

Abstract
Objectives
ω-3 Fatty acids (FAs), natural ligands for the peroxisome proliferator-activated receptor–α (PPAR-α), attenuate parenteral nutrition–associated liver disease (PNALD). However, the mechanisms underlying the protective role of ω-3 FAs are still unknown. The aim of this study was to determine the effects of ω-3 FAs on hepatic triglyceride (TG) accumulation in a murine model of PNALD and to investigate the role of PPAR-α and microsomal triglyceride transfer protein (MTP) in this experimental setting.
Methods
129S1/SvImJ wild-type or 129S4/SvJaePparatm/Gonz/J PPAR-α knockout mice were fed chow and water (controls); oral, fat-free PN solution only (PN-O); PN-O plus intraperitoneal (IP) ω-6 FA-predominant supplements (PN–ω-6); or PN-O plus IP ω-3 FA (PN–ω-3). Control and PN-O groups received sham IP injections of 0.9% NaCl. Hepatic histology, TG and cholesterol, MTP activity, and PPAR-α messenger RNA were assessed after 19 days.
Results
In all experimental groups, PN feeding increased hepatic TG and MTP activity compared with controls. Both PN-O and PN–ω-6 groups accumulated significantly greater amounts of TG when compared with PN–ω-3 mice. Studies in PPAR-α null animals showed that PN feeding increases hepatic TG as in wild-type mice. PPAR-α null mice in the PN-O and PN–ω-6 groups demonstrated variable degrees of hepatic steatosis, whereas no evidence of hepatic fat accumulation was found after 19 days of oral PN plus IP ω-3 FAs.
Conclusions
PN induces TG accumulation (steatosis) in wild-type and PPAR-α null mice. In PN-fed wild-type and PPAR-α null mice given IP ω-3 FAs, reduced hepatic TG accumulation and absent steatosis are found. Prevention of steatosis by ω-3 FAs results from PPAR-α–independent pathways.
Keywords: microsomal triglyceride transfer protein, ω-3-fatty acids, parenteral nutrition–associated liver disease, peroxisome proliferator-activated receptor–α, steatosis
Introduction
Parenteral nutrition–associated liver disease (PNALD) may be characterized by cholestasis, steatohepatitis, fibrosis, and cirrhosis. This complication of therapy is a common occurrence in children, particularly in premature infants, who receive long-term parenteral nutrition (PN).1–3 Studies in both rodent and piglet models of PNALD have shown that hepatic steatosis is a precursor to more severe liver damage.4–7 Studies of nonalcoholic fatty liver disease (NAFLD) have demonstrated that hepatic lipid accumulation results from an imbalance between de novo lipogenesis, fatty acid (FA) oxidation, and/or triglyceride (TG) secretion.8 These mechanisms may also be implicated in hepatic steatosis occurring as a consequence of long-term PN.
Several molecular mediators are known to be involved in the pathogenesis of hepatic steatosis. In particular, absent or decreased activity of microsomal triglyceride transfer protein (MTP), a rate-limiting intracellular chaperone responsible for the assembly and secretion of apoB100-containing very low-density lipoprotein (VLDL),9 has been correlated with lipid accumulation in a variety of conditions, including abetalipoproteinemia, drug-induced liver injury, and chronic hepatitis C.9–14 Both the expression and functional activity of MTP have increased via activation of the peroxisome proliferator-activated receptor–α (PPAR-α).15 This nuclear receptor regulates the transcription of genes for key enzymes promoting β-oxidation and lipid clearance from hepatocytes. Consistent with these findings, stimulation of PPAR-α by its synthetic agonist, Wy-14643, significantly reduces steatohepatitis in a murine model.16 In human NAFLD therapeutic trials, however, the effects of fibrates, PPAR-α agonists, have yielded mixed results.17–19 In recent years, emerging evidence has linked another naturally occurring ligand for PPAR-α, ω-3 polyunsaturated FA, to the regulation of lipogenesis. Accordingly, several animal studies of PNALD have demonstrated that intravenous (IV) or enteral supplementation with ω-3 FAs results in reduced hepatic steatosis and lower serum TG levels when compared with nutrition supplementation with standard IV lipid emulsions rich in ω-6 FAs.20–23 Although the anti-inflammatory role of ω-3 FAs, acting as a potent inhibitor of arachidonic acid–derived eicosanoids, has been well characterized, 24 the mechanisms for this protective effect in PN-induced hepatic steatosis are still not clear. One current hypothesis states that ω-3 FAs may enhance FA β-oxidation, as well as inhibit de novo lipogenesis.25–27 To date, however, the possible interrelationships among ω-3 FA, MTP, and PPAR-α in the etiopathogenesis of PNALD have not been elucidated.
In the present study, we tested the hypothesis that “protection” of hepatic parenchyma from PN-associated lipid accumulation by ω-3 FAs could, in part, be the consequence of an MTP and/or a PPAR-α–mediated pathway. Employing a murine model of PN-induced hepatic steatosis, we investigated the effects on intrahepatic lipid accumulation of oral feeding with a dextrose/amino acid solution, given alone and in concert either with intraperitoneal (IP) ω-3 FAs or with IP ω-6 FA-based lipid emulsions (a previously validated model mimicking IV administration28,29). These data were also correlated with hepatic MTP expression. Employing the same experimental model in PPAR-α knockout mice, hepatic steatosis was also examined as a consequence of these feeding regimens.
