MSCs) as therapy for MS to ameliorate symptoms and pathology. MSCs can be isolated from multiple tissues, including adipose and bone marrow, and are able to migrate to sites of pathology, release anti-inflammatory factors, and provide immunomodulatory and neuroprotective effects once administered. Numerous studies have demonstrated the beneficial effects of MSCs in experimental autoimmune encephalomyelitis (EAE), an induced model of MS. EAE can be induced in several species; however, the mouse is commonly used for therapeutic testing. In the following chapter, scientists will be able to learn how to prepare reagents and MSCs (e.g., isolate, culture, and expand) as well as skillfully execute induction of EAE in mice and administer stem cell-based treatments. Standard methods used to evaluate the disease progression and analyze postmortem tissues are also included.
Keywords: Multiple sclerosis, murine experimental autoimmune encephalomyelitis, EAE, T cell-mediate autoimmune disease, mesenchymal stem cells, myelin oligodendrocyte glycoprotein
1. Introduction
Multiple sclerosis (MS) is a neurodegenerative disease affecting over 300,000 people in the U.S. making it the leading cause of neurological impairment, especially in young adults (1). More specifically, MS is generated by an autoimmune response against constituents of the central nervous system (CNS) that manifests as various symptoms that distinguishes the 4 subtypes of the disease: relapsing-remitting, primary progressive, secondary progressive, and progressive relapsing (1, 2). Evidence suggests that T cells lose self-tolerance and attack their own nervous system due to exposure to a stimulatory molecule that structurally resembles myelin. Once stimulated to attack myelin or myelin-producing cells, the cytotoxic and pathogenic helper T cells initiate the inflammatory processes characteristic of MS (3). The induction phase of the disease is associated with an increase in cytotoxic CD8+T cells within the CNS that destroy oligodendrocytes, leading to inflammation, demyelination, and damage to the blood-brain barrier (BBB) (3-4). Compromise to the BBB allows infiltration of immune cells, such as CD4+T cells and macrophages, into the CNS. These cells further enhance the pathology to the CNS tissues by producing demyelinating lesions of the white matter, reactive gliosis, and increasing axonal loss (3, 5). Currently, there are several FDA approved drugs available for relapsing-remitting MS (1, 6); however, there is still no cure for MS and no effective treatments available for the progressive forms.
Mesenchymal stem cells (MSCs) have recently been tested in clinical trials as therapy for MS patients, and all trials have demonstrated the safety of MSCs in these patients using autologous MSCs without the use of immunosuppression (6-8). MSCs are isolated from adipose tissue and bone marrow and are capable of homing to damaged areas (9), providing immunomodulatory effects (10, 11), and inducing neuroregeneration through neuroprotective mechanisms (12). More specifically for MS, these stem cells have the ability to inhibit the migration of T cells from the periphery to the CNS (13-14), and multiple animal models of MS have had significant symptomatic alleviation, improved CNS pathology, and decreased inflammation after MSC transplantation (13, 15-17).
MS-like symptoms can be induced in many species (e.g., mouse, rat, guinea pigs, rabbits, and primates); however, the mouse is most commonly used for therapeutic screening for MS. The mouse can be induced with experimental autoimmune encephalomyelitis (EAE) with purified myelin, myelin protein, spinal cord homogenate, or a myelin oligodendrocyte glycoprotein (MOG) peptide in the presence of an adjuvant with Mycobacterium tuberculosis and pertussis toxin (13, 18). EAE results in widespread brain inflammation and demyelination throughout the CNS, making it the prototype for T cell-mediated autoimmune diseases (17), as well as an ideal model for progressive forms of MS. The administration of a myelin product stimulates the animal's T cells to recognize its own myelin as foreign, whereas the adjuvant and pertussis toxin are administered to generate inflammation and increase the permeability of the BBB, respectively (13, 17). After induction, EAE-induced mice have increased levels of infiltrating immune cells in the CNS and present with lesions similar to those seen in MS patients (17).
Several methods are used to evaluate the efficacy of stem cell-based treatments. Clinical scoring is a widely used method based on assessments of the motor functions of research animals. A concise description of scoring: Score 0 – no disease, Score 1 – tail atony, Score 2 – hind limb weakness, Score 3 – partial hind limb paralysis, Score 4 – complete hind limb paralysis and incontinence, Score 5 – moribund or dead. A more objective means to assess motor function capabilities is by recording the animals in an arena over a set time period. Videos can then be uploaded and analyzed with Noldus EthoVision XT7 software for quantification of several spatial parameters (19, 20).
