

Abstract
The native environment for a membrane protein is a phospholipid bilayer. Because the protein is immobilized on NMR timescales by the interactions within a bilayer membrane, solid-state NMR methods are essential to obtain high-resolution spectra. Approaches have been developed for both unoriented and oriented samples, however, they all rest on the foundation of the most fundamental aspects solid-state NMR, and the chemical shift and homo- and hetero-nuclear dipole-dipole interactions. Solid-state NMR has advanced sufficiently to enable the structures of membrane proteins to be determined under near-native conditions in phospholipid bilayers.
Keywords: Bilayers, Magic angle spinning, Dipolar coupling, Chemical shift anisotropy, Phospholipids, Structure determination
I. Introduction
Membrane proteins and solid-state NMR is an important, topical area of research. NMR is the most powerful form of spectroscopy, and solid-state NMR is particularly so because of the retention of the anisotropic character of the nuclear spin interactions in the experimental results. One-quarter of all proteins encoded in the human genome are membrane proteins, and many of them have distinctive structures and unique biological functions. Membrane proteins are generally distinguished from soluble, globular proteins by having a relatively high percentage of hydrophobic residues, and by their structures being dominated to a large extent by either α-helices of β-sheet in the two major classes of membrane proteins. However, it is their association with phospholipid bilayers that uniquely distinguishes them from soluble proteins. This not only means that they are influenced by a distinctive, asymmetric chemical environment, but also, more importantly for NMR, that it immobilizes the polypeptides on the timescale of the chemical shift and dipolar coupling spin interactions. It is the immobilization of membrane proteins by their interactions with phospholipids that intimately links membrane proteins and solid-state NMR.
In unoriented, immobile samples, the chemical shift anisotropy and heteronuclear dipole-dipole interactions yield powder pattern resonances with frequency breadths of 104–105 Hz, which is the critical factor for characterizing the dynamics of membrane proteins and defining the spectroscopic requirements for obtaining high-resolution spectra. Significantly, in modern NMR experiments the radiofrequency irradiations have magnitudes >105 Hz, the rates of magic angle sample spinning are typically > 104 Hz, and the receiver bandwidths can be >106 Hz. All of these factors combine to make high resolution solid-state NMR feasible, and are necessary for applications to membrane proteins in phospholipid bilayers.
Historically, high-resolution solid-state NMR spectroscopy and NMR of membrane proteins were developed in parallel, starting at essentially the same time, since the instrumentation and methods of solid-state NMR suitable for single crystals and powders of organic solids were also suitable for proteins and phospholipids in membrane bilayers. The initial papers describing the application of high resolution double resonance solid-state NMR to membranes [1, 2] were published within a year of those introducing proton-enhanced nuclear induction spectroscopy. Notably, 50 years after high-resolution NMR spectroscopy of solids was first developed in the Waugh laboratory at M.I.T. [3–5]; the experiments continue to be the principal spectroscopic tools for NMR studies of membranes.
The origins of high-resolution solid-state NMR are the same as for the field of NMR spectroscopy itself. Following the original descriptions of the nuclear resonance phenomena [6, 7], Pake demonstrated that both characteristic powder patterns and distinct doublets could be observed from solid samples of water bound to gypsum [8]. Subsequent studies showed the well-defined effects of motional averaging of powder patterns, which is an essential characteristic of many materials and especially membrane proteins in phospholipid bilayers. These motional averaging effects on homonuclear dipole-dipole [9] and chemical shift anisotropy [10] powder patterns are illustrated in Figure 1. The shape and breadth of a powder pattern that is averaged by motion along a single axis reflects the angle between the principal axis of the spin interaction tensor and the axis of motion [11].
Figure 1.
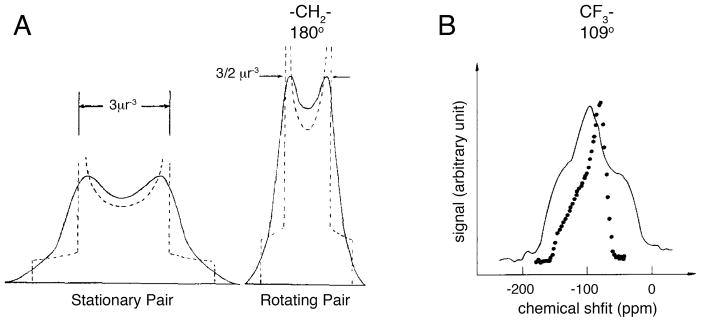
The effects of rotational motion on dipole-dipole coupling and chemical shift anisotropy powder patterns. A. Theoretical line shapes for a nuclear pair with spin ½ when stationary and when in motion about an axis perpendicular to the internuclear axis. From reference [9]. B. 19F powder spectra of silver trifluoroacetate at 107°K (.) and 40°K (-). The spectra are characteristic of rotating (107°K) and rigid (40°K) CF3 groups. From reference [10].
The Pake doublet [8] played several roles at the inception of NMR. It demonstrated experimentally the existence of the homonuclear dipole-dipole interaction between a pair of 1H nuclei. It also demonstrated that the interaction was anisotropic, since an unoriented sample gave a powder pattern and a single crystal sample gave a single doublet for each unique site that varied with the orientation of the crystal with respect to the direction of the magnetic field. In 1948 the instrumentation and experimental methods were extremely limited in their capabilities. Thus, the Pake experiment depended on isotopic dilution. The reason that resolved doublets could be observed at all was because each of the water molecules was relatively isolated from others on the surface of gypsum. In addition to the other principles, this was the advent of isotopic dilution to isolate and narrow resonances, an approach that was re-introduced for proteins by Jardetzky and coworkers in 1968 [12], and is widely used at the present time in both solution NMR and solid-state NMR studies of proteins [13].
Isotopic dilution was also the key to the demonstration of the nature of the chemical shift interaction. The characteristic chemical shift anisotropy rotation patterns were observed in calcite (CaCO3) [14]. Studies of dilute spin chemical shifts were transformed by the introduction of proton enhanced nuclear induction spectroscopy, which simultaneous increased the sensitivity and removed the severe broadening from bonded and surrounding 1H nuclei in organic and biochemical compounds. This is illustrated in Figure 2 where multiple, narrow resonances were obtained from a single crystal sample of an organic compound (durene) [15].
Figure 2.
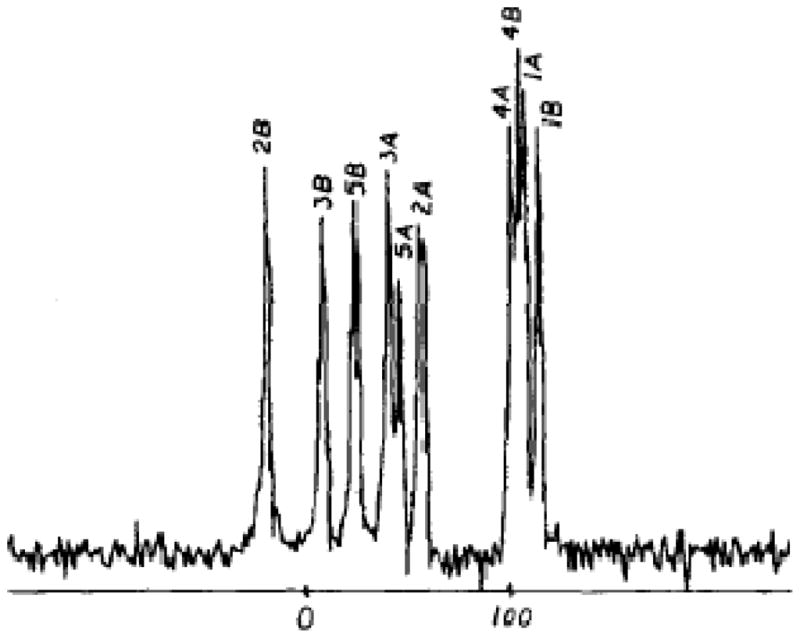
High-resolution solid-state 13C NMR spectra of a single crystal of durene. From reference [15].