Materials and Methods
Animals and Diet
The Animal Care and Use Committee of the State University of New York Downstate Medical Center approved the protocol. Six-week-old male 129S1/SvImJ mice (19–22 g; Jackson Laboratory, Bar Harbor, ME) were divided into 4 groups. Group 1 controls (C) were fed a standard chow diet (Purina 5008; Nestle Purina, St Louis, MO) containing 6.5% fat (17.3% of calories), 47% carbohydrates (55.1% of calories), and 23.5% protein (27.6% of calories), as well as water ad libitum. Group 2 (PN-O) received ad libitum PN solution in water bottles with no other source of hydration or nutrition. The PN solution was identical to that used in the neonatal intensive care unit and was composed of 20% dextrose, 2% essential and nonessential amino acids (TrophAmine; B. Braun Medical, Englewood, NJ), 30 mEq/L sodium, 20 mEq/L potassium, 15 mEq/L calcium gluconate, 10 mEq/L magnesium, 10 mmol/L phosphate, 5 mEq/L acetate, 30 mEq/L chloride, 0.2% pediatric trace elements (American Regent, Shirley, NY), and 0.5% pediatric multivitamins (MVI Pediatric; aaiPharma, Wilmington, NC). Bottles were replaced daily with fresh solution and the consumption measured. Group 3 (PN–ω6) received the same PN solution in water bottles ad libitum with IP injections every other day (6 mg/g body weight) of 20% Intralipid (Baxter Healthcare/Fresenius Kabi, Bod Homburg, Germany). The ratio of ω-6 FA to ω-3 FA in Intralipid was 5.5:1. Group 4 (PN– ω3) received the same PN solution ad libitum plus IP injections every other day of an ω-3 FA lipid emulsion (Omegaven; Fresenius Kabi; 6 mg/g body weight). To simulate all experimental conditions, animals in the control and PN-O groups received IP sham injections with the same volume of 0.9% NaCl. All animals were weighed every third day and received their designated diets for 19 days prior to study. For all studies, 3 sequential experiments were performed, therefore allowing repeated corroboration of results in similarly treated groups of 5–6 animals per group per experiment. These studies are summarized as follows:
Experiment 1 = C vs PN-O group
Experiment 2 = C vs PN-O vs PN–ω-6 groups
Experiment 3 = C vs PN-O vs PN–ω-6 vs PN–ω-3 groups
Animals were studied after 19 days. Prior to investigation, no IP injections were administered for 24 hours and animals were fasted for 6 hours. Mice were anesthetized with 0.1 mL of IP nembutal sodium (50 mg/mL), weighed, killed, and plasma and livers collected. Liver specimens were snap frozen in liquid nitrogen and stored at −70°C until use. For ease of comparison, data are combined for animals in each specific feeding group over all experiments. In a subsequent study, separate but identical experiments were conducted using 8-week-old male PPAR-α knockout mice (129S4/SvJaePparatm/Gonz/J; Jackson Laboratories). Four groups (each group n = 5) of PPAR-α knockout animals (control, PN-O, PN–ω-6, and PN–ω-3) were studied as described above.
Histopathology
Liver sections were stained with hematoxylin and eosin. A single pathologist (V.A.), blinded to the feeding regimens, scored steatosis according to the NASH Clinical Research Network30 as follows: grade 0 (<5% steatotic hepatocytes), grade 1 (<33%), grade 2 (33%–66%), and grade 3 (>66%). Hepatocyte ballooning was graded from 0–2 as follows: 0 = none, 1 = few balloon cells, and 2 = many/prominent balloon cells.
Plasma and Hepatic Lipid Measurements
Plasma total cholesterol and TG were measured using commercial kits, after precipitation of apoB lipoproteins.31 Hepatic lipids were extracted from tissue homogenates, using the Bligh and Dyer32 procedure, and quantified.
MTP Activity Determination
Liver sections (0.1 g) were washed, homogenized in a glass homogenizer using 1 mM Tris-HCl (pH 7.6) buffer with 1 mM EDTA plus 1 mM MgCl2, and centrifuged, and supernatants were collected to measure MTP activity as previously described33 using a standard kit (Chylos, Woodbury, NY).
RNA Quantification
Total RNA was isolated using TRIzol (Invitrogen, Carlsbad, CA). Purity and integrity were assessed by the A260/A280 ratio. RNAs with ratios >1.7 were used for complementary DNA (cDNA) synthesis. The first-strand cDNA was synthesized with an Omniscript RT kit (QIAGEN, Valencia, CA) and used for quantitative reverse transcription polymerase chain reaction (RT-PCR) (SYBR Green I; Eurogentec, Fremont, CA). Forward 5′-GTGGCTGCTATAATTTGCTGTG-3′ and reverse 5′-GAAGGTGTCATCTGGATGGGT-3′ primers were used for PPAR-α messenger RNA (mRNA) quantifications. Data were analyzed using the ΔΔCt method.
Statistical Analysis
Data are presented as mean ± SD. Statistical significance (P < .05) was determined using the Student t test for comparison between 2 individual groups and analysis of variance for comparisons of all groups.
Results
Diet and Body Weight Gain
Consistent with published standards of a mean daily calorie intake in 20-g mice,20 all groups consumed approximately 15 mL/d of PN solution, equivalent to 500 kcal/kg/d plus 10 g/kg/d of protein. All groups gained weight, and no differences in the percent weight change were found among groups over the 19 days of the protocol (Figure 1).
Figure 1.
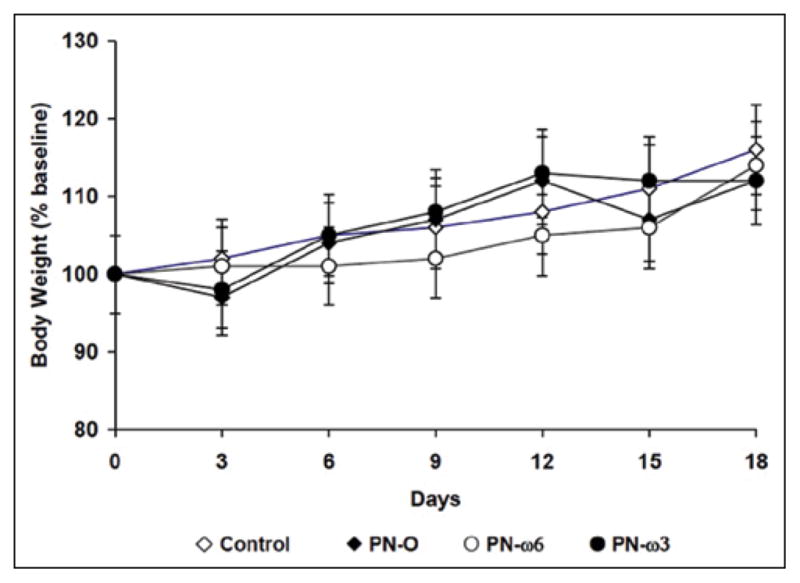
Body weight changes in chow-fed control, PN-O, PN–ω-6, and PN–ω-3 groups. Values are presented as % baseline weights. PN-O, oral, fat-free parenteral nutrition (PN) solution only; PN–ω-6, PN-O plus intraperitoneal (IP) ω-6 fatty acid (FA)–predominant supplements; PN–ω-3, PN-O plus IP ω-3 FA.
