

Abstract
Repeated ethanol exposure and withdrawal in mice increases voluntary drinking and represents an animal model of physical dependence. We examined time- and brain region-dependent changes in gene coexpression networks in amygdala (AMY), nucleus accumbens (NAC), prefrontal cortex (PFC), and liver after four weekly cycles of chronic intermittent ethanol (CIE) vapor exposure in C57BL/6J mice. Microarrays were used to compare gene expression profiles at 0-, 8-, and 120-hours following the last ethanol exposure. Each brain region exhibited a large number of differentially expressed genes (2,000-3,000) at the 0- and 8-hour time points, but fewer changes were detected at the 120-hour time point (400-600). Within each region, there was little gene overlap across time (~20%). All brain regions were significantly enriched with differentially expressed immune-related genes at the 8-hour time point. Weighted gene correlation network analysis identified modules that were highly enriched with differentially expressed genes at the 0- and 8-hour time points with virtually no enrichment at 120 hours. Modules enriched for both ethanol-responsive and cell-specific genes were identified in each brain region. These results indicate that chronic alcohol exposure causes global ‘rewiring‘ of coexpression systems involving glial and immune signaling as well as neuronal genes.
Introduction
Long-term alcohol use and dependence alter brain function and are linked to persistent changes in gene expression [1–3]. Gene expression profiling in human alcoholics [4–6] and rodent models of binge drinking [7–9] and dependence [10–12] have provided insight into the changes in the brain transcriptional landscape resulting from different drinking paradigms; however, to date it is not clear whether transcriptome changes found in animal models of excessive alcohol consumption are consistent with changes found in human alcoholics. Consilience in gene expression would be a key step toward validating animal models by determining commonalities in molecular plasticity between human and rodent brain.
Genomic approaches have successfully identified alcohol-mediated changes in gene expression in animal models of alcoholism [9,13,14]. These studies suggest that distinct patterns of gene expression underlie specific alcohol-related phenotypes. Animal models of excessive consumption have been developed to investigate different stages of the alcohol abuse cycle that ultimately lead to dependence, including continuous two-bottle choice [15,16], drinking in the dark (DID) [17–19] (a model of binge drinking), and chronic intermittent ethanol (CIE) exposure [12,20] (a model of dependence). In general, studies have focused on transcriptional changes at a single time point following alcohol treatment; thus, it is difficult to determine if the changes in expression patterns are transient or longer lasting. CIE vapor exposure can be used to achieve and maintain high blood ethanol concentrations (180–200 mg/dl) in C57BL/6J mice, and it results in increased self-administration of ethanol [21–23]. Transcriptome profiling immediately following CIE exposure, rather than after subsequent bouts of voluntary drinking, could reveal gene expression and gene network changes associated with induction of ethanol dependence and early withdrawal.
We defined global gene expression profiles in amygdala (AMY), nucleus accumbens (NAC), prefrontal cortex (PFC), and liver of C57BL/6J mice exposed to 4 cycles of intermittent ethanol vapor. Tissue was harvested at 3 time points following the last vapor treatment to assess time-dependent changes in gene expression. We identified time-dependent gene clusters in AMY and NAC that were enriched with astrocytes, microglia, and oligodendrocyte cell types. These sets of genes were primarily associated with inflammatory response function. In contrast, the PFC was enriched with neuronal genes and displayed a greater diversity in directional expression changes, suggesting that the PFC is under greater transcriptional regulatory control than the AMY and NAC.
Materials and Methods
Ethics Statement
All procedures were approved by the Medical University of South Carolina Institutional Animal Care and Use Committee and adhered to NIH Guidelines. The Medical University of South Carolina animal facility is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care.
Animals and Chronic Ethanol Inhalation Procedure
Chronic intermittent ethanol vapor exposure (or air) was delivered in Plexiglas inhalation chambers, as previously described [21,22,24] to drug-naïve C57BL/6J (B6) male mice (8 treated and 8 controls per group). B6 mice were utilized because they show significant escalation of drinking when given access to alcohol after vapor exposure [21–23]. Ethanol treatments were performed in the laboratory of Dr. H.C. Becker (Medical University of South Carolina, Charleston, SC, USA). Briefly, ethanol (95%) was volatilized, mixed with fresh air and delivered to the chambers at a rate of 10 L/min to maintain consistent ethanol concentrations (15–20 mg/L air) in the chamber. Before entry into the chambers for each 16-hour exposure period, mice were administered ethanol (1.6 g/kg; 8% w/v) and the alcohol dehydrogenase inhibitor pyrazole (1 mmol/kg; i.p.) in a volume of 20 ml/kg body weight. Control mice were handled similarly, but they received saline and pyrazole and were exposed to air rather than alcohol vapor. The housing conditions in the inhalation chambers were identical to those in the colony room. Chamber ethanol concentrations were monitored daily using a LifeLoc Breathalyzer and airflow was adjusted to maintain ethanol concentrations within the specified range (180–200 mg/dl). The chamber exposure (16 hr/day) was administered in 4 weekly cycles alternated with 1 week in between in which the mice were left undisturbed (mimicking drinking weeks). Animals were sacrificed at 3 time points: 0-, 8- and 120-hours following the last ethanol vapor or air treatment. Brain and liver samples were snap frozen in liquid nitrogen and stored at −80°C until being shipped (frozen) to the Mayfield/Harris lab at University of Texas at Austin.
Tissue Harvest and RNA Isolation
Frozen brains were placed in a plastic mold containing Optimal Cutting Temperature compound (OCT) and maintained in a mixture of powdered dry-ice and isopentane. A Microm HM550 cryostat (Thermo Scientific, Ontario, CA) was used for sectioning at a thickness of 300 μm. Micropunches were collected from amygdala (AMY; 1.25 mm; combined basolateral and central nucleus), nucleus accumbens (NAC; 1.25 mm; combined core and shell), and prefrontal cortex (PFC; 2.0 mm). See S1 Fig for tissue punch details. Approximately 100 mg of tissue was obtained from the lower lobes of the liver. Total RNA was isolated according to manufacturer’s instructions using the MagMAX-96 Total RNA Isolation Kit (Ambion, Austin, TX). Total RNAs were quantified on a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific Inc., Rockford, IL), assessed for quality on an Agilent 2200 TapeStation Instrument (Agilent Technologies, Santa Clara, CA), and amplified/biotin-labeled using the Illumina TotalPrep RNA-96 Amplification kit (Ambion, Austin, TX).