The third spin interaction that is used in solid-state NMR studies of membrane proteins is the heteronuclear dipole-dipole coupling, which is also an integral part of proton-enhanced nuclear induction spectroscopy. Because the severe broadening effects of the 1H homonuclear couplings obscure the presence of the heteronuclear dipolar couplings, they had to be detected indirectly [16]. Only through experiments that are able to separate the effects of homo- and hetero-nuclear couplings is it possible to characterize them. Two of the most popular are rotational-echo double resonance NMR (REDOR) [17] and separated local field (SLF) [18, 19] spectroscopy. More sophisticated versions of experiments that effectively decouple the homonuclear interactions while enabling the heteronuclear dipolar couplings to evolve are in current use, and are a major factor in providing high resolution multidimensional spectra of membrane proteins.
II. High resolution solid-state NMR
A universal aim of NMR spectroscopy, regardless of the state of matter, is to obtain “high resolution” spectra. This means not just narrow resonance lines, as attractive as they are, but also with regard to obtaining resonances from individual sites, assigning the resonance to a particular site in the protein, and observing spectral characteristics, i.e. frequencies, line shapes, splittings, etc., that are associated with a single spin-interaction. Progress in solid-state NMR as applied to membrane proteins now fully justifies that use of the term “high resolution.” Membrane proteins with as many as 350 residues have had their spectra essentially fully resolved and assigned [20].
Recent achievements are built on the fundamentals of NMR spectroscopy as summarized above. The three basic approaches to obtaining high resolution – motional averaging, spin dilution, and spin manipulation - are used in solid-state NMR studies of membrane proteins. It is worth noting that membrane proteins require the full range of solid-state NMR methods and instrumentation, and this accounts for the relatively long development period from the first double-resonance spectra of a liposome sample to the complete structures of membrane proteins that are now being determined [21].
There are multiple parts to the definition of high resolution in NMR, and one of the most important is that each resonance is associated with a single chemical site. It is essential to know which site this is. It is equally important that one spin interaction at a time is analyzed. The separation can occur by removal of interfering interactions, such as with 1H-13C heteronuclear decoupling. Another approach is through the use of multidimensional NMR, and the spectral separations and simplifications that result.
In general, magic angle spinning NMR [22, 23] yields high resolution spectra of unoriented “powder” solids when applied in combination with double-resonance methods [24]. To a first approximation, these spectra are equivalent to solution NMR spectra with a single line resonance for each 1H decoupled 13C site. There are two notable exceptions between direct comparisons of MAS spectra and solution NMR spectra. One is that when motions with frequencies near the spinning frequency are present, for example in the hop motions of amino acid sidechains, then broad or unusual lineshapes can be observed [25]. The second major source occurs at relatively low magnetic fields and that is that the coupling between 13C and 14N is not fully averaged by the spinning, yield asymmetric doublets for the 13C resonances bonded to nitrogens [26]. The substantial nuclear quadrupole interaction of the 14N tilts the magnetization away from the z-axis, interfering with the averaging performed by the magic angle spinning.
The combination of cross polarization, 1H decoupling and magic angle spinning is an extremely powerful approach to solid-state NMR [24] and is in wide use in studies of membrane proteins in phospholipid bilayers. The cost is the loss of structural information inherent in the spin interactions because so much effort is put into averaging (removing) this information from the spectra in order to achieve narrow well-resolved signals. Nonetheless, some experimental measurements are possible in the simplest magic angle spinning regime because the isotropic chemical shift frequencies, and in some cases lineshapes, are available for analysis.
For homonuclear couplings, dilute spin exchange [27, 28] is one of the most flexible methods. Variations of it are in wide use at the present time. Another technique that requires isotopic arrangement of the sample is homonuclear dipolar recoupling between appropriately arranged labeled sites [29]. In the sense of measuring distances between two labeled sites, there are strong similarities between dipolar recoupling [30] and REDOR [17] experiments. However, they both require spin dilution in order to yield quantitative measurements. Before the advent of high-speed magic angle spinning and triple-resonance methods, it was necessary to develop an alternative approach to obtain multiple measurements for structure determination. And this involved a fourth factor, the manipulation of the sample so that it was not a powder but rather was uniaxially oriented, which could be done in a number of ways.
III. Oriented sample solid-state NMR
Besides magic angle sample spinning, there is a second approach to obtaining high-resolution solid-state NMR spectra of proteins with single-line resonances, and that is the use of oriented samples. The archetypical example of an oriented sample, of course, is a single crystal, which gives single lines for each unique site in the unit cell. A solid-state NMR spectrum of a single crystal sample of an organic molecule illustrates this in Figure 2 [15].
It is also the case that a single line resonance results from uniaxial alignment of a polymeric sample parallel to the direction of the magnetic field. This was originally demonstrated with spectra of mechanically aligned polyethylene [31]. There is only a single type of chemical group in polyethylene, thus only one signal, as shown in Figure 4. Its importance lies in its simplicity, which establishes the basic spectroscopic characteristics of oriented sample solid-state NMR.
Figure 4.
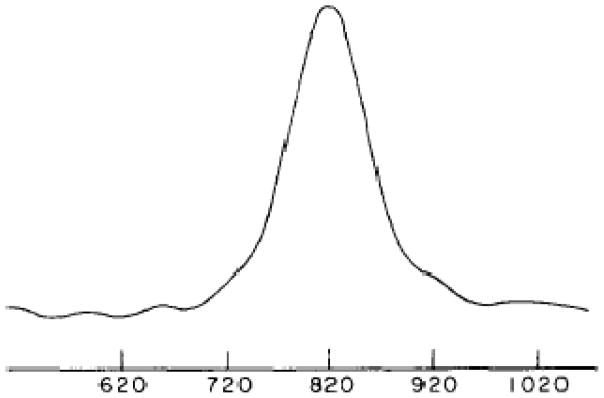
Solid-state NMR spectrum of an oriented sample of polyethylene with its long axis parallel to the direction of the magnetic field. From reference [31].
The same oriented sample method outlined above for a synthetic polymer can be applied to membrane proteins. In oriented sample (OS) solid-state NMR of stationary, aligned samples of membrane proteins there are two basic approaches to sample alignment. One is mechanical alignment between glass plates [32–34] and the other is magnetic alignment using bicelles [35, 36], which are mixtures of long chain lipids that form the planar bilayer for the protein, with short chain lipids or detergents to ‘cap’ the ends of the bilayer region. It is also possible to prepare magnetically-alignable ‘detergent-free’ bicelles through the use of an amphipathic peptide instead of short chain lipids, which are referred to as macrodiscs [37].
Very highly aligned samples of membrane proteins can be prepared with magnetic alignment in bicelles [38]. This is shown in Figure 5 for membrane proteins with a range of sizes, as illustrated with cartoons of the proteins on the top of the Figure. Figure 5D, E, and F are 31P NMR spectra of the phospholipids that form the bilayers in the samples. The 31P resonances are narrow, demonstrating that the bilayers are very highly aligned by the magnetic field. The spectra in Figure 5A, B, and C are of the uniformly 15N labeled samples of proteins embedded in the aligned bilayers. As can be seen for the smallest protein in Figure 5A, the resonance line widths are quite narrow.
Figure 5.