Hepatic Histology
Histologic examination of liver tissue showed changes consistent with hepatic steatosis in both PN-O and PN–ω-6 mice (Figure 2B,C), with lipid accumulation appearing less prominent in the PN–ω-6 group. By contrast, little hepatic lipid accumulation was observed in histopathologic sections from control and PN–ω-3 feeding groups (Figure 2A,D).
Figure 2.

Representative hematoxylin and eosin–stained sections from liver tissue of mice. (A) Control group shows normal hepatocytes with absence of either steatosis or ballooning degeneration. (B) The PN-O group demonstrates severe steatosis (grade 3), with ballooning degeneration (grade 2). (C) The PN–ω-6 group shows mild steatosis (grade 1) with ballooning degeneration of some hepatocytes. (D) The PN–ω-3 group shows normal hepatocytes with absence of either steatosis or ballooning degeneration. In these sections, examples of steatosis are indicated by the long arrows and ballooning degeneration by the short arrows (A, B, D = ×10 magnification; C = ×20 magnification). PN-O, oral, fat-free parenteral nutrition (PN) solution only; PN–ω-6, PN-O plus intraperitoneal (IP) ω-6 fatty acid (FA)–predominant supplements; PN–ω-3, PN-O plus IP ω-3 FA.
Effects of ω-3 FAs on Hepatic and Plasma Lipids
Significant increases in hepatic TG content were noted in all animals that received PN compared with controls (Figure 3). However, hepatic TG content was significantly lower in the PN–ω-3 group compared with the PN-O and PN–ω-6 groups. No differences in hepatic cholesterol levels were found among feeding groups receiving PN, whereas hepatic cholesterol levels in all experimental groups were higher than in controls. Plasma TG in all experimental groups trended higher when compared with controls. Plasma TG also trended lower in the PN–ω-3 group when compared with PN-O and PN–ω-6 animals (Figure 4). However, these differences did not reach statistical significance. No significant changes in plasma total cholesterol and non–high-density lipoprotein cholesterol levels were found among these feeding groups (data not shown).
Figure 3.

Quantitative comparisons of hepatic triglycerides and (top) and cholesterol (bottom). Values are presented as means ± SD for the 4 feeding groups (***P < .001, compared with controls; +++P < .001, compared with controls; and P < .005, compared with PN-O and PN–ω-6). PN-O, oral, fat-free parenteral nutrition (PN) solution only; PN–ω-6, PN-O plus intraperitoneal (IP) ω-6 fatty acid (FA)–predominant supplements; PN–ω-3, PN-O plus IP ω-3 FA.
Figure 4.
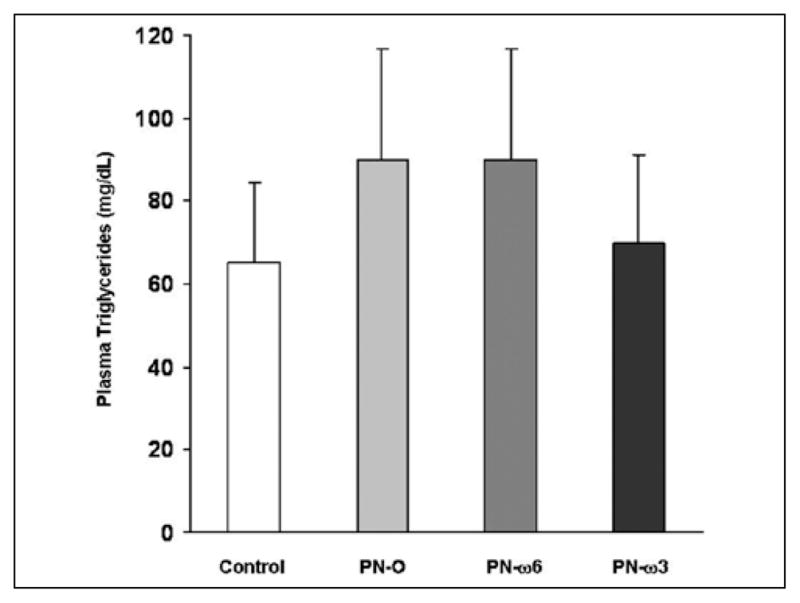
Quantitative comparisons of plasma triglycerides. Values are presented as means ± SD for the 4 feeding groups (P = not significant, among all feeding groups). PN-O, oral, fat-free parenteral nutrition (PN) solution only; PN–ω-6, PN-O plus intraperitoneal (IP) ω-6 fatty acid (FA)–predominant supplements; PN–ω-3, PN-O plus IP ω-3 FA.
Hepatic MTP Activity
Hepatic MTP activity was significantly increased in all PN groups compared with controls (Figure 5). MTP activities in PN–ω-6 and PN–ω-3 mice were similar, and both feeding groups demonstrated significantly increased MTP activity when compared with PN-O mice.
Figure 5.

Hepatic microsomal triglyceride transfer protein (MTP) activity in controls, PN-O, PN–ω-6, and PN–ω-3 groups. Values are shown as means ± SD (**P < .005, compared with controls; ***P < .001, compared with controls and PN-O). PN-O, oral, fat-free parenteral nutrition (PN) solution only; PN–ω-6, PN-O plus intraperitoneal (IP) ω-6 fatty acid (FA)–predominant supplements; PN–ω-3, PN-O plus IP ω-3 FA.
