

Abstract
Despite the high prevalence of chronic gastritis caused by H. pylori, the gastric mucosa has received little investigative attention as a unique immune environment. Here, we analyzed whether retinoic acid (RA), an important homeostatic factor in the small intestinal mucosa, also contributes to gastric immune regulation. We report that human gastric tissue contains high levels of the RA precursor molecule, retinol, and that gastric epithelial cells express both RA biosynthesis genes and RA response genes, indicative of active RA biosynthesis. Moreover, primary gastric epithelial cells cultured in the presence of retinol synthesized RA in vitro and induced RA biosynthesis in co-cultured monocytes through an RA-dependent mechanism, suggesting that gastric epithelial cells may also confer the ability to generate RA on gastric DCs. Indeed, DCs purified from gastric mucosa had similar levels of aldehyde dehydrogenase activity and RA biosynthesis gene expression as small intestinal DCs, although gastric DCs lacked CD103. In H. pylori-infected gastric mucosa, gastric RA biosynthesis gene expression was severely disrupted, which may lead to reduced RA signaling and thus contribute to disease progression. Collectively, our results support a critical role for RA in human gastric immune regulation.
Introduction
Despite the high prevalence of chronic gastritis caused by H. pylori and the serious complications that can arise from H. pylori infection, the gastric mucosa has received little investigative attention as a unique immunological compartment. H. pylori shares traits of both pathogenic and commensal bacteria (1), and we recently showed that soluble mediators present in gastric lamina propria suppress the adaptive response to H. pylori through downregulation of dendritic cell (DC) function (2). Moreover, increasing evidence indicates that the human gastric mucosa also harbors a diverse microflora of true gastric commensal bacteria that does not induce an inflammatory immune response (3, 4). These findings suggest that homeostatic immune mechanisms that support tolerance to colonizing microbes are likely present in human gastric mucosa.
Retinoic acid (RA) is a key homeostatic factor in human small intestine, and RA synthesis by small intestinal CD103+ DCs is considered essential for the induction of T cell expression of gut-homing receptors CCR9 and α4β7 and for the conversion of naïve T cells to FoxP3 regulatory T cells (5–8). Thus, DC RA production is thought to contribute to intestinal tolerance to commensal bacteria and dietary antigens. RA is generated from retinol (ROL, Vitamin A) through a two-step reaction, the first step involving oxidation of ROL to retinal by a retinol dehydrogenase, most importantly RDH10, and the second step involving further oxidation of retinal to all-trans RA by tissue-specific isoforms of retinaldehyde dehydrogenase, RALDH1, RALDH2 and RALDH3 (9). The ability of intestinal DCs to generate RA depends on tissue-specific crosstalk between epithelial cells and DCs. Thus, previous studies in the mouse have shown that intestinal DCs acquire the ability to synthesize RA from adjacent RA-producing intestinal epithelial cells through a positive feedback loop that involves the RALDH2 gene aldh1a2 (6, 10). In murine DCs, a retinoic acid response element (RARE) half-site was recently identified that mediates RA-dependent induction of Aldh1a2 through binding of the RA-RARα/RXRα receptor complex (11). RA biosynthesis in human gastric mucosa has been described previously (12), but the cells that produce RA have not been identified, and whether RA contributes to gastric mucosal immune regulation is not known.
The goal of the present study was to determine whether RA-dependent mechanisms could contribute to gastric homeostasis. Using primary human cells isolated from mucosal tissue samples, we show that both gastric epithelial cells and DCs were as efficient at RA biosynthesis as small intestinal epithelial cells and DCs, although gastric DCs lacked CD103 expression. Moreover, primary human gastric epithelial cells drove RA biosynthesis in co-cultured monocytes through an RA-dependent mechanism, indicating that gastric epithelial cells may confer the ability to synthesize RA on gastric DCs. Collectively, these data suggest a role for RA in human gastric immune regulation.
Results
Gastric epithelial cells synthesize RA
RA synthesis by small intestinal epithelial cells contributes to homeostasis in intestinal mucosa through RA-mediated differentiation of tolerogenic mucosal DCs that, in turn, induce regulatory T cells with mucosal homing capacity (6). To establish whether epithelial cells in human gastric mucosa likewise contribute to gastric immune regulation through RA synthesis, we first analyzed primary gastric epithelial cells for their ability to convert ROL to RA. Normal phase HPLC-analysis revealed that supernatants from gastric epithelial cells cultured for 24 h in the presence of ROL (2 μM) contained 43.4 ± 4.3 pmol/mL of RA, as well as low levels of retinal (Table 1). Notably, the total amount of RA synthesized by the epithelial cells was likely higher than the amount measured, since a proportion of the RA may have been metabolized or degraded prior to the analysis. In contrast, neither RA nor retinal synthesis was detected in epithelial cell cultures without exogenous ROL (Table 1) or where ROL was added to cell culture medium that did not contain epithelial cells (data not shown).
Table 1.
Primary human gastric epithelial cells synthesize RA from retinol
| Retinol (pmol/mL) | Retinal (pmol/mL) | Retinoic acid (pmol/mL) | |
|---|---|---|---|
| Epithelial cells | 44.0 ± 4.9 | n.d.* | n.d.* |
| Epithelial cells + retinol (2 μM) | 1,043.0 ± 97.9 | 6.8 ± 0.4 | 43.4 ± 4.3 |
Retinoids were detected by normal phase HPLC in supernatants of gastric epithelial cell monolayers cultured in the presence of 2 μM retinol for 24 h, n=5.