Standard techniques exist to quantitatively compare groups of research animals for postmortem histological analyses. Histological staining of CNS tissues can quantify (de)myelination and infiltration of immune cells using Luxol fast blue (LFB) and hematoxylin and eosin (H&E), respectively. Immunohistochemistry (IHC) uses antibody-mediated detection of markers to localize specific cells or tissues prepared on microscope slides. Detection of such antigens on CNS infiltrating immune cells allows identification and quantification of cell populations pertinent to this model.
2. Materials
2.1. Induction of EAE
| Mice | 1. | C57BL/6 female mice, 6-8 weeks old |
| Supplies | 2. | Lyophilized MOG35-55 peptide |
| 3. | Mycobacterium tuberculosis H37 RA, desiccated | |
| 4. | Lyophilized pertussis toxin | |
| 5. | Complete Freund's adjuvant (CFA) | |
| 6. | 5cc emulsifying glass syringes with metal Leur lock tip | |
| 7. | Metal micro-emulsifying needle with reinforcing bar, 13G 2-7/8″ | |
| 8. | 50ml polystyrene reagent reservoir | |
| 9. | 1ml Leur lock disposable syringes with 0.1ml graduations | |
| 10. | 27G ½″ needles | |
| 11. | Isoflurane | |
| Equipment | 12. | Biosafety laminar flow hood |
| 13. | Anesthesia induction chamber for small animals | |
| 14. | Anesthetic vaporizer machine with oxygen tank |
2.2. BMSC isolation
| Tissue | 1. | Femurs and tibias |
| Supplies | 2. | Scissors |
| 3. | 50ml conical tube | |
| 4. | PBS | |
| 5. | Complete expansion media (CEM) | |
| 6. | 25G 1″ needle | |
| 7. | 5ml disposable syringe with Leur lock tip | |
| 8. | 70μm mesh strainer | |
| 9. | Trypan blue | |
| 10. | Microcentrifuge tubes | |
| 11. | Hemocytometer | |
| 12. | Micropipettes and pipettes with tips | |
| 13. | 140×20mm cell culture dishes | |
| Equipment | 14. | Large centrifuge |
2.3. ASC isolation
| Tissue | 1. | Subcutaneous/inguinal fat tissue |
| Supplies | 2. | Scissors |
| 3. | Scale with measurements in grams | |
| 4. | 50ml conical tubes | |
| 5. | PBS | |
| 6. | 265units/mg Collagenase type-1 | |
| 7. | Bovine serum albumin (BSA) | |
| 8. | 2mM Calcium chloride in ddH2O (CaCl2) | |
| 9. | 70μm mesh strainer (optional) | |
| 10. | Parafilm | |
| 11. | Complete expansion media (CEM) | |
| 12. | 50ml Steriflip disposable vacuum filtration system with 0.22μm filter | |
| 13. | Vacuum tubing | |
| 14. | Timer | |
| 15. | Wedge-shaped object | |
| 16. | Trypan blue | |
| 17. | Microcentrifuge tubes | |
| 18. | Hemocytometer | |
| 19. | Micropipettes and pipettes with tips | |
| 20. | T225 cell culture flasks | |
| Equipment | 21. | Incubator shaker |
| 22. | Large centrifuge |
2.4. Cell culture, expansion, and injection preparation
| Supplies | 1. | Phosphate buffered saline (PBS) |
| 2. | Hank's balanced salt solution (HBSS) | |
| 3. | Dulbecco's Minimum Essential Media: Nutrient Mixture F-12 (DMEM/F-12 media) | |
| 4. | Fetal bovine serum (FBS) | |
| 5. | 2mM L-glutamine | |
| 6. | 10,000units/ml Penicillin – 10,000μg/ml streptomycin (Pen-Strep) | |
| 7. | 140×20mm cell culture dishes | |
| 8. | T225 cell culture flasks | |
| 9. | 0.25% Trypsin-EDTA | |
| 10. | Trypan blue | |
| 11. | Microcentrifuge tubes | |
| 12. | Cryovials | |
| 13. | Dimethylsulfoxide (DMSO) | |
| 14. | Stericup vacuum-mediated 0.22μm filter unit | |
| 15. | 1ml Leur lock disposable syringes with 0.1ml graduations | |
| 16. | 27G ½″ needle | |
| 17. | Micropipettes and pipettes with tips | |
| 18. | Hemocytometer | |
| 19. | Aspirator | |
| Equipment | 20. | Water bath |
| 21. | Large centrifuge | |
| 22. | CO2 Incubator | |
| 23. | Biosafety hood | |
| 24. | Liquid nitrogen dewar |
2.5. Motor function testing
| Supplies | 1. | Cylindrical arena |
| 2. | White paper | |
| 3. | Ring stand | |
| 4. | Webcam, video capture software, and computer | |
| 5. | Noldus EthoVision XT7 software | |
| Equipment | 6. | Biosafety hood |
3. Methods
3.1. Induction of EAE
3.1.1. Reagent Preparation
It is important to prepare an overestimated amount of reagents to compensate for loss during emulsification and transfer into syringes. For example, for induction of 20 mice, prepare reagents for 25 mice. (Figure 1 near here)
Figure 1. Materials for induction of EAE.