Effects of PPAR-α on Intrahepatic Lipid Accumulation
In studies comprising wild-type mice, PPAR-α mRNA levels trended higher in PN-O, PN–ω-6, and PN–ω-3 groups, although differences did not reach statistical significance (Figure 6). To directly examine the potential influence of PPAR-α expression in preventing or ameliorating PN-associated steatosis, an identical group of experiments was conducted in PPAR-α knockout mice. In these animals, as in wild-type mice, hepatic TG levels, when compared with controls, were significantly increased in PN-O, PN–ω-6, and PN–ω-3 groups. However, TG levels were significantly lower in PN–ω-3 animals when compared with PN-O and PN–ω-6 groups (Figure 7). No significant differences with respect to hepatic cholesterol levels were noted among the feeding groups. These studies indicate that PN induces hepatic TG accumulation in PPAR-α knockout mice as in wild-type mice and that ω-3 FA supplementation ameliorates hepatic steatosis both in wild-type and in PPAR-α null animals.
Figure 6.
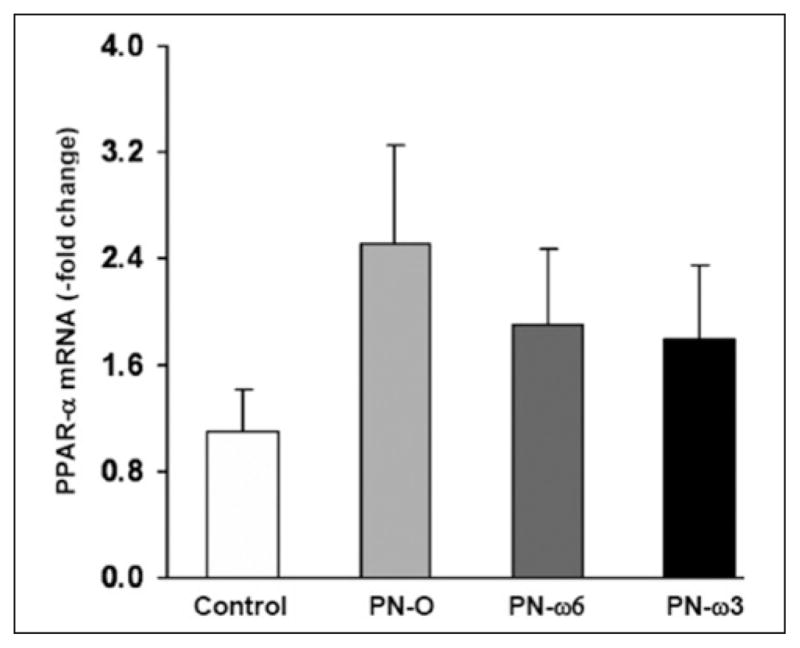
Hepatic peroxisome proliferator-activated receptor–α (PPAR-α) messenger RNA (mRNA) in controls, PN-O, PN–ω-6, and PN–ω-3 groups. Values are shown as means ± SD (P = not significant among all groups). PN-O, oral, fat-free parenteral nutrition (PN) solution only; PN–ω-6, PN-O plus intraperitoneal (IP) ω-6 fatty acid (FA)–predominant supplements; PN–ω-3, PN-O plus IP ω-3 FA.
Figure 7.

Hepatic triglyceride (TG) (top) and cholesterol (bottom) content in peroxisome proliferator-activated receptor–α knockout mice (*P < .05, compared with controls; **P < .01, compared with controls and PN–ω-3). PN-O, oral, fat-free parenteral nutrition (PN) solution only; PN–ω-6, PN-O plus intraperitoneal (IP) ω-6 fatty acid (FA)–predominant supplements; PN–ω-3, PN-O plus IP ω-3 FA.
Effects of PPAR-γ on Hepatic Lipid Accumulation
Because ω-3 polyunsaturated FAs and their metabolites have also been reported to be natural ligands and activate PPAR-γ in certain cells (eg, adipocytes),34,35 parallel studies were conducted to examine the potential role of PPAR-γ activation in affecting PN-associated hepatic lipid accumulation. Previous studies have shown that pioglitazone, a thiazolidinedione derivative, is a selective PPAR-γ agonist.36,37 Control (wild-type) and PPAR-α knockout mice groups received the PN–ω-6 regimen and were treated with 25 mg/kg pioglitazone every other day or vehicle by gavage feeding (20 μL total volume).38 Hepatic TG levels after 19 days were significantly higher in both PPAR-α knockout subgroups when compared with controls (Figure 8). However, pioglitazone treatment and, hence, PPAR-γ activation per se, both in control and in PPAR-α null mice, did not exert a significant influence on hepatic lipid accumulation (Figure 8).
Figure 8.

Hepatic triglyceride (TG) (top) and cholesterol (bottom) in wild-type (controls) and peroxisome proliferator-activated receptor–α (PPAR-α) knockout (KO) mice. Study groups received the PN–ω-6 regimen, either alone (PN–ω6) or supplemented with oral pioglitazone (PN–ω-6 + Pio) (*P < .05, **P < .01, compared with controls). PN-O, oral, fat-free parenteral nutrition (PN) solution only; PN–ω-6, PN-O plus intraperitoneal (IP) ω-6 fatty acid (FA)–predominant supplements; PN–ω-3, PN-O plus IP ω-3 FA.
Discussion
The data presented herein demonstrate that mice receiving oral PN solution (PN-O), without added lipid, exhibit marked hepatic steatosis after a 19-day feeding period. When this dietary regimen is combined with either IP ω-6 FAs or ω-3 FAs over the same study duration, reduced histopathologically identified steatosis, compared with PN-O animals, is noted. Importantly, the magnitude of this observed reduction in steatosis, associated with a significant reduction in hepatic TG content, is more profound in association with ω-3 FA administration.
PPAR-α activity was previously thought to be an essential factor involved in preventing or ameliorating steatohepatitis in similar experimental models. However, in parallel studies employing the PPAR-α knockout mouse model reported here, these findings suggest that the protective effects of ω-3 FAs are independent of changes in PPAR-α activity. Accordingly, these results suggest that ω-3 FA-rich lipid emulsions, in a PN regimen, prevent hepatic TG accumulation as a consequence of alternative mechanisms that remain to be elucidated. Although prior studies in mice, using comparable feeding protocols, have reported similar ω-3 FA effects on hepatic lipid deposition, our studies represent, to our knowledge, the first attempts to examine specific molecular mechanisms potentially involved in regulating these metabolic events.21
In these experiments, we did not employ a traditional animal model of PN, delivered via an indwelling IV catheter. Previous reports have shown that oral fat-free PN feeding causes hepatic steatosis in mice similar to that seen in animals receiving IV PN.21 This model is easy to manage, avoids complications of IV catheter placement and sepsis, and allows longer administration of PN, thus mimicking the treatment in human infants. To approximate the metabolic responses to parenteral lipid administration and, again, to limit complications related to IV catheter insertion and management, all lipid supplements were provided via the IP route. Following IP administration, lipid is absorbed directly into the bloodstream. This method of lipid supplementation has previously been reported to be a valid surrogate for IV lipid delivery in 2 animal models, while obviating the trauma and morbidity of indwelling central lines or tail-vein injections.28,29 Sham IP injections (0.9% NaCl) were carried out in control and PN-O mice, to confirm that IP injections per se did not influence nutrient consumption, weight gain, or other experimental findings.