RA synthesis requires conversion of ROL to retinal by a retinol dehydrogenase and, as a second step, the conversion of retinal to RA by (retin)aldehyde dehydrogenases (7). To determine whether gastric epithelial cells have access to a local source of ROL in vivo, fresh specimens of healthy human gastric mucosa were analyzed for the presence of ROL by HPLC. As a reference, donor-matched samples of small intestinal mucosa, which contains high levels of ROL (13), were analyzed in parallel. Surprisingly, ROL was present in gastric mucosa at significantly higher levels than in intestinal mucosa (224.3 ± 16.3 versus 138.1 ± 14.4 pmol/g, P=0.002, n=7, Fig. 1a). We next analyzed the expression of RDH10, the relevant retinol dehydrogenase (14), and of ALDH1A1, the epithelial cell-predominant isoform of ALDH, in freshly isolated human gastric epithelial cells and donor-matched small intestinal epithelial cells (Fig. 1b) and showed that both RDH10 and ALDH1A1 were expressed at significantly higher levels in gastric compared with intestinal epithelial cells (Fig. 1c). In addition, gastric epithelial cell expression of retinoic acid receptor (RAR)-β, RAR-γ and cellular retinoic acid binding protein (CRABP2)2 was also significantly higher than that of intestinal epithelial cells, whereas no difference was detected for expression of RAR-α (Fig. 2). RAR-α, RAR-β, RAR-γ and CRABP2 are RA target genes that are directly upregulated by RA (15); high expression levels are therefore indicative of high availability of RA in the gastric mucosa. Surprisingly, expression of TGM2, an RA-response gene important in macrophages (16, 17), was higher in intestinal than gastric epithelial cells (Fig. 2). However, the human TGM2 promoter is extremely responsive to a large variety of stimuli (18), including TGF-β, which is present at high levels in intestinal mucosa, but very low levels in gastric mucosa (2), offering a potential explanation for the discordant TGM2 expression in gastric vs. intestinal epithelium. Taken together, our data strongly suggest that human gastric epithelium has a similar capacity to generate RA as small intestinal epithelium.
Figure 1. RA biosynthesis by human gastric epithelial cells.
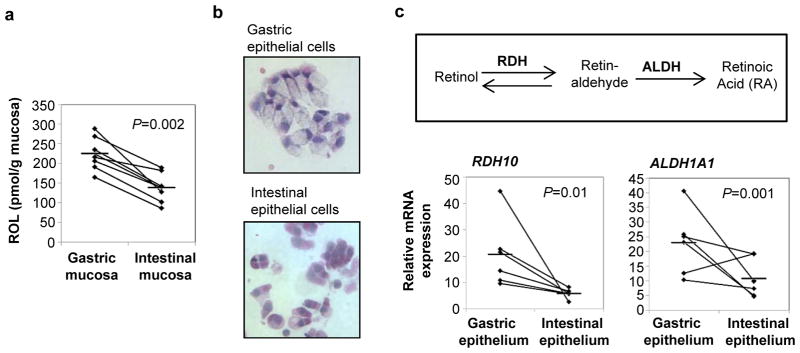
(a) Tissue levels of retinol in human gastric and small intestinal mucosa were measured by normal phase HPLC after lipid extraction (n=7). (b, c) Epithelial cells were recovered from fresh human gastric and intestinal tissue specimens by incubation with EDTA, and expression of genes involved in RA biosynthesis was analyzed by quantitative (q)RT-PCR using the random standard curve method. (b) Cytospins of H&E stained gastric and intestinal epithelial cells, original magnification 20x. (c) Relative gene expression for RDH10 and ALDH1A1. Diamonds: individual samples; bars: mean; lines connect donor-matched samples; n=6. Statistical significance was determined using the Student’s t- test.
Figure 2. Increased expression of RA response genes in primary human gastric compared with intestinal epithelial cells.
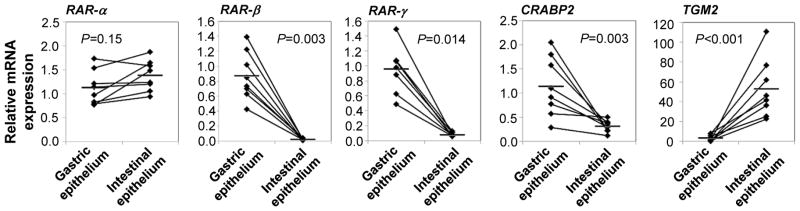
Epithelial cells were recovered from fresh human gastric and intestinal tissue specimens by incubation with EDTA, and gene expression of RAR-α, RAR-β, RAR-γ, CRABP2 and TGM2 was analyzed by qRT-PCR using the random standard curve method. Diamonds: individual samples; bars: mean; lines connect donor-matched samples; n=6–8. Statistical significance was determined using the Student’s t-test.
Retinoid metabolism differs between gastric and intestinal mucosa
The small intestine is generally considered a privileged site for RA synthesis, since intestinal epithelial cells have access to luminal ROL or retinal derived from bile and from intestinal digestion of dietary vitamin A (retinyl esters) and carotenoids (19). Moreover, studies in mice have shown that small intestinal mucosa contains higher levels of ROL than other organs such as spleen and peripheral lymph nodes (13). In contrast, dietary vitamin A is not metabolized to ROL in the human stomach (20), and an additional source of free ROL in the gastric mucosa has not been identified. Therefore, to determine whether the high gastric ROL levels (Fig. 1a) were due to a higher sequestration of ROL in the gastric mucosa, we analyzed the expression of cellular retinol binding proteins (CRBPs) in gastric and intestinal mucosa and isolated epithelial cells. CRBPs are cytoplasmic chaperones that bind and then channel ROL through reactions of RA and retinyl ester synthesis while protecting it from degradation through other pathways (21). CRBP2 facilitates ROL uptake by intestinal epithelial cells and has been implicated in enhancing local retinoid concentrations (19). Our experiments revealed that expression of CRBP2 was high in intestinal mucosa and epithelium but almost undetectable in gastric mucosa and epithelium (P<0.001, Supplementary Fig. 1). Compared to small intestine, gastric expression of CRBP1 was modestly but significantly higher in complete mucosa, but not in isolated epithelial cells (Supplementary Fig. 1). Thus, high local ROL retention by epithelial retinoid chaperones is not likely to be the cause for the high levels of ROL in gastric mucosa.
In the small intestine, free ROL can be either converted to RA or to retinyl esters, which are packaged in chylomicrons, transported to the liver through the lymphatics and then stored in stellate cells (Fig. 3a)(22). Analysis of human gastric and intestinal mucosa revealed significantly higher concentrations of retinyl esters in the small intestinal mucosa compared with the gastric mucosa (Fig. 3b), and gene expression of the major enzyme involved in retinyl ester biosynthesis, lecithin-retinol-acyltransferase (LRAT), also was significantly higher in small intestinal epithelial cells than in gastric epithelial cells (Fig. 3c). These data indicate that ROL in gastric mucosa is not converted to retinyl esters, whereas efficient LRAT-dependent conversion of ROL to retinyl esters in the small intestine may decrease the amount of free ROL in this compartment.
Figure 3. Small intestinal, but not gastric, epithelial cells are involved in biosynthesis of retinyl esters.