Induction of EAE requires administration of emulsified reagents i.e., myelin oligodendrocyte glycoprotein (MOG) peptide, complete Freund's adjuvant (CFA), and Mycobacterium tuberculosis, and also Pertussis toxin.
Preparing MOG/CFA emulsion:
Place all materials in a sterilized biosafety hood.
Add CFA to sterile reagent reservoir.
Reconstitute lyophilized M. tuberculosis powder in ddH2O to make 1mg/ml bacteria stock.
Add appropriate volume of bacteria stock to CFA to make 5mg/ml CFA/M. tuberculosis. Store bacteria stock in 4°Celsius.
Reconstitute lyophilized MOG35-55 peptide in PBS to make 2mg/ml solution.
Add (v/v) MOG35-55 peptide solution to 5mg/ml CFA/M. tuberculosis in reservoir.
Assemble one emulsification syringe with micro-emulsifying needle.
Place needle tip into a corner of the reservoir with gathered reagents.
Draw reagents into the syringe until completely collected in syringe.
Secure second syringe to free tip of the micro-emulsifying needle.
Emulsify reagents by pushing reagents into alternating syringes for about 45 minutes to 1 hour to ensure product is fully emulsified (see Note 1).
Collect MOG/CFA emulsion into one syringe.
Unscrew empty syringe from emulsifying needle.
Screw on 1ml Leur lock tip disposable syringe to free end of micro-emulsifying needle.
Securely fill disposable syringe by pushing glass syringe containing MOG/CFA emulsion (see Note 2).
Screw on 27G ½″needle
Repeat steps 14-16 until all MOG/CFA emulsion is transferred to disposable syringes.
Preparing 2ng/μl pertussis toxin solution:
Reconstitute lyophilized powder in ddH2O to make 100ng/μl stock solution.
Add appropriate volume of stock solution to PBS to make 2ng/μl pertussis toxin solution. Store stock solution in 4°Celsius.
3.1.2. Induction procedure
Each animal receives a total of 200μl of MOG/CFA emulsion. For example, administer two 100μl injections of MOG/CFA emulsion, one injection per flank. At the time of induction, each animal receives a single 100μl injection of 2ng/μl pertussis toxin solution, and a second 100μl injection of 2ng/μl pertussis toxin solution 48 hours after induction. (Figure 2 near here)
Figure 2. Administration of induction reagents.
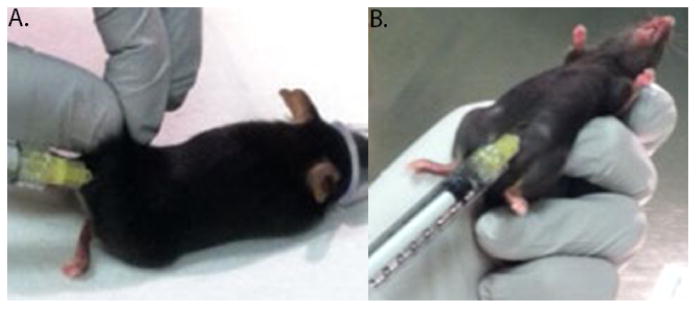
The research animal is anesthetized and prepared for the induction procedure. A. MOG/CFA emulsion via subcutaneous injection is administered to the flanks of the tail. B. Pertussis toxin is administered via intraperitoneal injection.
Fill anesthetic vaporizer machine with Isoflurane gas. Open oxygen dial to allow oxygen to combine with Isoflurane gas.
Set output to 4% Isoflurane gas in oxygen.
Place mouse in anesthesia induction chamber to induce anesthesia by inhalation.
Monitor respiratory rate. Once slowed, relocated mouse to auxiliary nosepiece.
Administer 100μl MOG/CFA emulsion via subcutaneous injection into right flank near tail.
Repeat step 5 for injection into left flank near tail.
Carefully handle mouse to secure on its backside.
Administer first dose of 100μl of 2ng/μl pertussis toxin solution (i.e., 200ng total) via intraperitoneal (IP) injection.
After 48 hours, administer second dose of 100μl of 2ng/μl pertussis toxin solution via IP injection.
3.2. Cell Isolation, Culture, Expansion, Cryopreservation, and Injection
3.2.1. Making complete expansion media (CEM)
Sterilize biosafety hood with UV-sterilization for 30 minutes prior to use.
Place all cell culture supplies in sterile hood.