Previous murine models have demonstrated that, similar to findings in the PN-O group, a high-carbohydrate, fat-free diet results in increased hepatic TG content.39 Thus, the findings of marked hepatic steatosis in this feeding group are not surprising. Administration of either fat-free diets (as in the PN-O group) or diets exceedingly low in γ-linolenic acid or in linoleic acid (the major lipid species provided to the PN–ω-6 group) may also precipitate essential FA (EFA) deficiency, a state that could represent a confounding variable in these investigations. This condition is mediated by the sterol regulatory element binding protein-1, a transcription factor activating genes involved in FA synthesis.40,41 Plasma FA profiles and, specifically, the triene to tetraene FA ratio as a measure of EFA deficiency were not measured in these studies. However, prior investigations have shown that the amount of lipid provided to both PN–ω-6 and PN–ω-3 groups meets the established murine dietary requirements for EFA,42 either as linoleic acid (PN–ω-6) or by supplying sufficient amounts of arachidonic acid (PN– ω-3),21 the FA metabolically downstream from linoleate. Nevertheless, the role of subclinical EFA deficiency cannot be ruled out, particularly in the PN-O feeding group. Published studies have demonstrated biochemical EFA deficiency in a murine model within a 19-day feeding period, employing a lipid-free oral PN solution.43,44 Furthermore, this feeding regimen, over the same feeding period, has been validated as a model for studying the role of EFA deficiency in the etiopathogenesis of hepatic steatosis.44
The role of MTP is thought to be pivotal in the intrahepatic transport of dietary lipid. MTP catalyzes the transfer of TGs to apoB100, a key step in the hepatocellular formation and secretion of VLDL particles.9–14 We hypothesized that increased hepatic deposition and decreased plasma apoB lipoproteins in mice fed oral PN solution, with or without IP lipid, could be a consequence of reduced MTP activity. However, in contrast to available human and animal studies showing decreased MTP activity in hepatic steatosis,10–14 we found that MTP activity was elevated in all experimental groups compared with chow-fed controls. In addition, no significant differences in MTP activity were noted between PN–ω-6 and PN–ω-3 groups, suggesting that ω-3 FA-associated prevention of hepatic steatosis was not specifically mediated by MTP upregulation. Thus, MTP deficiency does not play a role in PN-induced hepatosteatosis.
Several recent studies have proposed that ω-3 FA ameliorates hepatic steatosis and reduced hepatic TG content by activation of PPAR-α, an important transcription factor in MTP upstream signaling.45–47 PPAR-α acts as a sensor of FA levels in the hepatocyte, promotes FA oxidation in both peroxisomes and mitochondria, and increases hepatic FA secretion.45–47 PPAR-α–induced increases in the secretion of apoB100 have been shown to be mediated by increased MTP activity, in parallel with upregulation of MTP transcription.15 In our study, liver PPAR-α mRNA levels in wild-type mice trended higher in all 3 groups receiving PN, when compared with controls; however, these differences did not reach statistical significance. Most important in this regard is the observation that ω-3 FA-mediated inhibition of hepatic lipid accumulation is not abrogated in PN-fed, PPAR-α knockout mice. Consistent with these findings, no difference in liver mRNA for carnitine pal-mitoyl transferase-1 (CPT-1) was seen between PN–ω-6 and PN–ω-3 wild-type murine groups (data not shown). CPT-1 is a rate-limiting enzyme in FA oxidation that undergoes regulation by PPAR-α. Thus, these results suggest that activation of PPAR-α is unlikely to play a significant role in mediating the protective effect of ω-3 FA in this experimental model. Also, the observed increase in MTP activity in PN-fed groups appears more likely to represent a compensatory response to hepatocellular lipid accumulation.
ω-3 Polyunsaturated fatty acids have been shown to also activate PPAR-γ in specific tissue cells, including both adipocytes and hepatocytes. Additional studies were conducted to examine the possible influence on hepatic lipid accumulation of PPAR-γ upregulation by the thiazolidinedione derivative, pioglitazone. Hepatic lipid content both in wild-type and in PPAR-α knockout animals receiving PN–ω-6 was not significantly altered by pioglitazone treatment. Interestingly, recent studies have cast doubt on a protective role for PPAR-γ in hepatic steatosis. Thus, chronic activation of PPAR-γ by antidiabetic thiazolidinediones has, in fact, been shown to induce hepatic lipid accumulation,48 and, in leptin-deficient mice, hepatic steatosis is promoted by the PPAR-γ target gene, fatspecific protein 27.49
To clarify the pathogenesis of PNALD and the mechanisms underlying the protective role of ω-3 FA, future studies should focus on molecular pathways involved in hepatic lipid homeostasis, other than MTP, PPAR-α, and PPAR-γ. For example, 1 recent study has shown that experimental steatohepatitis, occurring as a consequence of lipoperoxide accumulation, cannot be prevented by a diet enriched with ω-3 FA but deficient in methionine and choline.50 This finding was noted despite ω-3 FA-associated activation of PPAR-α and suppression of de novo hepatic lipogenesis. Another nuclear receptor, liver xenobiotic receptor (LXR), has recently been characterized as countering PPAR-α–mediated effects on lipid metabolism.51 LXR switches on several genes involved in FA synthesis and thus increases hepatic TG-rich VLDL production.52,53 Activation of both LXR and PPAR-α may be dependent on their interactions with the same retinoid xenobiotic receptor.51 Considering the influences of these interactions on hepatocellular lipid metabolism, studies should investigate whether LXR signaling is influenced by ω-3 FA in this experimental model.