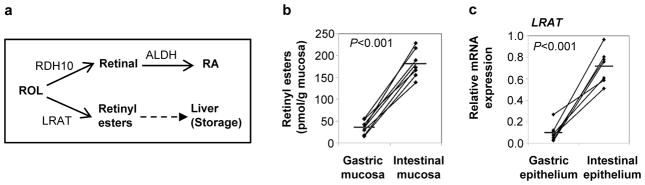
(a) Alternative metabolic pathways for retinol usage by epithelial cells. (b) Tissue levels of retinyl esters in human gastric and small intestinal mucosa were measured by reverse phase HPLC after lipid extraction (n=7). (c) Freshly isolated human gastric and intestinal epithelial cells were analyzed for their expression of LRAT by qRT-PCR using the standard curve method. Diamonds: individual samples; bars: mean; lines connect paired samples; n=7. Statistical significance was determined using the Student’s t-test.
Gastric epithelial cells deliver RA to co-cultured antigen-presenting cells and drive antigen-presenting cell RA biosynthesis
RA-dependent immune regulation in the small intestine involves the induction of RA biosynthesis gene expression in mucosal DCs by epithelial cell-derived RA through a positive feedback loop (6, 10, 11, 23). To determine whether RA-producing gastric epithelial cells could likewise confer the ability to generate RA upon gastric DCs, we performed co-culture experiments with primary gastric epithelial cells and blood monocytes, which are precursor cells for certain DC subsets and which have a relatively low baseline ALDH activity. Gastric epithelial cells were pulsed with ROL (2 μM) for 4 – 6 h to allow epithelial cell RA biosynthesis (Table 1), washed to remove free ROL, and blood monocytes were then added directly to the epithelial monolayers and harvested after overnight incubation (Fig. 4a). Recovered monocytes were >95% pure, as determined by FACS analysis of CD45 and CD11c (data not shown). Monocytes co-cultured with ROL-pulsed epithelial cells showed a >2-fold increased expression of ALDH1A2 compared with monocytes co-cultured with untreated epithelial cells (Fig. 4b, n=4, P=0.02, Kruskal-Wallis test with Bonferroni correction). In contrast, pre-treatment of gastric epithelial cells with retinyl palmitate (RP), a common dietary form of vitamin A that requires hydrolysis to ROL before it can enter the RA biosynthesis pathway, did not lead to increased ALDH1A2 expression in co-cultured monocytes (Fig. 4b). The induction of ALDH1A2 in monocytes co-cultured with ROL-treated epithelial cells was dependent on RA production, since addition of the RAR-receptor antagonist Ro-41-5253 (1 μM) to the co-cultures blocked monocyte ALDH1A2 expression. Moreover, exogenous addition of RA to the co-cultures also resulted in up-regulation of monocyte ALDH1A2 expression (P<0.001). In contrast, monocyte expression of RDH10 and of ALDH1A1, the epithelial cell predominant ALDH isoform was not significantly altered by any of the experimental conditions tested (data not shown).
Figure 4. ROL-treated gastric epithelial cells induce RA biosynthesis in co-cultured antigen-presenting cells.

(a) Experiment overview. (b) Monolayers of primary gastric epithelial cells were treated with ROL (2 μM) or retinyl palmitate (RP; 2 μM) for 4 – 6 h and washed. Blood monocytes were then added at 1 x106/well, together with medium alone, the RAR inhibitor Ro-41-5253 (Ro-41; 1 μM) or RA (50 nM). Monocytes were harvested after 16 h, and gene expression of ALDH1A2 in recovered monocytes was analyzed by qRT-PCR. ITGAX (CD11c) was used as a housekeeping gene. Data are from one representative experiment of four similar experiments. Mean±SEM of duplicates. (c) Monolayers of primary gastric epithelial cells were kept in medium (n=4) or fixed with 70% Ethanol (n=3) and then treated with ROL, ROL and Ro-41-5253, or RA. Blood monocytes were added at 1 x106/well for 6 – 8 h and then were harvested for analysis of RA production using the Sil-15 reporter cell line. Symbols: individual samples; bars: mean and SD, n=3–4; Kruskal-Wallis test with Bonferroni correction.
To determine whether induction of monocyte ALDH1A2 expression by epithelial cell-derived RA results in monocyte RA biosynthesis, we used the RA-reporter cell line Sil-15 (24). Monocytes were co-cultured for 6 – 8 h with gastric epithelial cells plus medium, ROL, ROL + Ro41-5253, or RA, harvested, and plated on top of the Sil-15 reporter cells for analysis of RA release (Fig. 4a). Monocytes cultured in the presence of epithelial cells plus ROL, but not monocytes cultured in the presence of epithelial cells plus ROL + RAR-inhibitor, released significant amounts of RA (Fig. 4c). In contrast, fixed epithelial cells provided with ROL were unable to drive monocyte RA release, indicating that conversion of ROL to RA by the epithelial cells is required to drive monocyte RA biosynthesis (Fig. 4c). Monocyte RA release was also observed in monocytes treated directly with RA in the presence of epithelial cells, consistent with the elevated expression of ALDH1A2 in these cultures. However, RA release by RA-treated monocytes was higher than expected based on the gene expression data, thus, we cannot rule out that additional passive carry-over of RA by the monocytes occurred in these samples, in spite of extensive washing steps. Collectively, our results show that gastric epithelial cells can drive ALDH1A2 expression and RA release in co-cultured monocytes through an RA-dependent pathway and support the notion that gastric epithelial cells provide RA to gastric DCs, thereby promoting DC RA biosynthesis.
Gastric DCs lack C D103 expression, but synthesize RA as efficiently as intestinal DCs
CD103+ DCs, the major DC subset in murine small intestinal lamina propria, have an enhanced capacity to generate RA (6, 25), but whether human gastric DCs express CD103 and synthesize RA is currently unknown. We have previously characterized human gastric DCs as HLA-DRhigh/CD13low cells that induce CD4+ T cell IFN-γ production but only weak proliferative responses (2, 26). Here, we show that freshly isolated human gastric DCs gated as live/lineage/HLA-DR high/CD45+ singlet cells (Fig. 5a) showed subset expression of CD11c (46±11%, n=6), BDCA1 (CD1c, 28±9%), DC-SIGN (CD209, 13±3%) and SIRP-α (CD172a, 27±10%), but expressed only low levels of CXCR1 (4±1%) and CD103 (3±1%) (Fig. 5b). In contrast, similarly gated DCs from human small intestine expressed significantly higher levels of CD103 protein (Fig. 5c, P=0.02, Kruskal-Wallis test with Bonferroni correction). The observed difference in CD103 expression between gastric and intestinal DCs was corroborated by quantitative RT-PCR analysis, which also revealed a significantly lower expression of ITGAE (CD103) mRNA in gastric compared with intestinal DCs (Fig. 5d, P=0.002). Importantly, FACS-purified gastric and intestinal DCs expressed similar levels of the DC-specific transcription factor ZBTB46, confirming that both cell populations both were DCs (27) (Fig. 5e). In comparison, control MoDCs displayed high expression of ITGAE and low expression of ZBTB46, whereas intestinal macrophages showed low expression of both CD103 and ZBTB46.