Add desired amount of DMEM/F-12 media to upper chamber of Stericup vacuum-mediated 0.22μm filter unit.
Add supplements to upper chamber: 10% FBS, 1% L-glutamine, and 1% Pen-strep.
Attach vacuum tubing to filter unit and vacuum port.
Filter CEM into receiver bottle.
Remove filter unit and secure lid to CEM bottle.
Store at 4°Celsius (see Note 3).
3.2.2. Cell Isolation
BMSC Isolation
Securely hold long bone at one end.
Carefully cut epiphysis off with scissors to expose the bone marrow in medullary cavity.
Assemble a 5ml disposable syringe with 25G 1″ needle containing PBS.
Insert tip of needle into the cavity of long bone.
Flush bone marrow out of the opposite end of long bone and collect in 50ml conical tube.
Flush long bone with ample PBS from each end until bone is visible cleared of bone marrow and collected in conical tube.
Break up and re-suspend bone marrow in PBS by pipetting up and down several times in conical tube.
Filter through a 70μm mesh strainer and collect in a new 50ml conical tube.
Centrifuge capped conical tube at 420Xg for 5 minutes at room temperature.
Carefully aspirate PBS leaving cell pellet intact.
Re-suspend pellet with small volume of warm CEM.
Determine cell viability and count: transfer 10μl sample of cell suspension to microcentrifuge tube, stain with (v/v) trypan blue, and count live cells using hemocytometer. Determine total cell count.
Plate cells on 140×20mm cell culture dish with a total volume of 20ml CEM (see Note 4).
Place culture dish(es) in incubator at 37°Celsius and 5%CO2 (see Note 5).
ASC Isolation
Collect fat tissue in 50ml conical tube.
Weigh fat.
Wash fat by adding ample PBS, vigorously shaking capped conical tube, removing PBS, and replacing with fresh PBS. Do this 3-4 times.
Remove fat tissue with forceps and transfer to new conical tube. Put aside conical tube containing fat tissue.
In a new 50ml conical tube, make digestion solution with (w/v) PBS (i.e., 1gram of fat to 1ml PBS).
Add supplements to PBS: 0.1% (w/v) collagenase type-1, 1% (w/v) BSA, and 1% (v/v) CaCl2 (see Note 6).
Gently rock conical tube to dissolve solutes; do not vortex.
Assemble Steriflip 50ml disposable vacuum filtration system with 0.22μm filter to conical tube with digestion solution.
Attach vacuum tubing to filter unit and vacuum port.
Filter digestion solution into attached collection tube.
Add filtered digestion solution to conical tube containing fat tissue. Start timer for 1 hour.
Quickly mince fat in digestion solution with scissors by downward motion cutting in conical tube. Mince until fat tissue is the size of small pebbles.
Add parafilm around top of conical tube to seal.
Place conical tube with fat tissue in digestion solution into incubator shaker (see Note 7).
Incubate fat in digestion solution at 37°Celsius in incubator for time remaining. Set shaker speed to 100rpm (see Note 8).
After 1 hour incubation, remove conical tube from incubator shaker.
Neutralize digestion solution by adding (v/v) complete expanding media (CEM) to conical tube.
Pipette up and down several times to break up tissue.
Centrifuge conical tube for 5 minutes at 300Xg at room temperature.
Remove and shake conical tube to assist in release of cells from tissue.
Centrifuge conical tube again for 5 minutes at 300Xg at room temperature.
Carefully remove conical tube from centrifuge. Be sure not to disturb layers.
Carefully aspirate top liquid layers leaving pellet of stromal vascular fraction (SVF) (Figure 2) (see Note 9).
Add CEM to conical tube to re-suspend cells of SVF by pipetting up and down several times.
(optional) Filter cell suspension through 70μm filter and collect in a new 50ml conical tube.
Determine cell viability and count: transfer 10μl sample of cell suspension to microcentrifuge tube, stain with (v/v) trypan blue, and count live cells using hemocytometer. Determine total cell count.
Plate cells into T225 flask at desired density (see Note 10).
Add CEM to flask and disperse cells evenly by gently rock side to side.
Place primary cell culture (passage 0) in incubator for 48 hours at 37°Celsius and 5%CO2.
After 48 hours, remove flask from incubator and decant CEM.
Wash culture with PBS.
Replace with fresh CEM (see Note 5).
3.2.3. Cell Expansion
Replace cell cultures with fresh CEM every 3-4 days until desired confluence is reached (≤ 70% confluence is recommended). Then, lift cells from plasticware, collect, and re-plate on multiple, new plasticware at a lower density where it is considered the next higher passage. When desired passage is achieved, cells can either be prepared for injections or frozen in liquid nitrogen until time of use.