Microarray Analysis
A web-based tool was used to determine the number of arrays required to detect meaningful statistical changes with a power of 0.8 (http://bioinformatics.mdanderson.org/MicroarraySampleSize/). Aliquots of labeled cRNA were sent to the Yale Center for Genome Analysis (West Haven, CT) where they were hybridized to Illumina MouseRef-8 v2 Expression BeadChips (Illumina, Inc., San Diego, CA) according to manufacturer protocols. Since each BeadChip contains 8 independent arrays, samples were hybridized in a group counter-balanced format to minimize batch effects. Each array was hybridized with material obtained from a single animal; thus, 192 arrays were included in the analysis (16 animals x 4 tissues x 3 time points). Each expression array contains approximately 25,600 transcripts representing over 19,100 unique genes. Transcript abundance was measured by fluorescent intensity after scanning. Microarray data have been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE60676.
Statistics and Bioinformatics
Unless otherwise noted, the data were analyzed using open source software packages from Bioconductor (http://bioconductor.org) designed for the statistical language R (http://www.r-project.org) and Microsoft Excel. The data were first filtered to include only genes with a detection p-value of ≤0.05 that were present on >80% of the arrays. Data pre-processing included a variance stabilization transformation [25] followed by a quantile normalization step [26] using the Bioconductor package lumi [27]. Expression value outliers were removed using Grubbs’ test [28]. Differential expression analysis for each time point was conducted using empirical Bayes moderated t-statistics from the Bioconductor package limma [29] to compare treated and control mice.
Weighted gene coexpression network analysis (WGCNA) was used to investigate the modular structure of the data at a gene network level. The general framework of WGCNA has been described in detail elsewhere [4,30]. This analysis was conducted as described previously [4] with modifications outlined below. Briefly, we constructed a signed network by calculating Pearson correlations for all pairs of genes (across all time points) and the signed similarity (Sij) matrix derived from Sij = (1+cor(xi,xj))/2, where gene expression profiles xi and xj consist of the expression of genes across multiple microarray samples. Sij was then raised to the power β to represent the connection strength. The goal of this step is to emphasize strong correlations and reduce the emphasis of weak correlations on an exponential scale. We chose a power of β = 8–9 so that the resulting networks exhibited approximate scale-free topology (Soft.R.sq = 0.68–070). All genes were hierarchically clustered based on a dissimilarity measure of topological overlap which measures interconnectedness for a pair of genes [4]. The resulting gene dendrogram was used for module detection using the dynamic tree cut method (minimum module size = 100, cutting height = 0.99). Module preservation scores (z-scores based upon a module membership size of 100) were calculated using the WGCNA package and are shown in S1 Table.
Gene ontology terms were identified using the Database for Annotation, Visualization and Integrated Discovery (DAVID) [31,32], and Ingenuity Pathways Analysis (IPA Ingenuity Systems, www.ingenuity.com) was used to identify overrepresented functional pathways of known gene networks and biological functions. Hypergeometric tests were used to evaluate modules and individual data sets for over-representation of cell type-specific genes. Datasets for neurons, astrocytes, microglia, and oligodendrocytes were obtained from previously published work [33,34]. The dataset used to identify enriched immune-related genes is included in S2 Table and was curated from SA Biosciences (QIAGEN). We then used an effect-size based approach to determine the direction and magnitude of ethanol-induced changes (adjusted p≤0.05) for each coexpression module. Mean t-values were calculated for each module, and this analysis was completed for each time point. Mean t-values were based on unique gene symbols within a module.
The Bioconductor package maSigPro [35] was used to identify clusters of genes showing different patterns of expression as a function of time. The method involves a two-step regression approach. First, a global regression model was used to identify differentially expressed genes, and second, a variable selection strategy was applied to identify differences between groups and time-related expression profiles. Prior to running maSigPro, a set of genes was identified in which gene expression was unchanged over time in control animals (AMY, n = 8098; NAC, n = 8323; PFC, n = 7246; Liver, n = 4157 genes). These genes were then used in the overall maSigPro analysis comparing gene expression in treated versus control animals. For data visualization, hierarchical clustering (4 clusters) was used to identify genes with similar expression patterns.
Historically it has been standard practice to verify a subset of microarray-generated gene expression changes using qRT- PCR. However, we did not include such confirmation in the present study because we have used the Illumina platform (including the particular array used in this study) extensively and ‘‘validated” expression differences with independent qRT- PCR experiments in the past. The level of correspondence between the microarray and RT-PCR results exceeds 80% [4,36,37].
Results
Gene Expression Changes
Time- and brain region-dependent changes in gene expression were detected in response to CIE vapor exposure in mice. These procedures do not result in overt behavioral signs of withdrawal at the exposure levels used this study. A similar number of genes were detected in each brain region (9,026–9,185), but fewer genes were detected in liver (7,616). CIE vapor elicited pronounced gene expression changes in all brain regions, as well as in liver (Fig. 1). However, after 120 hours, the transcriptional response to CIE had declined substantially in all tissues when compared with the 0- and 8-hour time points. Differential gene expression was not detected in any of the tissues at the 120-hour time point after multiple comparison correction. The 0- and 8-hour time points showed the greatest overlap of differentially expressed genes across tissues (Fig. 2; S3 Table; note that these data represent the top 500 differentially expressed genes regardless of multiple comparison correction). This overlap was less than 21% for each of the brain regions and about 50% for liver (data not shown). Differentially expressed genes at 0-hour were mostly (60–80%) regulated in a similar direction at 8 hours; however, the consistency of genes changing in the same direction at these time points was greatest for liver and least for PFC (Fig. 3).
Fig 1. Gene expression changes in brain and liver at three time points following CIE vapor.

Bars indicate the number of genes differentially expressed (p≤0.05) in each tissue at 0-, 8-, and 120-hours following CIE vapor treatment. The horizontal lines represent the number of differentially expressed genes after correcting for multiple comparisons (FDR≤0.05).
Fig 2. Overlap of the top 500 differentially expressed genes across time.
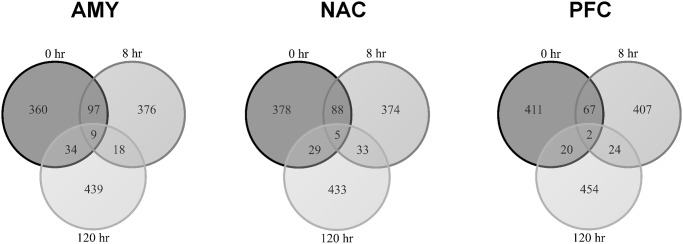
Each time point displays a relatively unique set of differentially expressed genes for each brain region. Times 0 and 8 share the greatest overlap of differentially expressed genes within each brain region; however, this overlap was less than 21%. The three time points combined exhibit very little overlap of differentially expressed genes within each brain region (AMY = amygdala; NAC = nucleus accumbens; PFC = prefrontal cortex).