Figure 5. Gastric DCs lack expression of CD103.

(a) Gating strategy for isolation of gastric and intestinal DCs. (b) Phenotype of isolated gastric DCs. Data are representative of n=6–8. (c) Intestinal DCs express higher levels of CD103 than gastric DCs. Diamonds: individual samples; bars: mean; P=0.02, Kruskal-Wallis test with Bonferroni correction; n=6–8. (d, e) Gastric DCs lack ITGAE (CD103) gene expression but express similar levels of ZBTB46. RNA was isolated from FACS-sorted gastric and intestinal dendritic cells (live HLA-DR+/CD45+/lineage− cells), and expression of (d) ITGAE and (e) ZBTB46 was analyzed by qRT-PCR. Monocyte-derived DCs (MoDCs; n=1) and elutriated intestinal macrophages (Macs, n=1) were analyzed for comparison. Diamonds: individual samples; bars: mean; lines connect paired samples; n=7. Statistical significance was determined using the Student’s t-test.
We next determined whether human gastric DCs had the ability to synthesize RA, using the flow cytometry-based Aldefluor® assay, which measures ALDH activity (28). Human gastric and intestinal DCs exhibited similar levels of ALDH activity (Fig. 6a), whereas Aldefluor® geometric mean fluorescence of blood monocytes was signifcantly lower. Furthermore, FACS-purified gastric and intestinal DCs expressed similar levels of the RA biosynthesis genes RDH10, ALDH1A1 and ALDH1A2 (Fig. 6b). Expression of RA response genes TGM2 and CRABP2 (Fig. 6c) and the RA receptors RAR-α, RAR-β, RAR-γ (Fig. 6d) also was similar or higher in gastric DCs compared with intestinal DCs (Fig. 6c), evidence of autocrine or paracrine RA signaling in gastric DCs. Taken together, these data suggest that gastric DCs are likely equivalent to small intestinal DCs in their ability to generate RA and provide RA to responsive T cells.
Figure 6. Gastric DC synthesize RA as efficiently as intestinal DCs.

(a) ALDH activity of isolated gastric and intestinal DCs and blood monocytes was analyzed with the Aldefluor® assay. Left panels: representative FACS data; grey histograms: DEAB-treated control cells, black outline: untreated samples. Middel panel: Combined data from 5–7 experiments shown as geometric mean fluorescence (normalized to geomean of the DEAB-treated control); Right panel: Combined data from 5–7 experiments shown as % positive cells. (b, c) RNA was isolated from FACS-sorted gastric and intestinal dendritic cells (live HLA-DRhigh/CD45+/lineage cells), and expression of (b) RA biosynthesis genes RDH10 and ALDH1A2, (c) RA response genes and TGM2 and CRABP2 and (d) RA receptors RAR-α, RAR-β and RAR-γ was analyzed by qRT-PCR using the random standard curve method. Diamonds: individual samples; bars: mean; lines connect paired samples; n=5–7. Statistical significance was determined using the Student’s t-test.
In the small intestine, RA-producing DCs direct T cells to the mucosa by inducing CCR9 and α4β7 (29). A previous report suggest that gastric T cell homing is α4β7-dependent (30). To determine whether CCR9 might also play a role in gastric T cell homing, we compared the expression of the cognate ligand for CCR9, CCL25, in gastric and intestinal mucosa. Immunofluorescence and gene expression analysis showed that CCL25 was highly expressed in small intestinal mucosa, particularly on the basolateral side of intestinal crypts and around intestinal blood vessels, but absent from gastric mucosa (Supplemental Fig. 2a, b). Therefore, we focused on α4β7 in our analysis of mucosal homing molecule induction by gastric DCs. Interestingly, SEB-pulsed gastric DCs induced only low levels of α4 and β7 co-expression on naïve CD4+ T cells, whereas SEB-pulsed small intestinal DCs induced high levels of T cell α4β7 expression (Supplemental Fig. 2c, d). This was paralleled by a low induction of proliferation by gastric DCs and a high induction of proliferation by intestinal DCs (data not shown), contrasting with our earlier published report (2). Surprisingly, T cell stimulation with anti-CD3/28-beads also increased the expression of T cell α4β7. Further investigations are necessary to elucidate how gastric DCs induce T cell homing to the stomach.
H. pylori infection disrupts RA biosynthesis in the gastric mucosa
H. pylori infection causes altered gastric epithelial cell and gastric DC function and an infiltration of the gastric mucosa with CD4+ T cells (26, 31). Infiltrating CD4+ T cells in H. pylori infected adults are predominantly Th1 cells (32), and low Treg numbers in the H. pylori infected gastric mucosa are associated with enhanced inflammation and increased frequency of peptic ulcer disease (33–35). Since gastric DC-derived RA is likely involved in the generation of these effector and regulatory T cell populations in H. pylori infection, we investigated whether H. pylori infection modulates gastric RA biosynthesis. RA biosynthesis gene expression in gastric mucosal biopsies obtained from H. pylori-infected subjects and healthy controls was analyzed by quantitative RT-PCR. Our experiments revealed that H. pylori infection was associated with a significantly increased gene expression of Rdh10 and a significantly decreased expression of Aldh1a1 and Aldh1a2 (Fig. 7a). We confirmed these observations in a mouse model of chronic H. pylori infection (Fig. 7b). C57/BL6 mice were gavaged with H. pylori strain SS1 to establish gastric infection, and, two months later, gastric tissue was analyzed for RA biosynthesis gene expression. Similar to our findings using human samples, H. pylori infection in mice was associated with a significant decrease in the expression of aldh1a1 and aldh1a2 (Fig. 7b). Moreover, H. pylori infection in mice also caused a significant decrease in rdh10 expression. These data indicate that chronic H. pylori infection disrupts RA biosynthesis in the gastric mucosa.