Other dietary factors associated with PN administration and possibly independent of PPAR activation may also exert important and, as yet, not fully characterized influence on hepatic lipid metabolism and the expression of PNALD. For example phytosterols, present in variable amounts in lipid emulsions, accumulate in patients receiving soya oil–based products.54 Biologic activities of phytosterols include disruption in nuclear receptor signaling, proinflammatory signal activation, and increased cellular apoptosis55; phytosterolemia also has been linked to cholestatic liver disease.54 The administration of lipid emulsions rich in ω-3 FAs has also been associated with significantly reduced oxidative stress in human infants. In a recent double-blind, randomized trial comparing infants receiving an ω-3 FA-rich PN regimen with those receiving a standard ω-6 FA-based lipid emulsion, total antioxidant potential and vitamin E levels were significantly increased in those receiving ω-3 FAs.56 Future investigations must address these, as well as the other dietary elements and endogenous molecular signaling species described herein, to improve our understanding of those factors involved in the development, promotion and, importantly, prevention of hepatic steatosis and steatohepatitis during long-term PN.
Clinical Relevancy Statement.
Parenteral nutrition–associated liver disease (PNALD) represents a common complication, especially in preterm infants, of long-term central venous alimentation. Although the precise factors underlying the progression from hepatic steatosis to steatohepatitis and, ultimately, to cirrhosis, have not been fully elucidated, available studies indicate that both excess carbohydrate and lipid calories play an etiopathogenic role. Currently available lipid emulsions for parenteral nutrition (PN) are rich in ω-6 fatty acids (FAs). Recent evidence, however, indicates that lipid supplements composed predominantly of ω-3 FAs may be protective against the development of PNALD in both animal models and humans. The present study aims to examine specific intrahepatocellular factors accounting for this protective effect, employing a murine model of PN.
Acknowledgments
We thank Dr Miguel Guzman and Mr Sujai Jalaj for their technical assistance.
Footnotes
Financial disclosures: Supported in part by USPHS grants DK-46700 and HL-095924 and a grant-in-aid from the American Heart Association (to M.M.H.).
References
- 1.Kelly DA. Liver complications of pediatric parenteral nutrition—epidemiology. Nutrition. 1998;14:153–157. doi: 10.1016/s0899-9007(97)00232-3. [DOI] [PubMed] [Google Scholar]
- 2.Levine A, Maayan A, Shamir R, et al. Parenteral nutrition–associated cholestasis in preterm neonates: evaluation of ursodeoxycholic acid treatment. J Pediatr Endocrinol Metab. 1999;12:549–553. doi: 10.1515/jpem.1999.12.4.549. [DOI] [PubMed] [Google Scholar]
- 3.Kaufman SS. Prevention of parenteral nutrition–associated liver disease in children. Pediatr Transplant. 2002;6:37–42. doi: 10.1034/j.1399-3046.2002.1o061.x. [DOI] [PubMed] [Google Scholar]
- 4.Wang H, Khaoustov VI, Krishnan B, et al. Total parenteral nutrition induces liver steatosis and apoptosis in neonatal piglets. J Nutr. 2006;136:2547–2552. doi: 10.1093/jn/136.10.2547. [DOI] [PubMed] [Google Scholar]
- 5.Loff S, Waag KL, Kranzlin B, et al. Long-term total parenteral nutrition– induced hepatobiliary dysfunction in a rabbit model. J Pediatr Surg. 1998;33:694–699. doi: 10.1016/s0022-3468(98)90189-0. [DOI] [PubMed] [Google Scholar]
- 6.Katayama T, Tanaka M, Tanaka K, et al. Alterations in hepatic mitochondrial function during total parenteral nutrition in immature rats. JPEN J Parenter Enteral Nutr. 1990;14:640–645. doi: 10.1177/0148607190014006640. [DOI] [PubMed] [Google Scholar]
- 7.Tazuke Y, Drongowski RA, Btaiche I, et al. Effects of lipid administration on liver apoptotic signals in a mouse model of total parenteral nutrition (TPN) Pediatr Surg Int. 2004;20:224–228. doi: 10.1007/s00383-003-1115-1. [DOI] [PubMed] [Google Scholar]
- 8.Musso G, Gambino R, Cassader M, et al. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (NAFLD) Prog Lipid Res. 2009;48:1–26. doi: 10.1016/j.plipres.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 9.Castro J, Amigo L, Miquel JF, et al. Increased activity of hepatic microsomal triglyceride transfer protein and bile acid synthesis in gallstone disease. Hepatology. 2007;45:1261–1266. doi: 10.1002/hep.21616. [DOI] [PubMed] [Google Scholar]
- 10.Letteron P, Sutton A, Mansouri A, et al. Inhibition of microsomal triglyceride transfer protein: another mechanism for drug-induced steatosis in mice. Hepatology. 2003;38:133–140. doi: 10.1053/jhep.2003.50309. [DOI] [PubMed] [Google Scholar]
- 11.Mirandola S, Realdon S, Iqbal J, et al. Liver microsomal triglyceride transfer protein is involved in hepatitis C liver steatosis. Gastroenterology. 2006;130:1661–1669. doi: 10.1053/j.gastro.2006.02.035. [DOI] [PubMed] [Google Scholar]
- 12.Letteron P, Fromenty B, Terris B, et al. Acute and chronic hepatic steatosis lead to in vivo lipid peroxidation in mice. J Hepatol. 1996;24:200–208. doi: 10.1016/s0168-8278(96)80030-4. [DOI] [PubMed] [Google Scholar]