Fig 3. Directionality of differentially expressed genes at 0- and 8-hours.

Bars show the percentage of differentially expressed genes (FDR ≤ 0.05) in the same (white) and opposite (black) direction at 0- and 8-hours. Percentages are based on the total number of differentially expressed genes at time 0. (AMY = amygdala; NAC = nucleus accumbens; PFC = prefrontal cortex).
WGCNA and Enrichment Analyses
For all tissues, there was distinct clustering of gene networks at 120-hours compared to other times, and this time point had a greater effect than treatment on sample clustering due to the lack of expression changes (data not shown). WGCNA identified 34–45 modules in brain and 24 modules in liver, with module sizes ranging from 78–1,412 transcripts. The Database for Annotation, Visualization and Integrated Discovery (DAVID) was used for over-representation analysis and to evaluate the biological function of each module.
These results were further substantiated by enrichment analysis using cell-specific and functional gene lists. Published cell-type gene lists (see Methods for details) were used to determine which cells might be disproportionately enriched with differentially expressed genes (adjusted p≤0.05) in response to CIE vapor as a function of time. Enrichment of microglia-specific genes was observed in all brain regions at the 8-hour time point while almost all brain regions and time points were enriched with astrocyte-specific genes (Table 1). The NAC was the only brain region enriched with oligodendrocyte-specific genes, and the PFC was the only region enriched with neuronal genes (Table 1). A list of 824 immune-related genes obtained from the SA Biosciences website (S2 Table) was used to identify over-represented differentially expressed immune genes. For this analysis, an FDR cutoff of 0.05 was used. Notably, there was consistent enrichment of immune-related genes in all brain regions at 8 hours (Table 2). However, there was no over-representation of immune genes in liver (data not shown).
Table 1. Enrichment of ethanol-responsive, cell type-specific genes at 0- and 8-hours after CIE vapor.
| Tissue | Time Point | Astrocyte | Microglial | Neuron | Oligodendrocyte |
|---|---|---|---|---|---|
| AMY | 0 hr | 5.96E-06 | 7.49E-02 | 2.17E-01 | 5.45E-01 |
| 8 hr | 7.67E-09 | 7.14E-07 | 9.97E-01 | 9.22E-02 | |
| NAC | 0 hr | 4.52E-01 | 4.52E-01 | 1.42E-01 | 4.38E-07 |
| 8 hr | 2.62E-03 | 2.53E-05 | 4.32E-01 | 7.42E-03 | |
| PFC | 0 hr | 4.80E-04 | 5.61E-02 | 4.80E-04 | 5.95E-01 |
| 8 hr | 2.83E-04 | 1.21E-02 | 2.39E-02 | 3.60E-01 |
Table shows hypergeometric q values for over-representation of cell-type specific genes in differentially expressed genes (adjusted p ≤ 0.05).
AMY = amygdala; NAC = nucleus accumbens; PFC = prefrontal cortex. Q values are shown (hypergeometric test) for differentially expressed genes (FDR≤0.05) that are enriched in specific cell types within each brain region.
Table 2. Enrichment of ethanol-responsive, immune-related genes at 0 and 8 hours after CIE vapor.
| Tissue | Time Point | P Value |
|---|---|---|
| AMY | 0 hour | 2.16E-01 |
| 8 hour | 1.23E-05 | |
| NAC | 0 hour | 7.05E-01 |
| 8 hour | 1.79E-02 | |
| PFC | 0 hour | 3.95E-03 |
| 8 hour | 1.49E-02 |
AMY = amygdala; NAC = nucleus accumbens;cPFC = prefrontal cortex. P values are shown (hypergeometric test) for differentially expressed immune-related genes (FDR≤0.05) in each brain region.
We also examined the overlap of cell-specific differentially expressed genes (FDR≤0.05) at the 0- and 8-hour time points. Microglial genes were the most highly conserved group of cell type-specific genes across these time points for all brain regions (Fig. 4). In addition, modules were evaluated for enrichment with differentially expressed genes. All brain regions exhibited a number of ethanol-responsive modules that were also enriched with genes from one or more of the cell-type/functional gene lists (Table 3). Microglia- and astrocyte-enriched modules showed the most persistent gene expression changes across time and were primarily associated with anti-apoptosis and immune response. In contrast, neuron- and oligodendrocyte-enriched modules showed more transient gene expression changes. For example, neuronal enrichment was only observed in one module (PFC/2) at 0- and 8-hours. A single module (NAC/3) was enriched with microglia and oligodendrocyte genes as well as ethanol-responsive genes at 0- and 8-hours. Table 3 also indicates differentially expressed module hub genes for each brain region and time point. Specific gene names and level of significance is provided in S4 Table.
Fig 4. Overlap of cell-specific, differentially expressed genes at 0- and 8-hours after CIE vapor.

Bars show the percentage of cell type-specific genes differentially expressed (FDR ≤ 0.05) at both the 0- and 8-hour time points. Percentages are based on the total number of differentially expressed, cell type-specific genes at time 0. (AMY = amygdala; NAC = nucleus accumbens; PFC = prefrontal cortex).
Table 3. Ethanol-responsive WGCNA co-expression modules enriched with cell type-specific genes.
| Astrocytes | Microglia | Neuron | Oligodendrocytes | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Tissue/ Module | Module Function Summary | 0 hour | 8 hour | 0 hour | 8 hour | 0 hour | 8 hour | 0 hour | 8 hour |
| AMY/1 | Anti-apoptosis | X, H FDR | X, H FDR | X, H FDR | X, H FDR | ||||
| AMY/2 | Antigen processing and presentation; adaptive immune response; major histocompatability complex | X, H | X, H FDR | ||||||
| AMY/3 | Neuron development; axonogenesis; glycoprotein | X, H FDR | |||||||
| AMY/4 | GPCR activity; nucleotide receptor activity | X, H FDR | X, H FDR | ||||||
| AMY/5 | Glutathione metabolism; drug metabolism | X, H | |||||||
| NAC/1 | GPCR activity; disulfide bond; immunoglobulin, extracellular matrix | X, H FDR | X, H FDR | ||||||
| NAC/2 | Glutathione metabolism; drug metabolism | X, H FDR | X, H FDR | ||||||
| NAC/3 | ras/rho GTPase signaling; anti-apoptosis | X, H FDR | X, H FDR | X, H FDR | X, H FDR | ||||
| NAC/4 | Antigen processing and presentation; adaptive immune response | X, H FDR | X, H FDR | ||||||
| NAC/5 | Fibronectin, type III subdomain; cell adhesion | X, H FDR | |||||||
| PFC/1 | Antigen processing and presentation; adaptive immune response; major histocompatability complex | X, H FDR | X, H FDR | ||||||
| PFC/2 | Small GTPase mediated signal transduction; circadian rhythm | X, H FDR | X, H FDR | ||||||
| PFC/3 | Ribosome; extracellular region; disulfide bond; binding (glycosaminoglycan, pattern, polysaccharide, heparin, carbohydrate); glycoprotein | X | |||||||
AMY = amygdala; NAC = nucleus accumbens; PFC = prefrontal cortex. Only modules enriched with both cell type-specific and ethanol-responsive genes (FDR ≤ 0.05) are shown. Functional annotations are shown for each module. [An "X" indicates enrichment of the module with ethanol-responsive and cell type-specific genes at the given time point, "H" indicates differentially expressed module Hub gene (unadjusted p-value ≤ 0.05), and HFDR indicates differentially expressed Hub gene (FDR ≤ 0.05)].