Sterilize biosafety hood with UV-sterilization for 30 minutes prior to use.
Place all cell culture supplies in sterile hood.
Remove culture dish/flask from incubator and transfer to sterile biosafety hood.
Carefully aspirate CEM. Be sure not to place aspirator tip near cell cultures (see Note 11).
Wash plate with PBS.
Aspirate PBS using same method in step 4.
Add minimum volume of 0.25% trypsin-EDTA sufficient to cover the surface of the culture dish/flask.
Transfer culture dish/flask to incubator for 4 minutes at 37°Celsius and 5%CO2.
In hood, neutralize digestion with (v/v) pre-warmed CEM.
Transfer cell suspension solution from culture dish/flask to 50ml conical tube(s).
Centrifuge conical tube(s) at 420Xg for 5 minutes at room temperature.
In hood, carefully aspirate liquid leaving cell pellet.
Re-suspend cell pellet by adding minimal volume of CEM and pipette up and down several times (see Note 12).
Determine cell viability and count: transfer 10μl sample of cell suspension to microcentrifuge tube, stain with (v/v) trypan blue, and count live cells using hemocytometer. Determine total cell count.
Re-plate cells at desired concentration in new culture dish/flask containing CEM for further expansion.
3.2.4. Cryopreservation of cells (continue from step 14)
In biosafety hood, prepare cryopreservation media: CEM with 5-10% DMSO (see Note 13).
Centrifuge conical tube at 420Xg for 5 minutes.
In hood, carefully aspirate CEM leaving cell pellet.
Add cryopreservation media and re-suspend cells (see Note 14).
Aliquot cryopreserved cells into cryovials.
Quickly transfer tubes to liquid nitrogen dewar.
3.2.5. Prepare cells for injection into EAE model (continue from step 14)
Centrifuge conical tube at 420Xg for 5 minutes.
In hood, carefully aspirate liquid leaving cell pellet.
Wash cells by adding pre-warmed PBS and re-suspend by gently pipetting up and down several times.
Centrifuge conical tube at 420Xg for 5 minutes.
In hood, carefully aspirate PBS leaving cell pellet.
Re-suspend cells in sterile HBSS at desired concentration (see Note 15).
3.3. Assessments of disease
Blinded researchers assess the motor functions of each EAE mouse as a method to quantify the severity of pathology during the disease course. It is important to conduct motor function testing for each animal daily for clinical scoring and weekly for video recordings, ideally at a set time of day for consistency. Clinical cores are based on observations of the animal's gait and hind limb spread while suspended (19, 20). (Figure 4 near here)
Figure 4. Clinical scoring over the disease course of the EAE model.
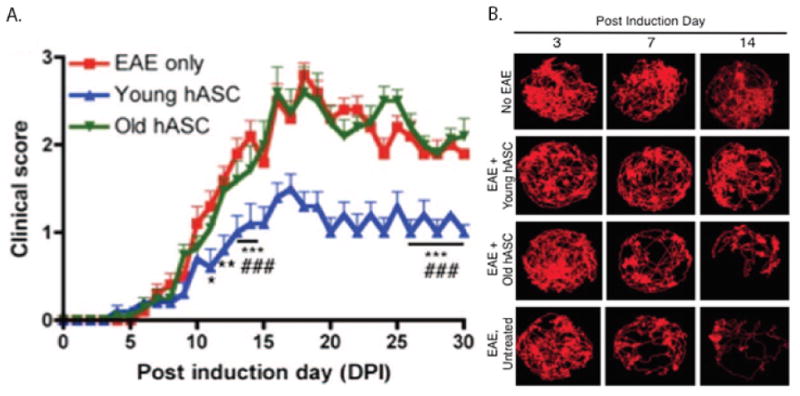
Adapted by permission from AlphaMed Press: SCTM. B.A. Scruggs et al. (2013). “Age of Donor Reduces the Ability of Human Adipose-Derived Stem Cells to Alleviate Symptoms in the Experimental Autoimmune Encephalomyelitis Mouse Model,” 2:797-807 (19). A. Clinical scoring is a method used by researchers to assess the pathological progression affecting the hind region of the EAE model throughout the course of disease. B. Track visualization generated by EthoVision XT7 of a research animal from multiple treatment groups that were video recorded over a 5 minute period in a cylindrical arena.
3.3.1 Clinical Scoring
Remove mice out of housing cage to open area (e.g., biosafety hood).
Allow mice to walk around freely.
Observe the tail and gait of each mouse.
Suspend each mouse by holding the base of the tail to confirm scoring category (see Note 16).
Record score.
3.3.2. Motor function testing using Noldus Ethovision
Setup up arena: place white paper on flat surface, place cylinder on top of paper (see Note 17).