- 13.Pan X, Hussain FN, Iqbal J, et al. Inhibiting proteasomal degradation of microsomal triglyceride transfer protein prevents CCl4 -induced steatosis. J Biol Chem. 2007;282:17078–17089. doi: 10.1074/jbc.M701742200. [DOI] [PubMed] [Google Scholar]
- 14.Sugimoto T, Yamashita S, Ishigami M, et al. Decreased microsomal triglyceride transfer protein activity contributes to initiation of alcoholic liver steatosis in rats. J Hepatol. 2002;36:157–162. doi: 10.1016/s0168-8278(01)00263-x. [DOI] [PubMed] [Google Scholar]
- 15.Ameen C, Edvardsson U, Ljungberg A, et al. Activation of peroxisome proliferator-activated receptor alpha increases the expression and activity of microsomal triglyceride transfer protein in the liver. J Biol Chem. 2005;280:1224–1229. doi: 10.1074/jbc.M412107200. [DOI] [PubMed] [Google Scholar]
- 16.Ip E, Farrell G, Hall P, et al. Administration of the potent PPAR-alpha agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology. 2004;39:1286–1296. doi: 10.1002/hep.20170. [DOI] [PubMed] [Google Scholar]
- 17.Basaranoglu M, Acbay O, Sonsuz A. A controlled trial of gemfibrozil in the treatment of patients with nonalcoholic steatohepatitis. J Hepatol. 1999;31:384. doi: 10.1016/s0168-8278(99)80243-8. [DOI] [PubMed] [Google Scholar]
- 18.Athyros VG, Mikhailidis DP, Didangelos TP, et al. Effect of multifactorial treatment on non-alcoholic fatty liver disease in metabolic syndrome: a randomised study. Curr Med Res Opin. 2006;22:873–883. doi: 10.1185/030079906X104696. [DOI] [PubMed] [Google Scholar]
- 19.Laurin J, Lindor KD, Crippin JS, et al. Ursodeoxycholic acid or clofibrate in the treatment of non-alcohol-induced steatohepatitis: a pilot study. Hepatology. 1996;23:1464–546. doi: 10.1002/hep.510230624. [DOI] [PubMed] [Google Scholar]
- 20.Chen WJ, Yeh SL, Huang PC. Effects of fat emulsions with different fatty acid composition on plasma and hepatic lipids in rats receiving total parenteral nutrition. Clin Nutr. 1996;15:24–28. doi: 10.1016/s0261-5614(96)80257-3. [DOI] [PubMed] [Google Scholar]
- 21.Alwayn IP, Gura K, Nose V, et al. Omega-3 fatty acid supplementation prevents hepatic steatosis in a murine model of nonalcoholic fatty liver disease. Pediatr Res. 2005;57:445–452. doi: 10.1203/01.PDR.0000153672.43030.75. [DOI] [PubMed] [Google Scholar]
- 22.Van Aerde JE, Duerksen DR, Gramlich L, et al. Intravenous fish oil emulsion attenuates total parenteral nutrition-induced cholestasis in newborn piglets. Pediatr Res. 1999;45:202–208. doi: 10.1203/00006450-199902000-00008. [DOI] [PubMed] [Google Scholar]
- 23.Chen WJ, Yeh SL. Effects of fish oil in parenteral nutrition. Nutrition. 2003;19:275–279. doi: 10.1016/s0899-9007(02)01009-2. [DOI] [PubMed] [Google Scholar]
- 24.Calder PC. N-3 polyunsaturated fatty acids, inflammation, and inflammatory diseases. Am J Clin Nutr. 2006;83:1505S–1519S. doi: 10.1093/ajcn/83.6.1505S. [DOI] [PubMed] [Google Scholar]
- 25.Neschen S, Moore I, Regittnig W, et al. Contrasting effects of fish oil and safflower oil on hepatic peroxisomal and tissue lipid content. Am J Physiol Endocrinol Metab. 2002;282:E395–E401. doi: 10.1152/ajpendo.00414.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burdge GC, Finnegan YE, Minihane AM, et al. Effect of altered dietary n-3 fatty acid intake upon plasma lipid fatty acid composition, conversion of [13C] alpha-linolenic acid to longer-chain fatty acids and partitioning towards beta-oxidation in older men. Br J Nutr. 2003;90:311–321. doi: 10.1079/bjn2003901. [DOI] [PubMed] [Google Scholar]
- 27.Willumsen N, Hexeberg S, Skorve J, et al. Docosahexaenoic acid shows no triglyceride-lowering effects but increases the peroxisomal fatty acid oxidation in liver of rats. J Lipid Res. 1993;34:13–22. [PubMed] [Google Scholar]
- 28.Moran Penco JM, Macia BE, Salas MJ, et al. Liver lipid composition and intravenous, intraperitoneal, and enteral administration of intralipid. Nutrition. 1994;10:26–31. [PubMed] [Google Scholar]
- 29.Kalfarentzos F, Spiliotis J, Christopoulos D, et al. Total parenteral nutrition by intraperitoneal feeding in rabbits. Eur Surg Res. 1988;20:352–357. doi: 10.1159/000128785. [DOI] [PubMed] [Google Scholar]
- 30.Kleiner DE, Brunt EM, Van NM, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 31.Iqbal J, Hussain MM. Evidence for multiple complementary pathways for efficient cholesterol absorption in mice. J Lipid Res. 2005;46:1491–1501. doi: 10.1194/jlr.M500023-JLR200. [DOI] [PubMed] [Google Scholar]
- 32.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 33.Athar H, Iqbal J, Jiang XC, Hussain MM. A simple, rapid, and sensitive fluorescence assay for microsomal triglyceride transfer protein. J Lipid Res. 2004;45:764–772. doi: 10.1194/jlr.D300026-JLR200. [DOI] [PubMed] [Google Scholar]
- 34.Edwards IJ, O’Flaherty JT. Omega-3 fatty acids and PPARgamma in cancer. PPAR Res. 2008;2008:358052. doi: 10.1155/2008/358052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deckelbaum RJ, Worgall TS, Seo T. n-3 Fatty acids and gene expression. Am J Clin Nutr. 2006;83:S1520–S1525. doi: 10.1093/ajcn/83.6.1520S. [DOI] [PubMed] [Google Scholar]
- 36.Sakamoto J, Kimura H, Moriyama S, et al. Activation of human peroxisome proliferator-activated receptor (PPAR) subtypes by pioglitazone. Biochem Biophys Res Commun. 2000;278:704–711. doi: 10.1006/bbrc.2000.3868. [DOI] [PubMed] [Google Scholar]