To further investigate ethanol-responsive modules, we used an effect-size based approach and determined the direction and magnitude of ethanol-induced changes (adjusted p≤0.05) for each coexpression module. Mean t-values were calculated for ethanol-responsive modules identified by WGCNA at each time point. The magnitude and direction of change were relatively consistent in AMY and NAC at 0- and 8-hours (Fig. 5A and 5B). In contrast, these parameters were quite different in the PFC (black and dark turquoise modules; Fig. 5C). The greatest consistency in t-value magnitude and direction was observed in liver (Fig. 5D).
Fig 5. Mean t values for ethanol-responsive modules in brain and liver at 0-, 8-, and 120-hours.

Bars show mean t values (±SEM) of ethanol-responsive modules for each tissue at each time point. Panel A = AMY (amygdala), Panel B = NAC (nucleus accumbens), Panel C = PFC (prefrontal cortex), and Panel D = Liver.
Time Series Analysis
A time series analysis was performed using the Bioconductor package maSigPro [35] to identify clusters of differentially expressed genes with similar patterns of expression. For each tissue, the gene clusters with the greatest overlap with WGCNA modules containing significantly enriched differentially expressed and cell type-specific genes are shown in Fig. 6. The complete sets of clusters for each tissue are included in S2–S5 Figs and S5 Table. In the AMY (Cluster 2), all 15 genes are included in WGCNA modules AMY/1 and AMY/2 (Table 3). These modules were significantly enriched with differentially expressed astrocyte and microglia genes (Table 3: AMY/2 and AMY/3). Similarly, in Cluster 3 of NAC, 20 out of 21 genes overlapped with modules enriched with microglia and oligodendrocytes (Table 3; NAC/3 and NAC/4). In contrast, the PFC cluster (Cluster 2) highly overlapped with a module (Table 3; PFC/2) enriched with neuronal genes. The genes included in each maSigPro cluster are shown in Table 4. Additional patterns of differentially expressed genes were detected but were not found to be enriched in WGCNA modules and are not shown (see S2–S5 Figs for the complete set of clusters).
Fig 6. Time series analysis (MaSigPro).

A two-step regression approach was used to identify clusters of differentially expressed genes with similar expression patterns across time. Each plot shows the hierarchical clustering (clusters = 4) of average expression profiles by time and tissue type (AMY = amygdala; NAC = nucleus accumbens; PFC = prefrontal cortex; Liver). Panels A-D show gene clusters that have the greatest overlap with WGCNA modules with significantly enriched differentially expressed genes and cell type-specific genes. The dots represent average expression values for each gene in the time series.
Table 4. Clusters of differentially expressed genes with similar expression patterns as a function of time (maSigPro time series analysis).
| Tissue | Cluster | Gene Symbol |
|---|---|---|
| AMY | 1 | Hba-a1, Asrgl1, Hbb-b1, Sox21, 1190002H23Rik, Pafah1b3, Hist1h4h, Mcm5, G3bp1, Mrp63, Trappc4, Slc30a10, Pcmt1 |
| 2 | B2m*, Cd74, Mfsd2, Fcgr2b, Psmb9, Pglyrp1, Htatip2, Pcsk4, Spp1, Sec63, EG630499, EG667977, Alox5ap, Ccl25 | |
| 3 | Dtna, Slc25a33 | |
| 4 | Rit1, Fkrp, Fam131a, Hes6 | |
| NAC | 1 | Elac2, Dtna, Snai3, Nrn1l, Mpp3, Fmo1, Hist1h1c, Vgf, Slc25a3, Sorcs3, Smap2 |
| 2 | Hba-a1, Heatr1, Hbb-b1, Sox21, AI428936, Aldh3b1, P2ry1, Pex6, Phldb1, Exdl2, H2afj | |
| 3 | H2-T23*, Mfsd2, Scg2, Psmb9, Pglyrp1, Gata2, Slc1a2, Jun, H2-M3, Htra1*, Cebpb, Il17rc, EG630499, EG667977, Cd151, H2-Q7, Ccl25, Cd74, Tsc22d3 | |
| 4 | Slco4a1, LOC100046883, Sdpr, Ppif | |
| PFC | 1 | Heatr1, Prmt2, Tsga14, Hbb-b1, Trib2, 4930455F23Rik, Dnajb6 |
| 2 | Bdnf, Rem2, Adcyap1, Sorcs3*, Pglyrp1, Fscn1, Cdkn1a*, Chst8, AI593442, Nrn1, Ttpal, Fam20a, Car12, Vgf, 2810452K22Rik | |
| 3 | LOC381629, Arpp21, Smpdl3b, Tprkb, Slc41a3, BC028528, Ccl25, Ppp4r4, R74862, Htra1*, BC031353, Snn, EG667977, Xpa, 0610007C21Rik* | |
| 4 | Wwp2, Cyhr1, Tmem132e, C230078M08Rik, Lsm10, Zmat4, Fchsd1 | |
| Liver | 1 | Mrpl48*, 2610528J11Rik, Arsb, Arsa, Pex5*, Dut, Dis3l, Smad2, Ipp, Acy3, Ndufaf1, Lman2, Ergic2, Adh4, Ube2l3, Tpk1, LOC100048313, Atpaf2, Snrk, Pygl*, Sh3glb2, Lrba, Tmem93, Mrpl22, Pnpla7, Muted, Sec14l2, Galt, Cyb5r3*, Clec11a, F2r, Mrps28, Mrpl53, Rngtt, Ppapdc2, Sacm1l, Trak1, Rtn4ip1, Zhx1, Nelf |
| 2 | Akr1b8, Vav1, Csf1r, Sftpd, Rcsd1, Rims3, Lgmn, Thy1, Clec4f, LOC100044439, Vcam1, Psap, Spic, Slc16a9, Clec4n, Btbd11 | |
| 3 | Ubl3, Soat2, Eps15, Rap1b, Tbk1, Adrb2, Epn2, Slc2a1, Rps9, Usp3, Nrbp2, Cyp2b10, Cyp3a13*, Acat1, Nr1i2, LOC100048445, Oas2, Gdpd1, Cd68, Eif3k, Sult1a1, Rnf166, Hgsnat, Cyp1a2, Tm4sf4, Nr1i3, Cyb5, Cyp2b23, Keap1, Samd4, Nrap | |
| 4 | 9630015D15Rik, Mgll**, Smarce1*, Cpt2 |
AMY = amygdala; NAC = nucleus accumbens; PFC = prefrontal cortex. Red text indicates clusters of genes overlapping with WGCNA modules that were enriched with differentially expressed and cell type-specific genes. Illumina Probe_Ids resulting in duplicate and triplicate Gene Symbols within the clusters are indicated by * and **, respectively.