(top) Schematic drawings of three membrane proteins aligned in planar “long-chain lipid”: Triton X-100 (q=5) bilayers. A.–C. One-dimensional solid-state 15N NMR spectra of uniformly 15N-labeled proteins. D.–F. One-dimensional solid-state 31P NMR spectra of the phospholipids in the same samples. A, D. The membrane-bound form of the 46-residue Pf1 coat protein. B, E. The 78-residue mercury transport protein MerE. C, F. The 350-residue G-protein-coupled receptor CXCR1. From reference [38].
The resonances in the larger proteins are also very narrow, but overlap due to the total number of similar, helical residues in the proteins.
Aligned samples give chemical shifts that directly contain angular information. This opened up the field when only single labeled samples were available. It remains useful where simple information about the alignment of helices is of value. It is very easy to tell the difference between a helix that is in the plane of the bilayer and one that is trans-membrane. This is now in wide use because of the interest in structure and orientation of antibiotic peptides [39, 40] [41].
IV. Separated local field spectroscopy
In the early days of high resolution solid-state NMR, the rotation patterns of the chemical shift lines were used primarily to determine chemical shift tensors [42]. In order to study more complex organic and biochemical molecules it was necessary to supplement the chemical shift frequencies. After the chemical shift, the next most useful interaction and associated measurements come from the heteronuclear dipolar interactions. The spectroscopic separation of effects from the chemical shift and heteronuclear dipolar interactions are manifested in several different ways in solid-state NMR experiments. The one-dimensional spectrum at the top of Figure 6 is of a single crystal of calcium formate, illustrating the benefits of sample orientation in giving high resolution in the chemical shift dimension, as do the spectra in Figures 2 and 4. Figure 6 also shows the first experimental two-dimensional separated local field spectrum [18]. Each signal associated with a 13C bonded to a single 1H is a doublet of the same fundamental origin as a Pake doublet [8]. In this case the local fields of the various carbon sites are separated from each other by the anisotropic chemical shift frequencies of the carbons. The use of multiple pulse homonuclear decoupling separates the heteronuclear dipolar couplings from the complications of the multiple couplings throughout the large network of 1H spins in the sample [19].
Figure 6.

Pure chemical shift spectrum (top) and SLF spectrum (bottom) for 13C in a single crystal of calcium formate. From reference [18].
The chemical shift dimension in separated local field spectra has intrinsically high resolution because continuous on resonance or modulated 1H irradiation decouples the heteronuclear dipolar interactions during the interval when the data are acquired. However, even with the use of homonuclear multiple pulse decoupling the doublets (or multiplets) in the dipolar frequency dimension are generally quite broad due to the rapid decay of the abundant 1H signals from incomplete decoupling of the homonuclear dipole-dipole interactions, short intrinsic T2 and cumulative effects of longer-range heteronuclear dipole-dipole interactions. Regardless, the two-dimensional separated local field spectrum in Figure 6 illustrates the ability of this approach to associate a single dipolar coupling with each resolved dilute spin resonance.
Polarization inversion spin-exchange at the magic angle (PISEMA) [43] is a high resolution version of separated local field spectroscopy. By integrating polarization inversion with phase and frequency alternated Lee-Goldburg homonuclear decoupling on the 1H nuclei following cross-polarization to the 13C or 15N nuclei during the t1 interval, very high resolution two-dimensional chemical shift/heteronuclear dipolar spectra of solid samples can be obtained. Line widths in the dipolar dimension are reduced by more than an order of magnitude compared to conventional SLF spectra obtained without homonuclear decoupling during the t1 interval. The combination of narrow lines and favorable scaling factor has a dramatic effect on the resolution. Indeed, it is now feasible to formulate solid-state NMR experiments where heteronuclear dipolar coupling frequencies provide a mechanism for resolution among similar chemical sites. Even more interestingly, dipolar coupling and chemical shift frequencies can be used in a number of complementary ways to enhance resolution in reduced or maximum dimensional experiments, and to provide qualitative and quantitative indices of molecular structure. The spectra enable dipolar couplings to complement chemical shifts as a mechanism for spectroscopic resolution, as well as provide measurements of orientationally dependent frequencies for individual molecular sites.
The two-dimensional spectrum in Figure 7 is from the same sample used to obtain the one-dimensional spectra in Figure 5A and D. It displays excellent resolution in both the chemical shift and heteronuclear dipolar coupling dimensions, and, as marked, all of the resonances have been assigned to specific sites in the protein. The most noticeable feature is the “wheel-like” pattern of resonances, which comes from the trans-membrane helix. This is because of the regularity of the helix [44, 45]. PISA (polarity index slant angle) wheels are extremely useful in the analysis of spectra of helical proteins
Figure 7.

Two-dimensional SLF spectrum of uniformly 15N-labeled Pf1 coat protein in DMPC:Triton X-100 bilayers obtained at 700 MHz. From reference [38].
In the meantime the technology of magic angle spinning was advancing. The most obvious need was for high speed spinning. This advanced incrementally and was especially impactful when it was possible to routinely possible to spin fast enough to average out 13C-13C dipolar interactions. This enabled the use of uniformly 13C labeled samples [46]. The other major advance was in the ability to recouple interactions averaged out by the high-speed magic angle spinning. These include the heteronuclear dipole-dipole coupling [47] and the chemical shift anisotropy [48]. These are very powerful experiments, and are incorporated into many of the multidimensional experiments used to acquire the frequencies that serve as constraints for structure determination.
The ability to acquire powder patterns associated with isotropic chemical shifts lies at the heart of rotationally aligned (RA) solid-state NMR. This makes it possible to observe the effects of rotational diffusion on the powder patterns, which yields the angle between the spin interaction tensor and the direction of rotation. In membrane proteins this direction is the bilayer normal, so there is an equivalence between the angles measured with aligned sample in the form of single lines and the parallel edge of the motionally average powder pattern [49].
V. Rotationally aligned solid-state NMR
Rotationally aligned solid-state NMR is specifically tailored for the unique properties of membrane proteins in liquid crystalline phospholipid bilayers. It combines features of magic angle spinning and oriented-sample solid-state NMR to resolve and assign resonances associated with each amino acid residue, measure site-specific orientation restraints relative to the bilayer, and calculate the three-dimensional structure of the protein and its orientation within the membrane bilayer. RA solid-state NMR differs from previously used OS solid-state NMR methods in that it relies on the inherent rotational diffusion of membrane proteins in phospholipid bilayers [50, 51] to provide orientation-dependent motional averaging of heteronuclear dipolar coupling and chemical shift anisotropy powder patterns relative to the bilayer normal, rather than the orientation-dependent frequencies of single-line resonances observed in OS NMR of stationary, uniaxially aligned samples. It differs from more standard MAS solid-state NMR approaches that typically rely on short-range distance and angle measurements within the polypeptide chain rather than to an externally defined axis, in this case the bilayer normal.
Phospholipids self-assemble to form extended bilayer membranes in liposomes. The polar head groups are exposed to water and the hydrophobic hydrocarbon chains face the membrane interior of the bilayer. On the molecular scale, these bilayers are infinitely long in two dimensions, but only two molecules thick in the third dimension. This is illustrated in Figure 8 by the representation of Singer and Nicholson’s fluid mosaic model of a biological membrane [52]. Both the phospholipids and the proteins undergo fast rotational diffusion about the bilayer normal, as indicated by the arrows, as well as translational diffusion in the plane of the bilayer.
Figure 8.

Schematic three-dimensional and cross-sectional views of the “Fluid mosaic model” of globular membrane proteins that are completely or partially embedded within a lipid matrix. Modified from reference [52].