Figure 7. Gastric H. pylori infection interferes with retinoid metabolism.

(a) RNA was isolated from human gastric biopsy specimens obtained from healthy or H. pylori-infected subjects. Gene expression of RDH10, ALDH1A1 and ALDH1A2 was determined by quantitative RT-PCR using the random standard curve method, with data normalized to GAPDH expression (n=9–11). (b) 6 – 8 week old female C57/BL6 mice (n=5) were infected with H. pylori (SS1) for 2 months or were mock-infected, stomachs were harvested, and gene expression of rdh10, aldh1a1 and aldh1a2 was determined by qRT-PCR. Data were normalized to hprt expression and analyzed using the ΔΔcT method. (a, b) Dots and squares: individual samples; bars: mean and SD; Statistical significance was determined using the Student’s t-test.
Discussion
The high prevalence of chronic gastritis as a result of H. pylori infection and the often lethal outcome of gastric adenocarcinoma that may develop as a consequence of chronic H. pylori gastritis highlight the importance of studying the gastric mucosa as a unique immunological compartment. Here, we investigated whether RA might contribute to immune regulation in human gastric mucosa. Our data provide evidence that both gastric epithelial cells and gastric DCs generate RA under steady-state conditions, similar to small intestinal epithelial cells and DCs (5, 36). Specifically, we show that (a) gastric mucosa contains ROL, the precursor molecule for RA, thus allowing RA biosynthesis; (b) primary human gastric epithelial cells have the ability to generate RA and confer the ability to generate RA to co-cultured antigen-presenting cells; and (c) gastric DCs have a similar capacity for RA biosynthesis as intestinal DCs, although, surprisingly, gastric DCs lack CD103 expression. These data support the hypothesis that RA-signaling contributes to immune homeostasis in human gastric mucosa.
In addition to the homeostatic and tolerogenic effects of RA in the intestinal immune system, RA has key regulatory functions for cell development, proliferation, survival and metabolism and for tissue morphogenesis (12, 37, 38). Therefore, RA metabolism and tissue concentrations are tightly controlled through auto- and paracrine positive and negative feedback mechanisms (15, 37, 39). Genes that are direct regulatory targets of RA include RAR-α, RAR-β, RAR-γ, CRABP2 and ALDH1A2, where binding of RA to RA receptor heterodimers allows interaction of the RA receptor complex with DNA response elements of the target genes (11, 15, 40). RA biosynthesis by small intestinal DCs is crucial for the induction of gut homing-properties and Foxp3 expression in responder T cells (29, 41, 42), and RA biosynthesis capacity of mucosal DCs is, in turn, dependent on the presence of RA, since small intestinal DCs from Vitamin A (ROL)-deficient mice have significantly decreased ALDH expression and activity (13, 28). In our study, increased expression of the RA biosynthesis genes RDH10 and ALDH1A1 in gastric compared to intestinal epithelial cells was paralleled by a higher expression of the RA response genes RAR-β, RAR-γ and CRABP2, indicating that the gastric epithelial cells were actively generating RA. Importantly, RA released by ROL-treated gastric epithelial cells significantly upregulated ALDH1A2 expression and RA production in co-cultured monocytes through an RA-dependent mechanism, evidence for a positive paracrine feedback loop.
Data from previous reports (10, 43) points to a crucial role for epithelial cells in imprinting DCs with RA biosynthesis capacity. Thus, direct co-culture of murine bone marrow-derived DCs with MODE-K cells and ICC12 cells, two murine small intestinal epithelial cell lines, resulted in an increased DC expression of ALDH1A2 and both ALDH1A1 and ALDH1A2, respectively. Our report is the first to demonstrate that primary human epithelial cells can confer RA-metabolizing activity upon primary human antigen-presenting cells and to show that the mechanism proposed for the imprinting of small intestinal DCs with RA-biosynthesis capacity also may apply to gastric DCs. Notably, several factors other than RA may drive ALDH1A2 expression in antigen-presenting cells, including TLR-engagement (44), PPAR-γ ligands (9, 45), GM-CSF and other cytokines (11, 28). In our study, induction of monocyte ALDH1A2 expression and RA production were dependent on RA signaling, since altered expression was not detected in the presence of the RAR-α inhibitor Ro41-5253. Interestingly, a recent study showed that, in murine DCs, GM-CSF may potentiate RA-induced Aldh1a2 gene expression through co-operative binding of GM-CSF-activated Sp1 and the RAR/RXR complex to the Aldh1a2 promoter (11). Since gastric epithelial cells have been reported to secrete GM-CSF (46), this mechanism may have contributed to the induction of ALDH1A2 expression and RA production in our experiments.
The small intestine was previously considered a privileged site for RA biosynthesis, since tissue levels of ROL are higher in small intestine than in most other tissues including spleen and colon (13). In this context, small intestinal epithelial cells are thought to have access to luminal ROL from dietary sources and bile (6) and express high levels of CRBP2, which may enhance luminal ROL uptake (19). Therefore, we were surprised that gastric mucosa contained significantly higher levels of ROL than small intestinal mucosa. Indeed, gastric mucosa also showed higher epithelial cell expression of RA biosynthesis genes and higher DC and epithelial cell expression of RA response genes, indicative of higher RA levels in stomach compared to small intestine. However, the source of this high ROL concentration in human gastric mucosa remains unclear. First, the gastric mucosa does not have direct access to free ROL from the diet, since dietary Vitamin A sources, i.e., retinyl esters or carotenes, require conversion to free ROL or retinal by small intestinal brush border enzymes or pancreatic enzymes that are not present in the stomach (47). Indeed, in our co-culture experiments, gastric epithelial cells were unable to utilize the retinyl ester retinyl palmitate (RP), but required free ROL in order to drive RA biosynthesis in co-cultured monocytes. Second, CRBP2, which may increase local ROL concentrations (19), is not expressed by gastric epithelial cells, as we have shown. However, our study revealed a significant difference in retinyl ester biosynthesis between gastric and small intestinal mucosa in that ROL is metabolized to retinyl esters by LRAT for subsequent liver transport and storage in the small intestine but not the stomach, thereby reducing ROL levels and the local availability of ROL for RA biosynthesis in the intestinal tissue. This difference between gastric and intestinal epithelial cell ROL metabolism may partly account for the higher levels of ROL observed in gastric compared with intestinal mucosa. Notably, stromal cells also may be involved in retinoid metabolism (48). Our analyses did reveal that total gastric mucosa, but not gastric epithelial cells, expressed high levels of the retinol chaperone CRBP1. Conceivably, CRBP1-expressing gastric stromal cells may elevate local ROL levels by sequestration of serum-derived ROL.