Discussion
The goal of the current study was to determine time-dependent transcriptional changes in brain and liver that result from administration of repeated ethanol vapor. This paradigm has been shown to escalate voluntary drinking in both rats and mice and represents a rodent model of dependence [38]. Our hypothesis was that time- and brain region-dependent changes in expression would be observed, and that at least a subset of genes, would be persistently changed in response to CIE vapor. Animals were sacrificed at 0-, 8-, and 120-hours after the last ethanol treatment to determine whether persistent changes in gene expression would be observed during protracted abstinence. At the 8-hour time point, mice are in acute withdrawal based on hypothalamic-pituitary-adrenocortical axis elevation [39], but they do not display seizures because they are a relatively seizure-resistant strain.
A multi-level analysis approach was utilized which included differential expression, network analysis, cell-type specificity, and time-series clustering analyses. These approaches include computational algorithms that enhance the analysis of gene coexpression networks existing in diverse expression datasets. In addition, since the brain transcriptome is organized into gene modules associated with major cell classes and specific synaptic and cellular functions [34], we classified sets of genes based upon subcellular localization. Neurons and glial cells are characterized by unique transcriptional signatures [33,40] and these signatures can be identified reliably from analysis of complex brain tissue without isolating homogeneous populations of cells [4,34]. Classifying significantly regulated genes into cell type-specific signatures improves the quality of inference and potentially leads to refined hypotheses [41].
A time-series analysis (Bioconductor package maSigPro) was performed to identify clusters of genes with similar expression patterns. Each brain region displayed distinct clusters overlapping with WGCNA modules enriched in differentially expressed and cell type-specific genes. In the AMY, a cluster was identified (Cluster 2: Fig. 6 and Table 4) in which all genes were included with modules enriched with differentially expressed, and enriched with astrocyte and microglia genes. The expression of this cluster of genes was significantly increased at 0- and 8-hours, but returned to baseline at 120-hours. IPA analysis indicated that this cluster was enriched in "Inflammatory Disease"-related genes (Alox5ap, B2m, Cd74, Fcgr2b, Hla-A, Pglyrp1, Psmb9, Spp1). Interestingly, B2m is known to be important for immune responses and has been shown to be alcohol responsive in multiple studies [13,42–44]. Blednov et al. [42] demonstrated that deletion of B2m reduced ethanol consumption in the limited access two-bottle choice test for ethanol intake, supporting a hypothesis that genes within this cluster may play a role in mediating voluntary drinking. In addition, the innate immune cytokine Cd74 was a member of this cluster. The expression of this gene is rapidly induced by alcohol and has been linked to the progression of cytokine responses during alcohol withdrawal [45] which is consistent with changes observed 8 hours post-treatment.
In NAC, a cluster of genes overlapped with modules enriched with microglia and oligodendrocytes (Cluster 3: Fig. 6 and Table 4). Similar to the AMY, IPA analysis indicated that this cluster was enriched in "Inflammatory Disease"-related genes (Cd74, Cebpb, Hla-A, Hla-E, Hla-G, Htra1, Il17rc, Pglyrp1, Psmb9, Slc1a2). Slc1a2 (a glial glutamate transporter) is highly expressed in microglia [46] and has been linked to neurodegenerative disorders [47] as well as drug dependence [48]. This gene is differentially expressed in human alcoholic post-mortem brain [4] and drugs acting on this target alter motivation to drink in dependent animals [49]. In addition, genetic variation in glutamate transporters confers risk-taking behavior in alcoholics [50]. Together, these findings support a role for Slc1a2 in alcohol intake and dependence. As in the AMY, the cytokine Cd74 was also identified in NAC, suggesting that it may have a role in innate immune responses in multiple brain regions. Other genes in this cluster that are differentially expressed in mouse models and human alcoholics include Htra1 [13] and Il17rc [4]. Tsc22d3 and Gata-2 are alcohol-responsive members of this gene set but are not members of inflammatory response pathways. Tsc22d3 functions as a transcriptional regulator and is differentially expressed in human alcoholics [4]. Tsc22d3 may be associated with neuroplastic changes in response to drugs of abuse, including ethanol in mouse striatum [51].
In contrast to the glial signature of many of the clusters, one PFC gene cluster (Cluster 2: Fig. 6 and Table 4) was highly overlapping with a WGCNA module enriched with differentially expressed neuronal genes. Mean t-value distributions (Fig. 5) indicated that the magnitude and direction of change were clearly different in the PFC compared to the other brain regions, showing both a significant down- and up-regulation of genes at the 0- and 8-hour time points. These data suggest that chronic ethanol vs withdrawal changes in neuronal gene regulation may reflect greater transcriptional control in PFC than in the enriched cell types in AMY and NAC. The most prominent pathway identified by IPA analysis indicated that this cluster was enriched in "Neurological Disease"-related genes (Adcyap1, Bdnf, CA12, Cdkn1a, Vgf). Bdnf has a well-documented role in synaptic plasticity [52,53] and addiction [54]. We recently demonstrated that Bdnf is significantly down-regulated in homogenized medial PFC tissue [11] as well as in purified synaptoneurosome preparations [55]. Reductions of Bdnf levels [56,57] and knockdown of Bdnf expression increase ethanol-drinking behavior [58]. Together, these data support a role for Bdnf in the modulation of ethanol intake. Alcohol consumption is known to be modulated by circadian-related cellular function [59]. In the current study, the circadian gene Adcyap1 was identified as differentially expressed member of an enriched neuronal module and may represent a regulatory mechanism involved in the time-dependent expression changes.