The rotational diffusion of membrane proteins in phospholipid bilayers, illustrated in Figure 8, was shown originally to occur by optical methods [50, 51]. Notably, the same phenomenon was observed in the averaging of lineshapes by NMR. McLaughlin and coworkers in 1975 [53] used 31P NMR of the phospholipids to show that they undergo fast rotational motion by two pertinent spectroscopic effects. One was the averaging of the powder pattern line shape and the other was the observation of single line spectra when the lipid bilayers were mechanically aligned on glass plates over a wide range of angles (not just with the normal parallel to the field). Ten years later, in a similar, complementary experiment Griffin and coworkers [54] demonstrated that the protein bacteriorhodopsin undergoes rotational diffusion in phosphatidyl choline bilayers because of the averaging of the 13C′ powder pattern. Contemporaneously, Cornell and coworkers [34, 55] showed that 13C′ labeled gramicidin undergoes the same type of motional averaging in phospholipid bilayers. In a highly definitive experiment [56], Cross and coworkers utilized gramicidin labeled at a single site to show with a two-dimensional SLF experiment that the motionally averaged chemical shift and dipolar coupling principal elements were collinear, while in a static sample they differ in alignment by about 17°. In an experiment very similar to the original McLaughlin and coworkers experiment, we were able to show single line resonances from tilted coil samples over a wide range of angles, again something that could only be observed in the presence of rotational diffusion [57]. The spectral manifestations of rotational averaging provide the experimental foundation for RA solid-state NMR.
In the structure determination of membrane proteins by RA solid-state NMR, it is essential to verify that the protein undergoes rapid rotational diffusion about the bilayer normal and that this rotation can be switched between slow and fast limit by changing the temperature. As shown in Figure 9, it is particularly convenient to monitor the effect of temperature on the 13C′ CSA powder pattern of a uniformly 13C/15N labeled membrane protein in proteoliposomes [54]. This powder pattern line shape is effective in demonstrating that a helical membrane protein is undergoing fast rotational diffusion, since the static powder pattern is highly asymmetric with a large frequency span. For transmembrane helices a large fraction of the backbone carbonyl bonds are approximately parallel to the bilayer normal (i.e., the axis of motional averaging); thus, when the protein undergoes fast rotational diffusion, the 13C′ powder pattern becomes axially symmetric and is significantly narrowed. This is shown in Figure 9 with experimental data from a helical membrane protein in DMPC bilayers [58]. A useful way to experimentally monitor the protein rotational diffusion is with slow (5 kHz) MAS as shown in Figure 9E (fast rotational diffusion of the protein) and Figure 9F (slow rotational diffusion of the protein). There is a family of sidebands when the 13C′ CSA has its full static breadth; in contrast, the motionally averaged CSA has such a small span that there are no observable spinning sidebands, even at the 5 kHz rate.
Figure 9.
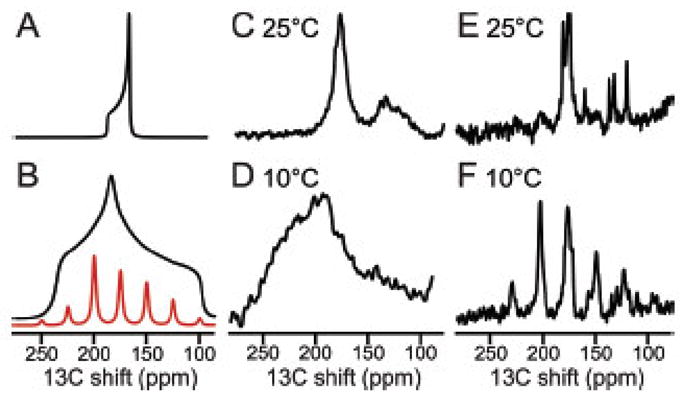
13C solid-state NMR spectra of uniformly 13C/15N labeled MerFt in DMPC proteoliposomes. The majority of resonance intensity centered near 175 ppm is from 13C′ backbone sites. A. Spectrum simulated for a single 13C′ group in a transmembrane helix undergoing rotational diffusion around the lipid bilayer normal. B. As in Panel A except for a static 13C′ group in a peptide bond. The family of sidebands in panel B (red) would be observed under slow (5 kHz) MAS. C. and D. Experimental spectra obtained for a stationary sample when the protein undergoes fast rotational diffusion about the phospholipid bilayer normal C. or where the protein is immobile on the time scale of the static 13C′ CSA powder pattern D. E. and F. Experimental spectra obtained from a sample undergoing slow (5 kHz) MAS where the 13C′ CSA powder pattern is motionally averaged E, or at where a family of sidebands spanning the width of the static 13C′ CSA powder pattern F is observed in the absence of protein rotational diffusion. Comparisons of the powder pattern frequency breadth (A vs. B; C vs. D) or the presence of spinning sidebands (E vs. F) are diagnostic for the presence of fast rotational diffusion of the protein under the experimental conditions used to measure the CSA and DC powder patterns. From reference [58].
The full two-dimensional 1H-15N dipolar coupling/13C chemical shift SLF spectrum shown in Figure 10A contains resonances from all of the protein’s 13Cα sites. Although there is considerable spectral overlap, an individual resonance assigned to Leu 31 can be identified, as marked in Figure 10A. The rotationally averaged 1H-15N dipolar coupling and 15N CSA powder patterns associated with each isotropic resonance are characterized with three-dimensional MAS solid-state NMR experiments [58]. Figure 10B and C contains two-dimensional 1H-15N dipolar coupling/13C chemical shift planes selected from a three-dimensional spectrum at the 15N shift frequency corresponding to Leu 31. 1H-13C heteronuclear dipolar couplings were measured with similar three-dimensional experiments (Figure 10C). Since we previously verified [49] that the measurement of orientation-dependent frequencies from the parallel edges of the recoupled, motionally averaged, axially symmetric powder patterns [8] are identical to those measured from the single line resonances observed in aligned, stationary samples, the frequencies measured from the spectra in Figure 10 can be used to determine the orientation of the peptide plane for Leu 31 and, in general, with sufficient data from other residues, to calculate the three-dimensional structure of the protein.
Figure 10.
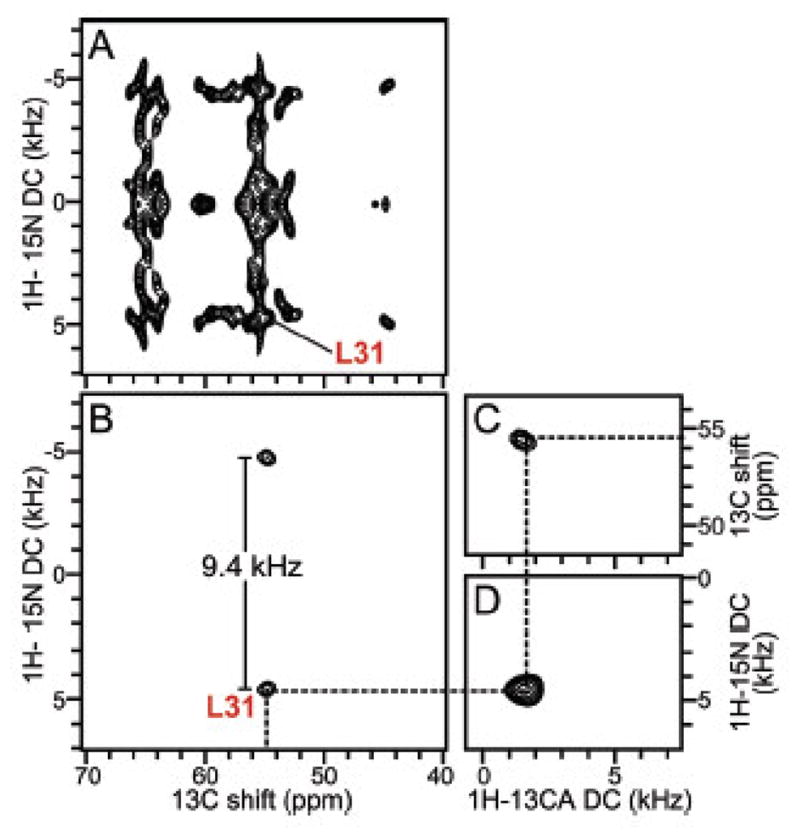
Examples of spectroscopic data for residue L31 obtained from MAS solid-state NMR spectra of uniformly 13C/15N labeled MerFt in DMPC proteoliposomes at 25°C: A. two-dimensional 1H-15N dipolar coupling/13C shift SLF spectrum, B. two-dimensional 1H-15N dipolar coupling/13C shift SLF spectral plane selected from a three-dimensional spectrum at an isotropic 15N chemical shift frequency of 118.6 ppm. C. Two-dimensional 1H-13CA dipolar coupling/13C shift SLF spectral plane selected from a three-dimensional spectrum at an isotropic 15N chemical shift frequency of 118.6 ppm. D. Two-dimensional 1H-13CA dipolar coupling/1H-15N dipolar coupling SLF spectral plane selected from a three-dimensional spectrum at an isotropic 13C chemical shift frequency of 54.6 ppm. All three spectral planes are associated with residue L31. The dashed line traces the correlations among the frequencies, which were obtained from three separate experiments. The dipolar coupling frequencies in the spectra correspond to the perpendicular edge frequencies of the corresponding powder patterns. Panel B shows that the 1H-15N dipolar coupling motionally averaged powder pattern for L31 has a perpendicular edge frequency of 4.7 kHz, corresponding to a splitting of 9.4 kHz, and a dipolar coupling value of 18.8 kHz. From reference [58].