Position ring stand with arm holding webcam overlooking center of circular arena at desired height.
Access live feed from webcam on computer with installed software.
Place animal in arena.
Start recording.
Stop recording after desired time interval.
Acquire video in EthoVision XT7 software.
Analyze spatial parameters and produce video tracking image output.
3.3.3. Postmortem histological analyses
Several methods are used to analyze harvested tissues, especially CNS tissues. Depending on the preferred method of analysis, tissues can be prepared in different fixatives in order to achieve the best results. Antibody detection is a widely used method for various methods of analysis.
For cell, protein, and/or transcription factor identification and/or quantification using flow cytometry:
Harvest tissue.
Homogenize fresh tissue.
Process homogenized tissue by fixation, permeabilization, and stained with antibodies (see Note 18).
Run sample on a flow cytometer to identify cell populations within tissue samples.
For RNA/DNA or protein quantification using techniques such as qPCRs or Western blot, respectively, tissues are collected and flash-frozen in liquid nitrogen to save until time of processing.
Harvest tissue.
Collect tissue in microcentrifuge, 15ml, or 50ml tube.
Submerge tube in liquid nitrogen. Transfer tube to -80°Celsius immediately.
When tissue sample is ready to be analyzed, thaw tissue sample.
Homogenize tissue sample using a hand held homogenizer with plastic pestles.
Process tissue according to manufacturer's protocol for extraction of component, e.g. RNA, DNA, or protein, of interest.
For histological examination using immunohistochemistry, different methods are used to fix tissues in order to embed on a microscope slide. For formalin-fixed, paraffin-embedded sections:
Harvest tissue.
Collect tissue in a tube containing 4% paraformaldehyde for 24-48 hours in 4°Celsius for fixation.
Remove fixed tissue and transfer to 30% sucrose solution in ddH2O for cryopreservation.
When tissue sinks to bottom of the tube, remove tissue.
Place tissue in cryo-mold.
Completely cover tissue in cryo-embedding compound.
Place mold with tissue on a platform in a container.
Add ethanol to container until platform is completely submerged in ethanol and mold is partially submerged in ethanol (see Note 19).
Place container in larger container that can withstand subzero temperatures.
Fill large container with liquid nitrogen until smaller container is partially submerged.
When tissue in mold is completely frozen, remove mold, and wrap in aluminum.
Store frozen mold at -80°Celsius until sectioning.
For spinal cord processing:
Remove entire spinal column and store in 10% neutral buffered formalin.
Allow tissue to fix for at least 48 hours.
Remove spinal cord from vertebral column using forceps and scissors.
Isolate the lumbar spinal cord.
Place lumbar spinal cord in tissue cassette.
Submerge cassette in 10% neutral buffered formalin.
Store at room temperature until sectioning.
Sectioning is performed on a microtome. Tissue sections are cut into 5 microns thick, paraffin-embedded.
For IHC on paraffin-embedded sections on a glass microscope slide (see Note 20):
Deparaffinize sections on a heating bed for at least 30 minutes.
Stepwise, rehydrate tissue samples stepwise with high to low dilutions of ethanol then water.
Use either heat-mediated or enzyme-mediated antigen retrieval method.
Wash sections with phosphate- or tris-buffered saline-based solutions.
Block section for 1 hour with blocking solution containing serum from secondary antibody's host and/or bovine serum albumin.
Dilute primary antibody in blocking solution.
Apply primary antibody either overnight at 4°Celsius or at room temperature for 1 hour in humidified chamber.
Wash sections.
Apply secondary antibody diluted in blocking solution for 1 hour at room temperature in humidified chamber.
Wash sections.
For chemiluminescence, apply chromogenic substrate, counterstain, dehydrate, mount with mounting solution containing nuclear stain, and apply coverslip. Let dry and visualize under high resolution microscope.
For fluorescence, apply mounting solution containing nuclear stain and coverslip. Let dry and visualize using deconvolution microscopy (see Note 21).
4. Notes
To test whether product is completely emulsified, add 1 drop of MOG/CFA emulsion to a beaker of water. If drop doesn't disperse in water, emulsion is complete.
Due to pressure from micro-emulsifying needle, hold thumbs firmly on blunt ends of disposable syringes while filling. Do not fill syringe completely. Air pressure will cause blunt end to eject from syringe.
Prior to use, warm CEM in water bath at 37°Celsius for at least 20 minutes.
A density of 100 cells/cm2 is ideal for 140×20mm cell culture dish.
Upon plating, a heterogeneous population of cells may be observed. After a few days of culturing in CEM, extraneous cells will deplete leaving a homogenous population of MSCs.
BSA protects cells during digestion. CaCl2 is a cofactor to collagenase to assist in digestion.