These marked changes in glial, and particularly microglial, genes at both the 0- and 8-hour time points support an emerging concept of neuroimmune changes during alcohol dependence [60,61]. Alterations in neuroimmune gene expression are seen in human alcoholics and also in rodent models with intermittent administration of high doses of ethanol [5,62,63]. It is interesting to note that chronic voluntary alcohol consumption (resulting in lower blood levels compared to inhalation or injection of ethanol) did not produce pronounced changes in expression of microglial or neuroimmune genes in a tissue homogenate [9], but such changes were more prevalent in a synaptoneurosome preparation that enriches for synaptic elements, including glia [55]. It will be interesting to examine in future studies whether voluntary alcohol consumption in this CIE model influences the gene expression profile reported in the present study.
The present study shows marked changes in gene expression between the 0- and 8-hour time points in brain neuronal compartments, but fewer differences in non-neuronal clusters, and very few in liver. This is consistent with marked changes in neuronal excitability during the first 8 hours following withdrawal of alcohol and emphasizes the importance of comparing neuronal and glial gene expression patterns, as the glial changes are much more stable during withdrawal than the neuronal clusters.
In conclusion, gene modules and time-dependent gene clusters were identified in AMY and NAC that were enriched with astrocytes, microglia, and oligodendrocyte cell types. These sets of genes were primarily associated with inflammatory response function and the changes in expression in this model of alcohol dependence is consistent with the proposed role of neuroimmune signaling in promoting alcohol consumption [42,60]. In contrast, the PFC was enriched with neuronal genes and displayed a greater diversity in directional expression changes, suggesting that PFC is under greater transcriptional regulatory control. Importantly, of the many expression changes identified at 0- and 8-hours after ethanol vapor treatment, the majority of changes returned to baseline levels after 120-hours. These results suggest that although intermittent ethanol vapor treatment increases alcohol consumption 120 hours after the last treatment, it is difficult to detect changes in gene expression that might be responsible for signs of protracted abstinence. It is possible that some of the weak changes observed in our study after 120 hours and by Tapocik et al. [10] after 3 weeks of abstinence are important but are diluted by the cellular heterogeneity of the brain. Analysis of cell-specific transcriptomes, or use of preparations such as synaptoneurosomes [55], may be needed to detect persistent changes in gene expression produced by alcohol dependence.
Supporting Information
Frozen brains were placed in a plastic mold containing OCT and maintained in a mixture of powdered dry-ice and isopentane. A Microm HM550 cryostat (Thermo Scientific, Ontario, CA) was used for sectioning at a thickness of 300 μm. Micropunches were collected from amygdala (AMY; 1.25 mm; combined basolateral and central nucleus), nucleus accumbens (NAC; 1.25 mm; combined core and shell), and prefrontal cortex (PFC; 2.0 mm).
(PDF)
A two-step regression approach was used to identify clusters of differentially expressed genes with similar expression patterns across time. Each plot shows the hierarchical clustering (clusters = 4) of average expression profiles by time in the amygdala (AMY). The dots represent average expression values for each gene in the time series.
(PDF)
A two-step regression approach was used to identify clusters of differentially expressed genes with similar expression patterns across time. Each plot shows the hierarchical clustering (clusters = 4) of average expression profiles by time in the nucleus accumbens (NAC). The dots represent average expression values for each gene in the time series.
(PDF)
A two-step regression approach was used to identify clusters of differentially expressed genes with similar expression patterns across time. Each plot shows the hierarchical clustering (clusters = 4) of average expression profiles by time in the prefrontal cortex (PFC). The dots represent average expression values for each gene in the time series.
(PDF)
A two-step regression approach was used to identify clusters of differentially expressed genes with similar expression patterns across time. Each plot shows the hierarchical clustering (clusters = 4) of average expression profiles by time in the Liver. The dots represent average expression values for each gene in the time series.
(PDF)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Acknowledgments
The authors would like to thank Dr. Jody Mayfield for thoughtful comments on the manuscript.
Data Availability
Microarray data have been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE60676.
Funding Statement
This work was supported by National Institute on Alcohol Abuse and Alcoholism (NIAAA) grants AA016648, AA012404, U01 AA014095, U01 AA020929, and P50 AA010761. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Farris SP, Wolen AR, Miles MF. Using expression genetics to study the neurobiology of ethanol and alcoholism. Int Rev Neurobiol. 2010;91: 95–128. 10.1016/S0074-7742(10)91004-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nestler EJ. Genes and addiction. Nat Genet. 2000;26: 277–81. [DOI] [PubMed] [Google Scholar]
- 3. Robison AJ, Nestler EJ. Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci. 2011;12: 623–37. 10.1038/nrn3111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ponomarev I, Wang S, Zhang L, Harris RA, Mayfield RD. Gene coexpression networks in human brain identify epigenetic modifications in alcohol dependence. Journal of Neuroscience. 2012;32: 1884–97. 10.1523/JNEUROSCI.3136-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu J, Lewohl JM, Harris RA, Iyer VR, Dodd PR, Randall PK, et al. Patterns of gene expression in the frontal cortex discriminate alcoholic from nonalcoholic individuals. Neuropsychopharmacology. 2006;31: 1574–82. [DOI] [PubMed] [Google Scholar]