Another unexpected finding in our study was the absence of CD103/integrin α E protein and mRNA expression in gastric DCs, in spite of the similar ALDH activity levels and RA biosynthesis gene expression in donor-matched gastric and small intestinal DCs. In mouse small intestinal lamina propria, the majority of DCs express CD103, and the CD103+ DCs express higher levels of aldh1a2 than CD103− DCs (5, 42). In our hands, although ITGAE mRNA levels were high, surface expression of CD103 on human small intestinal DCs was variable and considerably lower than that reported by Watchmaker et al. (49). CD103 expression is induced by TGF-β (50), raising the possibility that the significantly lower expression of CD103 by gastric DCs compared with small intestinal DCs is due to the lower concentration of TGF-β that we reported for gastric lamina propria compared with intestinal lamina propria (2). Notably, CD103− DCs in murine skin draining lymph nodes also had a high RA biosynthesis activity (51) and, in human intestinal DCs, both CD103− SIRPα+ DCs and CD103+ SIRPα+ had high levels of ALDH activity, whereas ALDH activity in CD103+ SIRPα− DCs was significantly lower (49). Thus, consistent with our observations, expression of CD103 does not seem to be a requirement for RA biosynthesis by DCs.
Our study has yielded novel data indicating that gastric epithelial cells and DCs generate RA in human gastric mucosa, thereby potentially contributing to the generation of tolerogenic responses to gastric commensals. However, H. pylori infection in human adults does not lead to tolerance, with only few FoxP3+ Tregs present in the H. pylori-infected gastric mucosa, as we have shown (33). To explain these findings, our data suggest that RA biosynthesis may be significantly disrupted in the H. pylori-infected stomach, reflected in the decreased expression of ALDH1A1 and ALDH1A2 in both human tissue samples from subjects naturally infected with H. pylori and in a mouse model of chronic H. pylori infection. Consistent with our observations, Matsumoto et al. previously determined that RA formation decreased with increased levels of gastric inflammation (12). Potential mechanisms for H. pylori-induced inhibition of RA biosynthesis include inhibition of RA biosynthesis gene expression by H. pylori or inflammatory mediators, or reduced dietary Vitamin A absorption due to altered gastrointestinal physiology in chronic H. pylori infection, as previously suggested (52). Disruption of RA pathways in the H. pylori-infected gastric mucosa may contribute to disease pathogenesis through at least two potential mechanisms. Lack of RA biosynthesis by DCs could lead to decreased induction of Foxp3+ T regs, thereby allowing increased pro-inflammatory responses and enhanced inflammation. Alternatively, lack of RA could lead to reduced recruitment of protective Th1 cells to the infected mucosa, thereby promoting chronic infection. In support of this hypothesis, H. pylori-infected children who lacked Vitamin A had higher bacterial counts than children with normal Vitamin A intake (53). Moreover, disrupted RA signaling in gastric epithelial cells could contribute to the development of gastric adenocarcinoma, since RA restricts cell cycle progression and proliferation and promotes cell differentiation, important anti-tumor mechanisms (38). Disruption of retinoid signaling including reduced ALDH activity is a common finding in many types of cancer (38). On the other hand, anti-tumor effects of all-trans RA treatment on human gastric cancer cells have been demonstrated both in vitro and in vivo (54, 55).
One crucial question that is the subject of ongoing investigations in our laboratory is whether RA-producing gastric DCs can direct lymphocyte homing to the gastric lamina propria. CCL25, the cognate ligand for CCR9, was not expressed in gastric mucosa, and our preliminary results did not support the hypothesis that gastric DCs induce T cell α4β7 expression. Thus, the mechanisms by which gastric DCs might induce T cell homing to the gastric mucosa remain to be elucidated.
In summary, we show that gastric epithelial cells are more effective than small intestinal epithelial cells in the ability to generate RA. Gastric DCs likewise are able to generate RA, likely due to induction of DC ALDH1A2 expression by epithelial cell-derived RA. These results suggest that RA biosynthesis by gastric epithelial cells and DCs may contribute to gastric homeostasis under steady state conditions, however, H. pylori infection may disrupt RA-dependent mechanisms that support gastric homeostasis.
Methods
Cells and tissues
Gastric tissue specimens from corpus and antrum and small intestinal tissue specimens from proximal jejunum were obtained with Institutional Review Board (IRB) approval and informed consent from non-H. pylori-infected adult subjects undergoing elective gastric bypass for obesity or diagnostic esophago-gastro-duodenoscopy at the University of Alabama at Birmingham (40 donors). One surgical sample (approximately 1 g of gastric mucosa) or 10 – 20 biopsies were obtained from each donor; data points in the figures represent cells derived from one subject. Additional gastric biopsy specimens from H. pylori-infected and non-infected subjects were obtained with local IRB approval from adult subjects with abdominal symptoms residing in Santiago, Chile (20 donors). H. pylori status was determined by rapid urease test and microscopic evaluation, and a study subject was judged colonized with H. pylori if one or both tests were positive for the bacteria. Exclusion criteria included (a) use of antibiotics, antacid, H2-blocker, proton-pump inhibitor, bismuth compound, non-steroidal anti-inflammatory drug or immunosuppressive agent during the two weeks prior to endoscopy; and (b) stool examination positive for ova or parasites.
Gastric epithelial cell cultures were established as previously described (56, 57). Briefly, tissue was minced with a scalpel blade and digested for 1 h at 37°C, 200 rpm, in RPMI1640 with collagenase (0.5 FALGPA units/mL; Sigma, St. Louis, MO), DNAse (0.2 mg/mL; Sigma) and BSA (0.3%; Fisher, Fair Lawn, NJ). Recovered cells were suspended in F12K medium containing 10% FBS, amphotericin (125 ng/mL), penicillin (100 U/mL), streptomycin (100 μg/mL) and gentamycin (50 μg/mL), plated on collagen-I-coated plates (Biocoat, Becton Dickinson, San Jose, CA), and non-adherent cells were removed after 18 h of culture (37°C; 5% CO2). Cultures were maintained for up to 5 days.