The frequencies measured from the spectra in Figure 10 are interpreted graphically in Figure 11. The experimentally measured, motionally averaged 1H-13C and 1H-15N heteronuclear dipolar couplings are compared to calculated static powder patterns on the right side of the Figure. On the left side, the resulting angular vectors derived from the experimental data are superimposed on the peptide plane of Leu 31.
Figure 11.

Orientation of the peptide plane associated with residue L31 based on the experimental data shown in Figure 10. From reference [85].
The 1H-15N and 1H-13Cα dipolar coupling restraints and 15N chemical shift anisotropy restraints are sufficient to determine the orientation of the associated peptide plane relative to the axis of alignment and yield the three-dimensional structure of the protein.
VI. Example: MerF of the bacterial mercury detoxification system
Bacteria that survive in mercury-polluted environments contain an operon whose proteins constitute a mercury detoxification system [59] that functions by importing highly toxic Hg(II) ions into the cytoplasm of the bacterial cell and enzymatically transforming it to its less toxic and volatile form Hg(0), which is passively eliminated. Imported Hg(II) binds to the periplasmic protein MerP, which delivers it to a membrane protein transporter such as MerF. The membrane protein is responsible for transporting Hg(II) across the cell membrane and delivering it to MerA, the cytoplasmic mercuric reductase, a multi-domain enzyme that reduces Hg(II) to Hg(0). Understanding the molecular mechanism of mercury detoxification is important for both environmental and biomedical applications of the components of the bacterial mercury detoxification system. Atomic resolution structures of MerP and MerA have been determined [60, 61]. Neither MerF nor any other mercury transport membrane proteins, e.g. MerT, MerC, from various isolates of bacterial mercury detoxification systems have been crystallized. Therefore, at present, NMR is the only viable approach to the structure determination of this key component of the detoxification system. As shown above, solid-state NMR has now been developed sufficiently to determine the structure a mercury transport membrane protein in phospholipid bilayers.
MerF [62], with 81 residues and two transmembrane helices connected by a short, structured interhelical loop, is the smallest of the mercury transport membrane proteins. Notably, a pair of vicinal Cys residues that bind Hg(II) are associated with each transmembrane helix. Both Cys pairs are located on the cytoplasmic side of the protein, one pair in the middle of the N-terminal helix and the other near the C-terminal end of the second helix. They are in relatively close spatial proximity and one can envision a mechanism whereby the Hg(II) is exchanged between the pairs.
The full two-dimensional 1H-15N dipolar coupling/13C chemical shift SLF spectrum shown in Figure 10 contains resonances from all the 13C sites from a truncated form of MerF [58], which is missing its N- and C-terminal regions that are mobile and unstructured in micelle preparations [63], Although there is considerable spectral overlap, an individual resonance assigned to Leu 31 can be identified, as marked in Figure 10A. The rotationally averaged 1H-15N dipolar coupling and 15N CSA powder patterns associated with each isotropic resonance are characterized with three-dimensional MAS solid-state NMR experiments. Since we previously verified that the measurement of orientation-dependent frequencies from the parallel edges of the recoupled, motionally averaged, axially symmetric powder patterns are identical to those measured from the single line resonances observed in aligned, stationary samples [49], the frequencies measured from the spectra in Figure 10 can be used to determine the orientation of the peptide plane for Leu 31 and, in general, with sufficient data from other residues, to calculate the three-dimensional structure of the protein.
Full length MerF has two hydrophobic transmembrane helices and a relatively short, structured inter-helical loop. Four residues at the N-terminus and eleven residues at the C-terminus are mobile and unstructured in phospholipid bilayers. The three-dimensional structure [64] shows that the two pairs of Cys residues involved in binding Hg(II) are in close proximity on the cytoplasmic side of the phospholipid bilayer. The details of the transport mechanism of Hg(II) from MerP to MerA are still unknown. However, the finding of the pairs of Cys residues of MerF is an important step towards synthesizing a functional model for metal ion transfer in the bacterial mercury detoxification system.
VII. Enhancements of solid-state NMR experiments
Solid-state NMR and membrane proteins is a rapidly advancing field. It benefits from improvements in many areas, in particular ultra fast magic angle spinning [65, 66] and dynamic nuclear polarization [67–69], the addition of paramagnetic reagents to alter relaxation properties for accelerating signal averaging [70] and for distance measurements [71–73]. Virtually all of the experiments work better at higher magnetic fields. Therefore, as ultra high magnetic fields become available all of the experimental measurements improve and higher sensitivity is obtained.
Much of the advancement is a direct outgrowth of the development of solid-state NMR spectroscopy as applied to proteins [46] [74–76]. Examples of applications to membrane proteins include the drug transporter EmrE [77], the calcium ATPase SERCA [78], cytochrome P 450 oxidoreductase [79], the M2 protein of Influenza virus [80, 81], and G-protein coupled receptors (GPCRs) [20, 82–84].
Importantly, initial studies of side chains and of complexes have been reported. This indicates that very high resolution and biologically relevant studies will be feasible in the near future. The ability to determine the structures of unmodified membrane proteins in liquid crystalline phospholipid bilayers under physiological conditions of temperature and pH is an enormous advantage over competitive methods. The field of NMR studies of membrane proteins in their native environment is poised for rapid advances.
Figure 3.
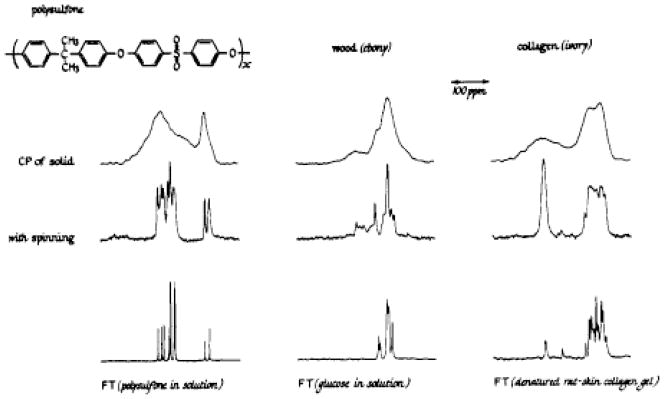
Dipolar-decoupled natural abundance 13C NMR spectra of some solids obtained using single Hartmann-Hahn cross-polarization contacts of 1 ms duration. The cross-polarization spectra, obtained both with and without magic-angle spinning, are compared to some standard Fourier transform 13C NMR spectra of various materials in solution. Each spectrum is 8 kHz wide (at 22.6 MHz). The magnetic field increases from left to right. From reference [24].