- 6. Farris SP, Mayfield RD. RNA-Seq Reveals Novel Transcriptional Reorganization in Human Alcoholic Brain. Int Rev Neurobiol. 2014;116: 275–300. 10.1016/B978-0-12-801105-8.00011-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Agrawal RG, Owen JA, Levin PS, Hewetson A, Berman AE, Franklin SR, et al. Bioinformatics analyses reveal age-specific neuroimmune modulation as a target for treatment of high ethanol drinking. Alcohol Clin Exp Res. 2014;38: 428–37. 10.1111/acer.12288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Iancu OD, Oberbeck D, Darakjian P, Metten P, McWeeney S, Hitzemann R. Selection for drinking in the dark alters brain gene coexpression networks. Alcohol Clin Exp Res. 2013;37: 1295–303. 10.1111/acer.12100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Osterndorff-Kahanek E, Ponomarev I, Harris RA. Gene expression in brain and liver produced by three different regimens of alcohol consumption in mice: comparison with immune activation. PLoS ONE. 2013;8: e59870 10.1371/journal.pone.0059870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tapocik JD, Solomon M, Flanigan M, Meinhardt M, Barbier E, Schank JR, et al. Coordinated dysregulation of mRNAs and microRNAs in the rat medial prefrontal cortex following a history of alcohol dependence. The Pharmacogenomics Journal. 2013;13: 286–96. 10.1038/tpj.2012.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Melendez RI, McGinty JF, Kalivas PW. Brain region-specific gene expression changes after chronic intermittent ethanol exposure and early withdrawal in C57BL/6J mice. Addict Biol. 2012;17: 351–64. 10.1111/j.1369-1600.2011.00357.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lopez MF, Becker HC. Operant ethanol self-administration in ethanol dependent mice. Alcohol. 2014;48: 295–9. 10.1016/j.alcohol.2014.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mulligan MK, Ponomarev I, Hitzemann RJ, Belknap JK, Tabakoff B, Harris RA, et al. Toward understanding the genetics of alcohol drinking through transcriptome meta-analysis. Proc Natl Acad Sci USA. 2006;103: 6368–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mayfield RD, Harris RA, Schuckit MA. Genetic factors influencing alcohol dependence. Br J Pharmacol. Blackwell Publishing Ltd; 2008;154: 275–87. 10.1038/bjp.2008.88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wahlsten D, Bachmanov A. Stability of inbred mouse strain differences in behavior and brain size between laboratories and across decades. Proc Natl Acad Sci USA. 2006;103: 16364–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hopf FW, Simms JA, Chang S-J, Seif T, Bartlett SE, Bonci A. Chlorzoxazone, an SK-Type Potassium Channel Activator Used in Humans, Reduces Excessive Alcohol Intake in Rats. Biol Psychiatry. 2011;69: 618–24. 10.1016/j.biopsych.2010.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hopf FW, Lesscher HMB. Rodent models for compulsive alcohol intake. Alcohol. 2014;48: 253–64. 10.1016/j.alcohol.2014.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thiele TE, Boehm SL II. “Drinking in the Dark (DID)”: A Simple Mouse Model of Binge-Like Alcohol Intake. Hoboken, NJ, USA: John Wiley & Sons, Inc; 2001. 10.1002/0471142301.ns0949s68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barkley-Levenson AM. High Drinking in the Dark Mice: A genetic model of drinking to intoxication. Alcohol. 2014;48: 217–23. 10.1016/j.alcohol.2013.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Becker HC. Animal models of excessive alcohol consumption in rodents Curr Top Behav Neurosci. Berlin, Heidelberg: Springer Berlin Heidelberg; 2013;13 (Chapter 203): 355–77. [DOI] [PubMed] [Google Scholar]
- 21. Becker HC, Lopez MF. Increased ethanol drinking after repeated chronic ethanol exposure and withdrawal experience in C57BL/6 mice. Alcohol Clin Exp Res. 2004;28: 1829–38. [DOI] [PubMed] [Google Scholar]
- 22. Lopez MF, Becker HC. Effect of pattern and number of chronic ethanol exposures on subsequent voluntary ethanol intake in C57BL/6J mice. Psychopharmacology. 2005;18: 688–96. [DOI] [PubMed] [Google Scholar]
- 23. Griffin WC III, Lopez MF, Becker HC. Intensity and duration of chronic ethanol exposure is critical for subsequent escalation of voluntary ethanol drinking in mice. Alcohol Clin Exp Res. Blackwell Publishing Ltd; 2009;33: 1893–900. 10.1111/j.1530-0277.2009.01027.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Becker HC, Hale RL. Repeated episodes of ethanol withdrawal potentiate the severity of subsequent withdrawal seizures: an animal model of alcohol withdrawal "kindling". Alcohol Clin Exp Res. 1993;17: 94–8. [DOI] [PubMed] [Google Scholar]
- 25. Lin SM, Du P, Huber W, Kibbe WA. Model-based variance-stabilizing transformation for Illumina microarray data. Nucleic Acids Res. 2008;36: e11–1. 10.1093/nar/gkm1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19: 185–93. [DOI] [PubMed] [Google Scholar]
- 27. lumi: a pipeline for processing Illumina microarray. Oxford University Press; 2008;24: 1547–8. 10.1093/bioinformatics/btn224 [DOI] [PubMed] [Google Scholar]
- 28. Sample Criteria for Testing Outlying Observations. Institute of Mathematical Statistics; 1950;21: 27–58. [Google Scholar]
- 29. Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. 2004;3: Article3 [DOI] [PubMed] [Google Scholar]
- 30. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9: 559 10.1186/1471-2105-9-559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4: 44–57. 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- 32. Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. Oxford University Press; 2009;37: 1–13. 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cahoy JD, Ben Emery, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. Journal of Neuroscience. 2008;28: 264–78. 10.1523/JNEUROSCI.4178-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, et al. Functional organization of the transcriptome in human brain. Nat Neurosci [Internet]. 2008;11: 1271–82. Available: http://www.nature.com/doifinder/10.1038/nn.2207 10.1038/nn.2207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Conesa A, Nueda MJ, Ferrer A, Talón M. maSigPro: a method to identify significantly differential expression profiles in time-course microarray experiments. Bioinformatics. 2006;22: 1096–102. [DOI] [PubMed] [Google Scholar]
- 36. Ponomarev I, Rau V, Eger EI, Harris RA, Fanselow MS. Amygdala transcriptome and cellular mechanisms underlying stress-enhanced fear learning in a rat model of posttraumatic stress disorder. Neuropsychopharmacology. 2010;35: 1402–11. 10.1038/npp.2010.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Harris RA, Osterndorff-Kahanek E, Ponomarev I, Homanics GE, Blednov YA. Testing the silence of mutations: Transcriptomic and behavioral studies of GABA(A) receptor α1 and α2 subunit knock-in mice. Neurosci Lett. 2011;488: 31–5. 10.1016/j.neulet.2010.10.075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Becker HC, Ron D. Animal models of excessive alcohol consumption: recent advances and future challenges. Alcohol. 2014;48: 205–8. 10.1016/j.alcohol.2014.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Becker HC. Effects of alcohol dependence and withdrawal on stress responsiveness and alcohol consumption. Alcohol Res. 2012;34(4):448–58. [PMC free article] [PubMed] [Google Scholar]
- 40. Sugino K, Hempel CM, Miller MN, Hattox AM, Shapiro P, Wu C, et al. Molecular taxonomy of major neuronal classes in the adult mouse forebrain. Nat Neurosci. 2005;9: 99–107. [DOI] [PubMed] [Google Scholar]