For best results, securely prop conical tube at 45° angle by taping conical tube to wedge-shaped object in incubator shaker.
Digestion of fat tissue is for a total of 1 hour which includes mincing and incubation period.
Be sure not to aspirate near SVF. Leave thin liquid layer on top of SVF layer to ensure SVF layer remains intact.
Density of 5,000cells/cm2 is ideal in T225 flask.
Tilt cell culture dish/flask at 30° angle where CEM collects at a corner. Aspirate at corner so aspirator tip will not come into contact with cell cultures.
Re-suspend pellet in minimal volume of CEM for ideal concentration for cell counting.
Increasing concentration of FBS is optional.
Ideal concentration for freezing is 1-2 million cells/ml.
Store suspended cells on ice until 30 minutes prior to injection, then keep at room temperature. At time of injection, re-suspend cells in HBSS with micropipette, collect in 1ml syringe, and assemble with needle.
During the early stages of pathology, mice are capable of extending hind limbs in a full, even spread. As symptoms progress, legs spread unevenly and/or adduct while mouse is suspended.
EthoVision XT7 software detects animal by contrast to background. White paper provides contrast to dark-colored animals.
Kits from manufacturers provide the necessary buffers and reagents along with detailed methods depending on preferred method of analysis.
Be sure mold is immobile on platform.
Optimization of protocol must be conducted for each antibody.
When using fluorescently-labeled antibodies, always process slides away from direct light. Light will cause depletion of the fluorescence molecules.
Figure 3. Adipose-derived stem cell (ASC) isolation from fat tissue.
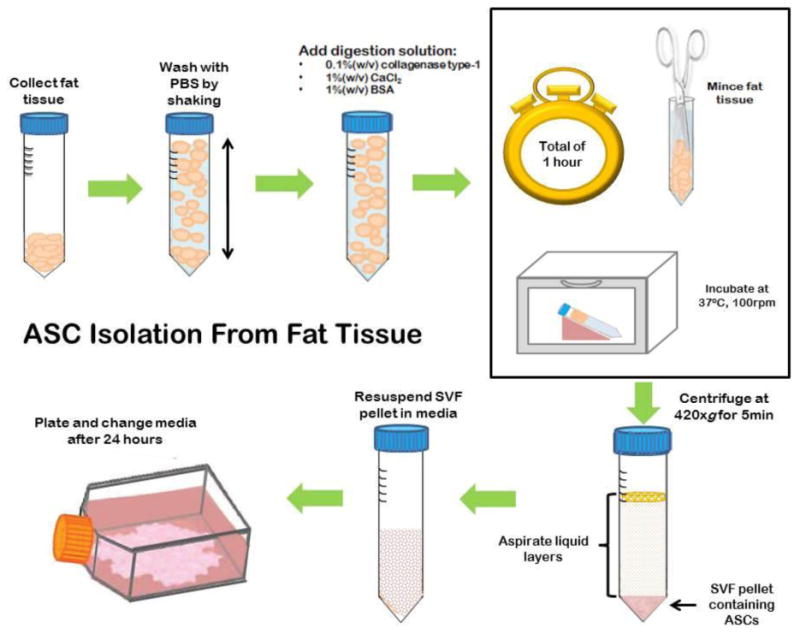
ASCs are isolated from fat tissue after digestion, isolation of stromal vascular fraction (SVF), and plating of SVF. Initial plate will contain a heterogeneous population of cells that will disintegrate leaving ASCs after culturing and passaging.