Figure 12.

The three-dimensional structure of MerF is shown as a ribbon diagram in aqua. Both termini of the protein are in the cytosol. The structure was calculated using the data from reference [64].
Highlights.
The development of solid-state NMR of proteins is summarized.
Solid-state NMR is essential for membrane proteins in phospholipid bilayers.
It is possible to determine structures with magic angle spinning approaches.
It is possible to determine structures with aligned sample approaches.
Acknowledgments
The research was supported by grant P41EB002031, R01EB005161, R01GM099986, R01GM066978, and P01AI074805 from the National Institutes of Health. It utilized the Biotechnology Resource Center for NMR Molecular Imaging of Proteins at the University of California, San Diego.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Urbina J, Waugh JS. Application of proton-enhanced nuclear induction spectroscopy to the study of membranes. Annals of the New York Academy of Sciences. 1973;222:733–739. doi: 10.1111/j.1749-6632.1973.tb15300.x. [DOI] [PubMed] [Google Scholar]
- 2.Urbina J, Waugh JS. Proton-enhanced 13C nuclear magnetic resonance of lipids and biomembranes. Proceedings of the National Academy of Sciences of the United States of America. 1974;71:5062–5067. doi: 10.1073/pnas.71.12.5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pines A, Gibby MG, Waugh JS. Proton-Enhanced Nuclear Induction Spectroscopy. A Method for High Resolution NMR of Dilute Spins in Solids. J Chem Phys. 1972;56:1776–1777. [Google Scholar]
- 4.Pines A, Gibby MG, Waugh JS. Proton-enhanced NMR of dilute spins in solids. J Chem Phys. 1973;59:569–590. [Google Scholar]
- 5.Waugh JS, Huber LM, Haeberlen U. Approach to high-resolution NMR in solids. Phys Rev Lett. 1968;20:180–182. [Google Scholar]
- 6.Bloch F, Hansen WW, Packard M. Nuclear Induction. Phys Rev. 1946;69:127. [Google Scholar]
- 7.Purcell EM, Torrey HC, Pound RV. Resonance absorption by nuclear magnetic moments in a solid. Phys Rev Lett. 1946:37–38. [Google Scholar]
- 8.Pake GE. Nuclear Resonance Absorption in Hydrated Crystals: Fine Structure of the Proton Line. J Chem Phys. 1948;16:327–336. [Google Scholar]
- 9.Gutowsky HS, Pake GE. Structural investigations by means of nuclear magnetism. II. Hindered rotation in solids. J Chem Phys. 1950;18:162–170. [Google Scholar]
- 10.Mehring M, Griffin RG, Waugh JS. 19F Shielding tensors from coherently narrowed NMR powder spectra. J Chem Phys. 1971;55:746–755. [Google Scholar]
- 11.Opella SJ. Protein dynamics by solid state nuclear magnetic resonance. Methods in Enzymology. 1985;131:327–361. doi: 10.1016/0076-6879(86)31048-6. [DOI] [PubMed] [Google Scholar]
- 12.Markley JL, Putter I, Jardetzky O. High-resolution nuclear magnetic resonance spectra of selectively deuterated staphylococcal nuclease. Science. 1968;161:1249–1251. doi: 10.1126/science.161.3847.1249. [DOI] [PubMed] [Google Scholar]
- 13.Asami S, Reif B. Proton-detected solid-state NMR spectroscopy at aliphatic sites: applicatin to crystalline systems. Acc Chem Res. 2013;46:2089–2097. doi: 10.1021/ar400063y. [DOI] [PubMed] [Google Scholar]
- 14.Lauterbur P. Anisotropy of the C13 chemical shift in calcite. Phys Rev Lett. 1958;1:343–344. [Google Scholar]
- 15.Pausak S, Pines A, Waugh JS. Carbon-13 chemical shielding tensors in single-crystal durene. J Chem Phys. 1973;59:591–595. [Google Scholar]
- 16.Kaplan DE, Hahn EL. Experiences de double irradiation en resonance magnetique par la methode d’impulsions. Journal de Physique et le Radium. 1958;19:821–825. [Google Scholar]
- 17.Gullion T, Schaefer J. Rotational-echo double-resonance NMR. J Magn Reson. 1989;81:196–200. doi: 10.1016/j.jmr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 18.Hester RK, Ackerman JL, Neff BL, Waugh JS. Separated local field spectra in NMR: determination of structure of solids. Phys Rev Lett. 1976;36:1081–1083. [Google Scholar]
- 19.Waugh JS. Uncoupling of local field spectra in nuclear magnetic resonance: determination of atomic positions in solids. Proceedings of the National Academy of Sciences of the United States of America. 1976;73:1394–1397. doi: 10.1073/pnas.73.5.1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park SH, Das BB, Casagrande F, Tian Y, Nothnagel HJ, Chu M, Kiefer H, Maier K, De Angelis AA, Marassi FM, Opella SJ. Structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature. 2012;491:779–783. doi: 10.1038/nature11580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Radoicic J, Lu GJ, Opella SJ. NMR structures of membrane proteins in phospholipid bilayers. Q Rev Biophys. 2014;47:249–283. doi: 10.1017/S0033583514000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andrew ER, Bradbury A, Eades RG. Nuclear magnet resonance spectra from a crystal rotated at high speed. Nature. 1958;182:1659. [Google Scholar]
- 23.Lowe IJ. Free induction decays of rotating solids. Phys Rev Lett. 1959;2:285–287. [Google Scholar]
- 24.Schaefer J, Stejskal EO. C-13 Nuclear Magnetic Resonance of Polymers Spinning At the Magic Angle. J Am Chem Soc. 1976;98:1031–1032. [Google Scholar]
- 25.Frey MH, DiVerdi JA, Opella SJ. Dynamics of phenylalanine in the solid state by NMR. J Am Chem Soc. 1985;107:7311–7315. [Google Scholar]
- 26.Hexem JG, Frey MH, Opella SJ. Influence of 14N on 13C NMR spectra of solids. J Am Chem Soc. 1981;103:224–226. [Google Scholar]
- 27.Szeverenyi NM, Sullivan MJ, Maciel GE. Observation of spin exchange by two-dimensional Fourier transform 13C cross-polarization-magic angle spinning. J Magn Reson. 1982;47:462–475. [Google Scholar]
- 28.Frey MH, Opella SJ. 13C spin exchange in amino acids and peptides. J Am Chem Soc. 1984;106:4942–4945. [Google Scholar]
- 29.Griffin RG. Dipolar recoupling in MAS spectra of biological solids. Nature Structural Biology. 1998;5(Suppl):508–512. doi: 10.1038/749. [DOI] [PubMed] [Google Scholar]
- 30.Jaroniec CP, Tounge BA, Rienstra CM, Herzfeld J, Griffin RG. Recoupling of heteronuclear dipolar interactions with rotational-echo double-resonance at high magic-angle spinning frequencies. J Magn Reson. 2000;146:132–139. doi: 10.1006/jmre.2000.2128. [DOI] [PubMed] [Google Scholar]
- 31.Opella SJ, Waugh JS. Two-dimensional 13C NMR of highly oriented polyethylene. J Chem Phys. 1977;66:4919–4924. [Google Scholar]
- 32.Nicholson LK, Moll F, Mixon TE, LoGrasso PV, Lay JC, Cross TA. Solid-state 15N NMR of oriented lipid bilayer bound gramicidin A′. Biochemistry. 1987;26:6621–6626. doi: 10.1021/bi00395a009. [DOI] [PubMed] [Google Scholar]