To obtain gastric and small intestinal epithelial cells for PCR analysis, tissue from bypass patients was dissected to remove the serosa and muscularis layers and then washed twice for 20 min at 37° C, 200 rpm, in HBSS with DTT (0.2 mg/mL, Sigma, St. Louis, MO) to remove adherent mucus. Epithelial cells then were isolated by incubation (30 min, 37° C, 200 rpm) in HBSS/DTT supplemented with EDTA (1.25 mM).
To obtain gastric and intestinal DCs (DCs), mucosal tissue was subjected to three rounds of EDTA treatment and then digested with collagenase solution, as described previously (26). Gastric and intestinal DCs were pre-enriched for HLA-DR+ cells by MACS (Miltenyi Biotec, Auburn, CA), and viable (propidiumiodide-negative) CD45pos/lineageneg/HLA-DRhigh DCs were purified by FACS sorting on a FACSAria II sorter (Becton Dickinson).
Animal studies
6–8 week old female C57/BL6 mice (Jackson Laboratory, Bar Harbor, ME) housed in the animal maintenance facility at the University of Michigan Health System were orally gavaged with H. pylori (strain SS1, 109 bacteria/dose) 3 times over 1 week, as described previously (58). After 2 months, the mice were euthanized, and stomachs were harvested and processed for RNA isolation. All animal experiments were approved by the University Committee on Use and Care of Animals at the University of Michigan.
Quantitative RT-PCR analysis
For gene expression analysis in human samples, RNA was isolated from epithelial cells or dissected mucosa using the RNeasy Minikit (Qiagen, Valencia, CA). Alternatively, to obtain RNA from gastric and intestinal DCs, cells were directly sorted into Trizol® LS (Invitrogen, Carlsbad, CA), and RNA was obtained using the Direct-zol™ RNA Mini Prep Kit (Zymo Research, Irvine, CA). cDNA was generated with iScript Reverse Transcriptase (Biorad, Hercules, CA). ALDH1A1 (Hs00180254_m1),3ALDH1A2 (Hs00180254_m1), ITGAX (CD11c, Hs01015064_m1), ITGAE (CD103, Hs01025372_m1), CCL25 (Hs_00608373_m1), CRBP2 (Hs00188160_m1), RAR-α (Hs00940446_m1), TGM2 (Hs00190278_m1), ZBTB46 (Hs01008168_m1), 18s rRNA (Ref. Seq. X03205.1), and GAPDH (Ref. Seq. NM_002046.3) were amplified for 40 cycles (15 sec 95°C, 60 sec 60°C) in 25 PL reactions containing TaqMan Universal PCR Master Mix and appropriate primer probe sets, all from Applied Biosystems, Foster City, CA. RDH10, RAR-β, CRBP1, CRABP2 and LRAT, were amplified for 45 cycles (15 sec 94°C, 15 sec 62°C, 15 sec 72°C) in 25 PL reactions containing SYBR® Green ER™ qPCR Super Mix (Life Technologies, Carlsbad, CA). Sequences of primers used for SYBR green reactions are available upon request. Real-time PCR reactions were run on a Chromo4 PCR system (Biorad), and analyzed with Opticon Monitor™ software, version 3.1. Gene expression was calculated using the standard curve method or the Pfaffl method (59), with data normalized to the geometric mean of GAPDH and 18s rRNA, unless stated otherwise. ITGAX gene expression was used to normalize monocyte gene expression in epithelial cell-monocyte co-culture experiments in order to exclude contaminating epithelial cells from the analysis; CD11c expression was stable across different treatments. Gene expression analysis in murine gastric samples was performed with SYBR Green as described previously (58), with the house keeping gene Hypoxanthine-guanine phosphoribosyltransferase (HPRT) used as an internal control and gene expression calculated using the Δ ΔcT method. Primer sequences are available upon request.
Retinoid detection by HPLC
Tissue samples and culture supernatants for retinoid analysis were prepared under red light, wrapped in aluminum foil, frozen on dry ice and stored at −80° C until further processing. Retinoids were extracted from cell culture medium or tissue homogenates as described before (60). The organic phase was evaporated under a stream of nitrogen. Normal or reverse phase HPLC were used as indicated in the respective figure legends. For normal phase HPLC analysis, the dry residue was dissolved in a mobile phase consisting of hexane/ethyl acetate/acetic acid (95:4.975:0.025, v/v/v). Samples were separated on Spherisorb S3W column (4.6 mm×100 mm) (Waters Corp., Milford, MA) at a flow rate of 0.7 mL/min using Waters Alliance 2695 HPLC system equipped with a Waters 2996 photodiode array. For reverse phase HPLC analysis, samples were dissolved in acetonitrile : dichloromethane (70:30, v/v) and separated on a SUPELCOSIL™ Suplex™ pKb-100 column (Sigma-Aldrich) with a mobile phase consisting of a gradient of solvents A (acetonitrile : 2% ammonium acetate : glacial acetic acid : methanol as 79:16:3:2) and B (acetonitrile : dichloromethane as 90:10) as follows: 0–24 min 100% A, 25–54 min 100% B, 55–60 min 100% B. Peaks were identified by comparison to retention times of retinoid standards and evaluation of wavelength maxima and quantified by comparing their peak areas to a calibration curve constructed from peak areas of a series of standards.
Retinoid treatment of gastric epithelial cells and co-culture experiments
ROL, RA and RP were obtained from Sigma, and stock solutions were maintained in 100% ethanol (ROL), DMSO (RA), or tetrahydrofurane (RP), respectively, at −80° C. All reagents were pre-diluted in FBS and vortexed vigorously before addition to cell culture media. Integrity and concentration of the reagents were checked periodically by comparing UV-Vis absorption spectra, which were obtained using a Nanodrop II spectrophotometer (Thermo Scientific, Waltham, MA), to reference spectra. All retinoid treatments were performed under red light illumination to avoid disintegration of the reagents. To analyze RA synthesis by gastric epithelial cells, 3-day old gastric epithelial cell monolayers were treated with 2 PM ROL overnight (16 h). RA, ROL and retinal concentrations in cell-free culture supernatants were analyzed by normal phase HPLC as described above. Co-culture experiments of primary human gastric epithelial cells and blood monocytes were performed to investigate the influence of epithelial cell-derived RA on antigen-presenting cell RA biosynthesis. Blood monocytes were isolated from PBMCs using anti-CD14 MACS beads (Miltenyi-Biotec, Auburn, CA). Live gastric epithelial cell monolayers or dead control monolayers prepared by 30 sec fixation with 70% ethanol were treated with 2 μM of ROL or RP or were left untreated. Monocytes were added at 1x106/well, either immediately or after 4 – 6 h pretreatment of the epithelial cells with retinoids. Control co-culture wells contained RA (50 nM) or the RA receptor inhibitor Ro41-5253 (1 μM, Enzo, Life Sciences, Farmingdale, NY). After co-culture, monocytes were recovered by vigorous pipetting and processed for further analysis.