- 37.Orasanu G, Ziouzenkova O, Devchand PR. The peroxisome proliferator-activated receptor–γ agonist pioglitazone represses inflammation in a peroxisome proliferator-activated receptor–α–dependent manner in vitro and in vivo in mice. J Am Coll Cardiol. 2008;52:869–881. doi: 10.1016/j.jacc.2008.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kubota N, Terauchi Y, Kubota T, et al. Pioglitazone ameliorates insulin resistance and diabetes by both adiponectin-dependent and -independent pathways. J Biol Chem. 2006;281:8748–8755. doi: 10.1074/jbc.M505649200. [DOI] [PubMed] [Google Scholar]
- 39.Chen WJ, Yeh SL, Huang PC. Effects of fat emulsions with different fatty acid composition on plasma and hepatic lipids in rats receiving total parenteral nutrition. Clin Nutr. 1996;15:24–28. doi: 10.1016/s0261-5614(96)80257-3. [DOI] [PubMed] [Google Scholar]
- 40.Ntambi JM, Miyazaki M. Regulation of stearoyl-CoA desaturases and role in metabolism. Prog Lipid Res. 2004;43(2):91–104. doi: 10.1016/s0163-7827(03)00039-0. [DOI] [PubMed] [Google Scholar]
- 41.Sekiya M, Yahaqi N, Matsuzaka T, et al. Polyunsaturated fatty acids ameliorate hepatic steratosis in obese mice by SREBP-1 suppression. Hepatology. 2003;38:1529–1539. doi: 10.1016/j.hep.2003.09.028. [DOI] [PubMed] [Google Scholar]
- 42.Javid PJ, Greene AK, Garza J, et al. The route of lipid administration affects parenteral nutrition-induced hepatic steatosis in a mouse model. J Pediatr Surg. 2005;40:1446–1453. doi: 10.1016/j.jpedsurg.2005.05.045. [DOI] [PubMed] [Google Scholar]
- 43.Le HD, Fallon EM, Kalish BT, et al. The effect of varying ratios of docosahexaenoic acid and arachidonic acid in the prevention and reversal of biochemical essential fatty acid deficiency in a murine model. Metabolism. 2013;62:499–508. doi: 10.1016/j.metabol.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alwayn IPJ, Javid PJ, Gura KM, et al. Do polyunsaturated fatty acids ameliorate hepatic steatosis in obese mice by SREPB-1 suppression or by correcting essential fatty acid deficiency? Hepatology. 2004;39:1176–1177. doi: 10.1002/hep.20189. [DOI] [PubMed] [Google Scholar]
- 45.Levy JR, Clore JN, Stevens W. Dietary n-3 polyunsaturated fatty acids decrease hepatic triglycerides in Fischer 344 rats. Hepatology. 2004;39:608–616. doi: 10.1002/hep.20093. [DOI] [PubMed] [Google Scholar]
- 46.Ip E, Farrell GC, Robertson G, et al. Central role of PPAR alpha–dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology. 2003;38:123–132. doi: 10.1053/jhep.2003.50307. [DOI] [PubMed] [Google Scholar]
- 47.Kersten S, Seydoux J, Peters JM, et al. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J Clin Invest. 1999;103:1489–1498. doi: 10.1172/JCI6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bedoucha M, Atzpodien E, Boelsterli UA. Diabetic KKAy mice exhibit increased hepatic PPARgamma1 gene expression and develop hepatic steatosis upon chronic treatment with antidiabetic thiazolidinediones. J Hepatol. 2001;35:17–23. doi: 10.1016/s0168-8278(01)00066-6. [DOI] [PubMed] [Google Scholar]
- 49.Matsusue K, Kusakabe T, Noguchi T, et al. Hepatic steatosis in leptin-deficient mice is promoted by the PPARγ target gene, fat-specific protein 27. Cell Metab. 2008;7:302–311. doi: 10.1016/j.cmet.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Larter CZ, Yeh MM, Cheng J, et al. Activation of peroxisome proliferator-activated receptor alpha by dietary fish oil attenuates steatosis, but does not prevent experimental steatohepatitis because of hepatic lipoperoxide accumulation. J Gastroenterol Hepatol. 2008;23:267–275. doi: 10.1111/j.1440-1746.2007.05157.x. [DOI] [PubMed] [Google Scholar]
- 51.Grefhorst A, Elzinga BM, Voshol PJ, et al. Stimulation of lipogenesis by pharmacological activation of the liver X receptor leads to production of large triglyceride-rich very low density lipoprotein particles. J Biol Chem. 2002;277:34182–34190. doi: 10.1074/jbc.M204887200. [DOI] [PubMed] [Google Scholar]
- 52.Mitro N, Mak PA, Vargas L, et al. The nuclear receptor LXR is a glucose sensor. Nature. 2007;445:219–223. doi: 10.1038/nature05449. [DOI] [PubMed] [Google Scholar]
- 53.Yoshikawa T, Ide T, Shimano H, et al. Cross-talk between peroxisome proliferator-activated receptor (PPAR) alpha and liver X receptor (LXR) in nutritional regulation of fatty acid metabolism, I: PPARs suppress sterol regulatory element binding protein-1c promoter through inhibition of LXR signaling. Mol Endocrinol. 2003;17:1240–1254. doi: 10.1210/me.2002-0190. [DOI] [PubMed] [Google Scholar]
- 54.Clayton PT, Whitfield P, Iyer K. The role of phytosterols in the pathogenesis of liver complications of pediatric parenteral nutrition. Nutrition. 1998;14:158–164. doi: 10.1016/s0899-9007(97)00233-5. [DOI] [PubMed] [Google Scholar]
- 55.Su K, Nadezhda SS, Lu J. The ABCG5 ABCG8 sterol transporter opposes the development of fatty liver disease and loss of glycemic control independent of phytosterol accumulation. J Biol Chem. 2012;287:28564–28575. doi: 10.1074/jbc.M112.360081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Skouroliakou M, Konstantinou D, Koutri K, et al. A double-blind, randomized clinical trial of the effect of omega-3 fatty acids on the oxidative stress of preterm neonates fed through parenteral nutrition. Eur J Clin Nutr. 2010;64:940–947. doi: 10.1038/ejcn.2010.98. [DOI] [PubMed] [Google Scholar]