- 33.Bechinger B, Kim Y, Chirlian LE, Gesell J, Neumann JM, Montal M, Tomich J, Zasloff M, Opella SJ. Orientations of amphipathic helical peptides in membrane bilayers determined by solid-state NMR spectroscopy. J Biomol NMR. 1991;1:167–173. doi: 10.1007/BF01877228. [DOI] [PubMed] [Google Scholar]
- 34.Cornell BA, Separovic F, Smith R, Baldassi AJ. Conformation and orientation of gramicidin A in oriented phospholipid bilayers measured by solid state carbon-13 NMR. Biophys J. 1988;53:67–76. doi: 10.1016/S0006-3495(88)83066-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanders CR, Hare BJ, Howard KP, Prestegard JH. Magnetically-oriented phospholipid micelles as a tool for the study of membrane-associated molecules. Progress in Nuclear Magnetic Resonance Spectroscopy. 1994;26:421–444. [Google Scholar]
- 36.Howard KP, Opella SJ. High-resolution solid-state NMR spectra of integral membrane proteins reconstituted into magnetically oriented phospholipid bilayers. J Magn Reson B. 1996;112:91–94. doi: 10.1006/jmrb.1996.0116. [DOI] [PubMed] [Google Scholar]
- 37.Park SH, Berkamp S, Cook G, Chan MK, Viadiu H, Opella SJ. Nanodiscs vs. Macrodiscs for NMR of membrane proteins. Biochemistry. 2011;50:8983–8985. doi: 10.1021/bi201289c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park SH, Opella SJ. Triton X-100 as the “short-chain lipid” improves the magnetic alignment and stability of membrane proteins in phosphatidylcholine bilayers for oriented-sample solid-state NMR spectroscopy. J Am Chem Soc. 2010;132:12552–12553. doi: 10.1021/ja1055565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bechinger B, Zasloff M, Opella SJ. Structure and interactions of magainin antibiotic peptides in lipid bilayers: a solid-state nuclear magnetic resonance investigation. Biophys J. 1992;62:12–14. doi: 10.1016/S0006-3495(92)81763-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Strandberg E, Zerweck J, Wadhwani P, Ulrich A. Synergistic insertion of antimicrobial magainin-family peptides in membranes depends on the lipid spontaneous curvature. Biophys J. 2013;104:L09–L11. doi: 10.1016/j.bpj.2013.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perrin B, Tian Y, Fu R, Grant C, Chekmenev E, Wieczorek W, Dao A, Hayden R, Burzynski C, Benable R, Sharma M, Opella SJ, Pastor R, Cotten M. High-resolution structures and orientations of antimicrobial peptides piscidin 1 and piscidin 3 in fluid bilayers reveal tilting, kinking, and bilayer immersion. J Am Chem Soc. 2014;136:3491–3504. doi: 10.1021/ja411119m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Griffin RG, Pines A, Pausak S, Waugh JS. 13C chemical shielding in oxalic acid, oxalic acid dihydrate, and diammonium oxalate. J Chem Phys. 1975;63:1267–1271. [Google Scholar]
- 43.Wu CH, Ramamoorthy A, Opella SJ. High-resolution heteronuclear dipolar solid-state NMR spectroscopy. J Magn Reson A. 1994;109:270–272. [Google Scholar]
- 44.Marassi FM, Opella SJ. A solid-state NMR index of helical membrane protein structure and topology. J Magn Reson. 2000;144:150–155. doi: 10.1006/jmre.2000.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang J, Denny J, Tian C, Kim S, Mo Y, Kovacs F, Song Z, Nishimura K, Gan Z, Fu R, Quine JR, Cross TA. Imaging membrane protein helical wheels. J Magn Reson. 2000;144:162–167. doi: 10.1006/jmre.2000.2037. [DOI] [PubMed] [Google Scholar]
- 46.McDermott A. Structural and dynamic studies of proteins by solid-state NMR spectroscopy: rapid movement forward. Curr Opin Struct Biol. 2004;14:554–561. doi: 10.1016/j.sbi.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 47.Zhao X, Eden M, Levitt MH. Recoupling of heteronuclear dipolar interactions in solid-state NMR using symmetry-based pulse sequences. Chem Phys Lett. 2001;342:353–361. [Google Scholar]
- 48.Chan JCC, Tycko R. Recoupling of chemical shift anisotropies in solid-state NMR under high-speed magic-angle spinning and in uniformly 13C-labeled systems. J Chem Phys. 2003;118:8378–8389. [Google Scholar]
- 49.Park S, Das BB, DeAngelis AA, Scrima M, Opella SJ. Mechanically, magnetically, and ‘rotationally aligned’ membrane proteins in phospholipid bilayers give equivalent angular constraints for NMR structure determination. J Chem Phys B. 2010;114:13995–13003. doi: 10.1021/jp106043w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cone RA. Rotational diffusion of rhodopsin in the visual receptor membrane. Nature New Biology. 1972;236:39–43. doi: 10.1038/newbio236039a0. [DOI] [PubMed] [Google Scholar]
- 51.Cherry RJ. Measurement of protein rotational diffusion in membranes by flash photolysis. Methods Enzymol. 1978;54:47–61. doi: 10.1016/s0076-6879(78)54007-x. [DOI] [PubMed] [Google Scholar]
- 52.Singer SJ, Nicholson GL. The fluid mosaic model of the structure of cell membranes. Science. 1972;175:720–731. doi: 10.1126/science.175.4023.720. [DOI] [PubMed] [Google Scholar]
- 53.McLaughlin AC, Cullis PR, Hemminga MA, Hoult DI, Radda GK, Ritchie GA, Seeley PJ, Richards RE. Application of 31P NMR to model and biological membrane systems. FEBS Lett. 1975;57:213–218. doi: 10.1016/0014-5793(75)80719-8. [DOI] [PubMed] [Google Scholar]
- 54.Lewis BA, Harbison GS, Herzfeld J, Griffin RG. NMR structural analysis of a membrane protein: bacteriorhodopsin peptide backbone orientation and motion. Biochemistry. 1985;24:4671–4679. doi: 10.1021/bi00338a029. [DOI] [PubMed] [Google Scholar]
- 55.Smith R, Cornell BA. Dynamics of the intrinsic membrane polypeptide gramicidin A in phospholipid bilayers. Biophys J. 1986;49:117–118. doi: 10.1016/S0006-3495(86)83617-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tian F, Song Z, Cross TA. Orientational constraints derived from hydrated powder samples by two-dimensional PISEMA. J Magn Reson. 1998;135:227–231. doi: 10.1006/jmre.1998.1544. [DOI] [PubMed] [Google Scholar]
- 57.Park SH, Mrse AA, Nevzorov AA, De Angelis AA, Opella SJ. Rotational diffusion of membrane proteins in aligned phospholipid bilayers by solid-state NMR spectroscopy. J Magn Reson. 2005;178:162–165. doi: 10.1016/j.jmr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 58.Das BB, Nothnagel HJ, Lu GJ, Son WS, Tian Y, Marassi FM, Opella SJ. Structure determination of a membrane protein in proteoliposomes. J Am Chem Soc. 2012;134:2047–2056. doi: 10.1021/ja209464f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barkay T, Miller SM, Summers AO. Bacterial mercury resistance from atoms to ecosystems. FEMS Microbiol Rev. 2003;27:355–384. doi: 10.1016/S0168-6445(03)00046-9. [DOI] [PubMed] [Google Scholar]
- 60.Steele RA, Opella SJ. Structures of the reduced and mercury-bound forms of MerP, the periplasmic protein from the bacterial mercury detoxification system. Biochemistry. 1997;36:6885–6895. doi: 10.1021/bi9631632. [DOI] [PubMed] [Google Scholar]