- 41. Ponomarev I, Maiya R, Harnett MT, Schafer GL, Ryabinin AE, Morikawa H, et al. Transcriptional signatures of cellular plasticity in mice lacking the alpha1 subunit of GABAA receptors. Journal of Neuroscience. 2006;26: 5673–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ponomarev I, Geil C, Bergeson S, Harris RA. Neuroimmune regulation of alcohol consumption: behavioral validation of genes obtained from genomic studies. Addict Biol. 2012;17: 108–20. 10.1111/j.1369-1600.2010.00284.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mulligan MK, Rhodes JS, Crabbe JC, Mayfield RD, Harris RA, Ponomarev I. Molecular profiles of drinking alcohol to intoxication in C57BL/6J mice. Alcohol Clin Exp Res. 2011;35: 659–70. 10.1111/j.1530-0277.2010.01384.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kimpel MW, Strother WN, McClintick JN, Carr LG, Liang T, Edenberg HJ, et al. Functional gene expression differences between inbred alcohol-preferring and -non-preferring rats in five brain regions. Alcohol. 2007;41: 95–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Freeman K, Brureau A, Vadigepalli R, Staehle MM, Brureau MM, Gonye GE, et al. Temporal changes in innate immune signals in a rat model of alcohol withdrawal in emotional and cardiorespiratory homeostatic nuclei. J Neuroinflammation. 2012;9: 97 10.1186/1742-2094-9-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. López-Redondo F, Nakajima K, Honda S, Kohsaka S. Glutamate transporter GLT-1 is highly expressed in activated microglia following facial nerve axotomy. Brain Res Mol Brain Res. 2000;76: 429–35. [DOI] [PubMed] [Google Scholar]
- 47. Thai DR. Excitatory amino acid transporter EAAT-2 in tangle-bearing neurons in Alzheimer's disease. Brain Pathol. 2002;12: 405–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nakagawa T, Satoh M. Involvement of glial glutamate transporters in morphine dependence. Ann N Y Acad Sci. 2004;1025: 383–8. [DOI] [PubMed] [Google Scholar]
- 49. Griffin WC III, Haun HL, Hazelbaker CL, Ramachandra VS, Becker HC. Increased extracellular glutamate in the nucleus accumbens promotes excessive ethanol drinking in ethanol dependent mice. Neuropsychopharmacology. 2014;39: 707–17. 10.1038/npp.2013.256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sander T, Ostapowicz A, Samochowiec J, Smolka M, Winterer G, Schmidt LG. Genetic variation of the glutamate transporter EAAT2 gene and vulnerability to alcohol dependence. Psychiatr Genet. 2000;10: 103–7. [DOI] [PubMed] [Google Scholar]
- 51. Piechota M, Korostynski M, Solecki W, Gieryk A, Slezak M, Bilecki W, et al. The dissection of transcriptional modules regulated by various drugs of abuse in the mouse striatum. Genome Biol. 2010;11: R48 10.1186/gb-2010-11-5-r48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Im H-I, Hollander JA, Bali P, Kenny PJ. MeCP2 controls BDNF expression and cocaine intake through homeostatic interactions with microRNA-212. Nat Neurosci. 2010;13: 1120–7. 10.1038/nn.2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schratt GM, Nigh EA, Chen WG, Hu L, Greenberg ME. BDNF regulates the translation of a select group of mRNAs by a mammalian target of rapamycin-phosphatidylinositol 3-kinase-dependent pathway during neuronal development. Journal of Neuroscience. 2004;24: 7366–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lobo MK, Covington HE, Chaudhury D, Friedman AK, Sun H, Damez-Werno D, et al. Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science. 2010;330: 385–90. 10.1126/science.1188472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Most D, Ferguson L, Blednov Y, Mayfield RD, Harris RA. The synaptoneurosome transcriptome: a model for profiling the emolecular effects of alcohol. Pharmacogenomics J. 2014; 10.1038/tpj.2014.43. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hensler JG, Ladenheim EE, Lyons WE. Ethanol consumption and serotonin-1A (5-HT1A) receptor function in heterozygous BDNF (+/–) mice. J Neurochem. 2003;85: 1139–47. [DOI] [PubMed] [Google Scholar]
- 57. McGough NNH, He D-Y, Logrip ML, Jeanblanc J, Phamluong K, Luong K, et al. RACK1 and brain-derived neurotrophic factor: a homeostatic pathway that regulates alcohol addiction. Journal of Neuroscience. 2004;24: 10542–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jeanblanc J, He D-Y, Carnicella S, Kharazia V, Janak PH, Ron D. Endogenous BDNF in the dorsolateral striatum gates alcohol drinking. Journal of Neuroscience. 2009;29: 13494–502. 10.1523/JNEUROSCI.2243-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Spanagel R, Rosenwasser AM, Schumann G, Sarkar DK. Alcohol consumption and the body's biological clock. Alcohol Clin Exp Res. 2005;29: 1550–7. [DOI] [PubMed] [Google Scholar]
- 60. Mayfield J, Ferguson L, Harris RA. Neuroimmune signaling: a key component of alcohol abuse. Curr Opin Neurobiol. 2013;23: 513–20. 10.1016/j.conb.2013.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Crews FT. Immune function genes, genetics, and the neurobiology of addiction. Alcohol Res. 2012;34: 355–61. [PMC free article] [PubMed] [Google Scholar]
- 62. Crews FT, Qin L, Sheedy D, Vetreno RP, Zou J. High mobility group box 1/Toll-like receptor danger signaling increases brain neuroimmune activation in alcohol dependence. Biol Psychiatry. 2013;73: 602–12. 10.1016/j.biopsych.2012.09.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zou JY, Crews FT. Release of neuronal HMGB1 by Ethanol through decreased HDAC activity activates brain neuroimmune signaling. PLoS ONE. 2014;9: e87915 10.1371/journal.pone.0087915 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Frozen brains were placed in a plastic mold containing OCT and maintained in a mixture of powdered dry-ice and isopentane. A Microm HM550 cryostat (Thermo Scientific, Ontario, CA) was used for sectioning at a thickness of 300 μm. Micropunches were collected from amygdala (AMY; 1.25 mm; combined basolateral and central nucleus), nucleus accumbens (NAC; 1.25 mm; combined core and shell), and prefrontal cortex (PFC; 2.0 mm).
(PDF)
A two-step regression approach was used to identify clusters of differentially expressed genes with similar expression patterns across time. Each plot shows the hierarchical clustering (clusters = 4) of average expression profiles by time in the amygdala (AMY). The dots represent average expression values for each gene in the time series.
(PDF)
A two-step regression approach was used to identify clusters of differentially expressed genes with similar expression patterns across time. Each plot shows the hierarchical clustering (clusters = 4) of average expression profiles by time in the nucleus accumbens (NAC). The dots represent average expression values for each gene in the time series.
(PDF)
A two-step regression approach was used to identify clusters of differentially expressed genes with similar expression patterns across time. Each plot shows the hierarchical clustering (clusters = 4) of average expression profiles by time in the prefrontal cortex (PFC). The dots represent average expression values for each gene in the time series.
(PDF)
A two-step regression approach was used to identify clusters of differentially expressed genes with similar expression patterns across time. Each plot shows the hierarchical clustering (clusters = 4) of average expression profiles by time in the Liver. The dots represent average expression values for each gene in the time series.
(PDF)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Data Availability Statement
Microarray data have been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE60676.