References
- 1.Goldenberg M. Multiple sclerosis review. P T. 2012;37:175–184. [PMC free article] [PubMed] [Google Scholar]
- 2.Ascherio A, Munger KL, Lünemann JD. The initiation and prevention of multiple sclerosis. Nat Rev Neurol. 2012;8:602–612. doi: 10.1038/nrneurol.2012.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Batoulis H, Addicks K, Kuerten S. Emerging concepts in autoimmune encephalomyelitis beyond the CD4/T(H)1 paradigm. Ann Anat. 2010;192:179–193. doi: 10.1016/j.aanat.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 4.Lassmann H, van Horssen J. The molecular basis of neurodegeneration in multiple sclerosis. FEBS Lett. 2011;585:3715–3723. doi: 10.1016/j.febslet.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 5.Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol. 2012;8:647–656. doi: 10.1038/nrneurol.2012.168. [DOI] [PubMed] [Google Scholar]
- 6.Uccelli A, Laroni A, Freedman MS. Mesenchymal stem cells as treatment for MS - progress to date. Mult Scler. 2012;19:515–519. doi: 10.1177/1352458512464686. [DOI] [PubMed] [Google Scholar]
- 7.Karussis D, Karageorgiou C, Vaknin-Dembinsky A, Gowda-Kurkalli B, Gomori JM, Kassis I, et al. Safety and immunological effects of mesenchymal stem cell transplantation in patients with multiple sclerosis and amyotrophic lateral sclerosis. Arch Neurol. 2010;67:1187–1194. doi: 10.1001/archneurol.2010.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Connick P, Kolappan M, Crawley C, Webber DJ, Patani R, Michell AW, et al. Autologous mesenchymal stem cells for the treatment of secondary progressive multiple sclerosis: an open-label phase 2a proof-of-concept study. Lancet Neurol. 2012;11:150–156. doi: 10.1016/S1474-4422(11)70305-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Payne NL, Sun G, McDonald C, Layton D, Moussa L, Emerson-Webber A, et al. Distinct immunomodulatory and migratory mechanisms underpin the therapeutic potential of human mesenchymal stem cells in autoimmune demyelination. Cell Transplant. 2012;22:1409–1425. doi: 10.3727/096368912X657620. [DOI] [PubMed] [Google Scholar]
- 10.Foust KD, Wang X, McGovern VL, Braun L, Bevan AK, Haidet AM, et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat Biotechnol. 2010;28:271–274. doi: 10.1038/nbt.1610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Krampera M, Cosmi L, Angeli R, Pasini A, Liotta F, Andreini A, et al. Role for interferon-gamma in the immuno-modulatory activity of human bone marrow mesenchymal stem cells. Stem Cells. 2006;24:386–398. doi: 10.1634/stemcells.2005-0008. [DOI] [PubMed] [Google Scholar]
- 12.Freedman MS, Bar-Or A, Atkins HL, Karussis D, Frassoni F, Lazarus H, et al. The therapeutic potential of mesen-chymal stem cell transplantation as a treatment for multiple sclerosis: consensus report of the International MSCT Study Group. Mult Scler. 2010;16:503–510. doi: 10.1177/1352458509359727. [DOI] [PubMed] [Google Scholar]
- 13.Kassis I, Grigoriadis N, Gowda-Kurkalli B, Mizrachi-Kol R, Ben-Hur T, Slavin S, et al. Neuroprotection and immunomodulation with mesenchymal stem cells in chronic experimental autoimmune encephalomyelitis. Arch Neurol. 2008;65:753–761. doi: 10.1001/archneur.65.6.753. [DOI] [PubMed] [Google Scholar]
- 14.Bai L, Lennon DP, Eaton V, Maier K, Caplan AI, Miller SD, et al. Human bone marrow-derived mesenchymal stem cells induce Th2-polarized immune response and promote endogenous repair in animal models of multiple sclerosis. Glia. 2009;57:1192–1203. doi: 10.1002/glia.20841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zappia E, Casazza S, Pedemonte E, Benvenuto F, Bonanni I, Gerdoni E, et al. Mesenchymal stem cells ameliorate experimental autoimmune encephalomyelitis inducing T-cell anergy. Blood. 2005;106:1755–1761. doi: 10.1182/blood-2005-04-1496. [DOI] [PubMed] [Google Scholar]
- 16.Constantin G, Marconi S, Rossi B, Angiari S, Calderan L, Anghileri E, et al. Adipose-derived mesenchymal stem cells ameliorate chronic experimental auto-immune encephalomyelitis. Stem Cells. 2009;27:2624–2635. doi: 10.1002/stem.194. [DOI] [PubMed] [Google Scholar]
- 17.Morando S, Vigo T, Esposito M, Casazza S, Novi G, Principato MC, et al. The therapeutic effect of mesen-chymal stem cell transplantation in experi-mental autoimmune encephalomyelitis is mediated by peripheral and central mecha-nisms. Stem Cell Res Ther. 2012;3:3. doi: 10.1186/scrt94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bernard CC, Carnegie PR. Experimental autoimmune encephalo-myelitis in mice: immunologic response to mouse spinal cord and myelin basic proteins. J Immunol. 1975;114:1537–1540. [PubMed] [Google Scholar]
- 19.Scruggs BA, Semon JA, Zhang X, Zhang S, Bowles AC, Pandey AC, et al. Age of Donor Reduces the Ability of Human Adipose-Derived Stem Cells to Alleviate Symptoms in the Experimental Autoimmune Encephalo-myelitis Mouse Model. SCTM. 2013;2:797–807. doi: 10.5966/sctm.2013-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scruggs BA, Bowles AC, Zhang X, Semon JA, Kyzar EJ, Myers L, et al. High-throughput screening of stem cell therapy for globoid cell leukodystrophy using automated neuro-phenotyping of twitcher mice. Behav Brain Res. 2013;236:35–47. doi: 10.1016/j.bbr.2012.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]