- 61.Schiering N, Kabsch W, Moore MJ, Distefano MD, Walsh CT, Pai EF. Structure of the detoxification catalyst mercuric ion reductase from Bacillus sp. strain RC607. Nature. 1991;352:168–172. doi: 10.1038/352168a0. [DOI] [PubMed] [Google Scholar]
- 62.Wilson JR, Leang C, Morby AP, Hobman JL, Brown NL. MerF is a mercury transport protein: different structures but a common mechanism for mercuric ion transporters? FEBS Lett. 2000;472:78–82. doi: 10.1016/s0014-5793(00)01430-7. [DOI] [PubMed] [Google Scholar]
- 63.Howell SC, Mesleh MF, Opella SJ. NMR structure determination of a membrane protein with two transmembrane helices in micelles: MerF of the bacterial mercury detoxification system. Biochemistry. 2005;44:5196–5206. doi: 10.1021/bi048095v. [DOI] [PubMed] [Google Scholar]
- 64.Tian Y, Lu GJ, Marassi FM, Opella SJ. Structure of the membrane protein MerF, a bacterial mercury transporter, improved by the inclusion of chemical shift anisotropy constraints. J Biomol NMR. 2014;60:67–71. doi: 10.1007/s10858-014-9852-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barbet-Massin E, Pell A, Retel J, Andreas L, Jaudzems K, Franks W, Nieuwkoop A, Hiller M, Higman V, Guery P, Pertarello A, Knight M, Felletti M, Le Marchand T, Kotelovica S, Akopjana I, Tars K, Stoppini M, Bellotti V, Bolognesi M, Ricagno S, Chou J, Griffin R, Oschkinaat H, Lesage A, Emsley L, Herrmann T, Pintacuda G. Rapid proton-detected NMR assignment for proteins with fast magic angle spinning. J Am Chem Soc. 2014;136:12489–12497. doi: 10.1021/ja507382j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Agarwal V, Penzel S, Szekely K, Cadalbert R, Testori E, Oss A, Past J, Samoson A, Ernst M, Bockmann A, Meier B. De novo 3D struccture determination from sub-milligram protein samples by solid-state 100 kHz MAS NMR spectroscopy. Angew Chem Int Ed Engl. 2014;53:12256–12256. doi: 10.1002/anie.201405730. [DOI] [PubMed] [Google Scholar]
- 67.Ni Q, Daviso E, Can T, Markhasin E, Jawla S, Swager T, Temkin R, Herzfeld J, Griffin R. High frequncy dynamic nuclear polarization. Acc Chem Res. 2013;46:1933–1941. doi: 10.1021/ar300348n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jakdetchai O, Denysenkov V, Becker-Baldus J, Dutagaci B, Prisner TF, Glaubitz C. Dynamic nuclear polarization-enhanced NMR on aligned lipid bilayers at ambient temperature. J Am Chem Soc. 2014;136:15533–15536. doi: 10.1021/ja509799s. [DOI] [PubMed] [Google Scholar]
- 69.Siaw TA, Fehr M, Lund A, Latimer A, Walker SA, Edwards DT, Han SI. Effect of electron spin dynamics on solid-state dynamic nuclear polarization performance. Physical chemistry chemical physics: PCCP. 2014;16:18694–18706. doi: 10.1039/c4cp02013h. [DOI] [PubMed] [Google Scholar]
- 70.Parthasarathy S, Nishiyama Y, Ishii Y. Sensitivity and Resolution Enhanced Solid-State NMR for Paramagnetic Systems and Biomolecules under Very Fast Magic Angle Spinning. Acc Chem Res. 2013;46:2127–2135. doi: 10.1021/ar4000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Balayssac S, Bertini I, Bhaumik A, Lelli M, Luchinat C. Paramagnetic shifts in solid-state NMR of proteins to elicit structural information. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:17284–17289. doi: 10.1073/pnas.0708460105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Otting G. Protein NMR Using Paramagnetic Ions. Annual Review of Biophysics. 2010;39:387–405. doi: 10.1146/annurev.biophys.093008.131321. [DOI] [PubMed] [Google Scholar]
- 73.Sengupta I, Nadaud PS, Jaroniec CP. Protein structure determination with paramagnetic solid-state NMR spectroscopy. Acc Chem Res. 2013;46:2117–2126. doi: 10.1021/ar300360q. [DOI] [PubMed] [Google Scholar]
- 74.Wang S, Ladizhansky V. Recent advances in magic angle spinning solid-state NMR of membrane proteins. Progress in Nuclear Magnetic Resonance Spectroscopy. 2014;82:1–26. doi: 10.1016/j.pnmrs.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 75.Yan S, Suiter C, Hou G, Zhang H, Polenova T. Probing structure and dynamics of protein assemblies by magic angle spinning NMR spectroscopy. Acc Chem Res. 2013;46:2047–2058. doi: 10.1021/ar300309s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tycko R. Solid-state NMR studies of amyloid fibril structure. Annu Rev Phys Chem. 2011;62:279–299. doi: 10.1146/annurev-physchem-032210-103539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cho MK, Gayen A, Banigan J, Leninger M, Traaseth N. Intrinsic conformational plasticity of nataive EmrE provides a pathway for multidrug resistance. J Am Chem Soc. 2014;136:8072–8080. doi: 10.1021/ja503145x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.DeSimone A, Mote K, Veglia G. Structural dynamics and conformational equilibria of SERCA regulatory proteins in membranes by solid-state NMR restrained simulations. Biophys J. 2014;106:2566–2576. doi: 10.1016/j.bpj.2014.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huang R, Yamamoto K, Zhang M, Popovych N, Hung I, Im SC, Gan Z, Waskell L, Ramamoorthy A. Probing the transmembrane structure and dynamics of microsomal NADPH-cytochrome P 450 oxidoreductase by solid-state NMR. Biophys J. 2014;106:2126–2133. doi: 10.1016/j.bpj.2014.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Can T, Sharma M, Hung I, Gor’kov P, Brey W, Cross T. Magic angle spinning and oriented sample solid-state NMR structural restraints combine for influenza a M2 protein functional insights. J Am Chem Soc. 2012;134:9022–9025. doi: 10.1021/ja3004039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liao S, Fritzsching K, Hong M. Conformational analysis f the full-length M2 protein of the influenza A virus using solid-state NMR. Protein Science. 2013;22:1623–1638. doi: 10.1002/pro.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ding X, Zhao X, Watts A. G-protein-coupled receeptor structure, ligand binding and activation as studied by solid-state NMR spectroscopy. Biochem J. 2013;450:443–457. doi: 10.1042/BJ20121644. [DOI] [PubMed] [Google Scholar]
- 83.Kira, Ackerman JL, Javkhlantugs N, Miyamori T, Sasaki Y, Eguchi M, Kawamura I, Ueda K, Naito A. Interaction of extracellular loop II o K-Opioid receptor (196–228) with opioi peptide dynorphin in membrane environments as revealed by solid state nuclear magnetic resonance, quartz crystal microbalance and molecular dynamics simulation. J Phys Chem B. 2014;118:9604–9612. doi: 10.1021/jp505412j. [DOI] [PubMed] [Google Scholar]
- 84.Goncalves J, Eilers M, South K, Opefi CA, Laissue P, Reeves PJ, Smith SO. Magic angle spinning nuclear magnetic resonance spectroscopy of G protein-coupled receptors. Methods Enzymol. 2013;522:365–389. doi: 10.1016/B978-0-12-407865-9.00017-0. [DOI] [PubMed] [Google Scholar]