RA bioassay
The Sil-15 cell line (24), kindly provided by Dr. Michael Wagner, SUNY Downstate Medical Center, Brooklyn, NY, was used to assess RA production by monocytes recovered from epithelial cell co-culture experiments. Sil-15 cells were grown on gelatin-coated 96-well plates (BD Labware, Bedford, MA) in DMEM supplemented with 20% FBS and 1% G418 (Mediatech, Inc., Manassas, VA). Cellular populations to be tested were added at 100,000/well to confluent monolayers of Sil-15 cells. After overnight incubation, supernatants were removed, and Sil-15 cells were lysed by three freeze-thaw cycles in PBS. β-Galactosidase activity in Sil-15 lysates was then determined using X-Gal (1 mg/mL; Thermo Scientific, Waltham, MA) in developer solution made of 5 mM K3[Fe(CN)6], 5 mM K4[Fe(CN)6] and 2 mM MgCl2 in PBS, and color development was measured at 630 nm. RA production was calculated as RA equivalents based on a standard curve generated with known amounts of RA (detection range 50 pM – 100 nM).
DC–T cell co-cultures
DC-T cell co-cultures were established using FACS-purified gastric and intestinal DCs that were pulsed with SEB (1 μg/mL, Toxin Technology, Sarasota, FL) and autologous naïve CD4+ T cells, as described previously (2). T cell proliferation was determined using the CFSE dilution assay.
Immunofluorescence and flow cytometry
Antibodies directed against the following human surface proteins were used: HLA-DR (L243), CD11c (B-ly6), DC-SIGN (DCN46),3CXCR1 (5A12), β7 (FIB504), α4 (9F10), cytokeratin (CAM5.2), CD45 (2D1, all from Becton Dickinson, San Jose, CA); BDCA-1/CD1c (L161), SIRP-α (SE5A5, both from Biolegend, San Diego, CA); CCL25 (rabbit polyclonal, from AbD Serotec, Raleigh, NC); and CD103 (B-Ly7, eBioscience, San Diego, CA). Our lineage cocktail contained antibodies to CD3, CD14, CD19, and CD20 (all from Becton Dickinson). A LIVE/DEAD® yellow dye (Life Technologies, Carlsbad, CA) or propidiumiodide were used to exclude dead cell populations. The capacity of live cells to convert retinaldehyde to RA was measured using the flow cytometry-based Aldefluor™ assay according to the manufacturer’s protocol (Stemcell Technologies, Vancouver, BC, Canada). The Aldefluor™ assay employs a fluorescent non-toxic aminoacetaldehyde, which freely diffuses into intact and viable cells and is converted by ALDH into an aminoacetate that is retained inside the cells. An LSRII flow cytometer (Becton Dickinson) and FlowJo 7.6.5 software (TreeStar, Ashland, OR) were used for analysis. Immunofluorescence staining of frozen gastric and intestinal sections was performed as described previously (26).
Statistical analysis
Data were analysed using Microsoft® Excel 2003 and Analyse-it® for Excel, version 1.73, or GraphPad Prism 6.04. Results are presented as mean ±SEM. Differences between values were analyzed for statistical significance by the two-tailed Student’s t-test, unless stated otherwise. Differences were considered significant at P<0.05.
Supplementary Material
Acknowledgments
Our sincerest thanks to Dr. Natalia Kedishvili and Dr. Olga Belyaeva, Department of Biochemistry and Molecular Genetics, University of Alabama at Birmingham, for performing HPLC analyses, providing critical feedback on our study and for sharing their knowledge on retinoid metabolism, function and detection with us. We would also like to thank Marion Spell for performing FACSAria cell sorts, Dr. Michael Wagner for providing the Sil-15 RA reporter cells, and Dr. Heidi J. Nick for helpful discussions. Funding for our study was provided by the National Institutes of Health grants: DK-54495 (PDS); DK-084063 (PDS); AI-083539 (LES); DK-097144 (DB); DK-087708 (JYK); the UAB Autoimmunity, Immunology and Transplantation Steering Committee Pilot Program (DB); the UAB CCTS Pilot Program UL1 TR000165 (DB); the Broad Medical Research Program of the Broad Foundation (LES); the Crohn’s and Colitis Foundation of American (LES) and the Research Service of the Veterans Administration (PDS). Services from the Analytic and Preparative Cytometry Facility (P30 AR48311) and the DDRDC Human Cell/Tissue Core (DK-64400) were used for our study.
Footnotes
Supplementary material is linked to the online version of the paper at http://www.nature.com/mi.
Disclosure The authors declared no conflicts of interest.
References
- 1.Atherton JC, Blaser MJ. Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. J Clin Invest. Sep;119(9):2475–87. doi: 10.1172/JCI38605. Epub 2009/09/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bimczok D, Grams JM, Stahl RD, Waites KB, Smythies LE, Smith PD. Stromal regulation of human gastric dendritic cells restricts the Th1 response to Helicobacter pylori. Gastroenterology. 2011;141(3):929–38. doi: 10.1053/j.gastro.2011.06.006. Epub 2011/06/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang I, Nell S, Suerbaum S. Survival in hostile territory: the microbiota of the stomach. FEMS Microbiol Rev. 2013;37(5):736–61. doi: 10.1111/1574-6976.12027. Epub 2013/06/25. [DOI] [PubMed] [Google Scholar]
- 4.Bik EM, et al. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci U S A. 2006;103(3):732–7. doi: 10.1073/pnas.0506655103. Epub 2006/01/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iwata M. Retinoic acid production by intestinal dendritic cells and its role in T-cell trafficking. Semin Immunol. 2009;21(1):8–13. doi: 10.1016/j.smim.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 6.Agace WW, Persson EK. How vitamin A metabolizing dendritic cells are generated in the gut mucosa. Trends Immunol. 2012;33(1):42–8. doi: 10.1016/j.it.2011.10.001. Epub 2011/11/15. [DOI] [PubMed] [Google Scholar]
- 7.Pino-Lagos K, Benson MJ, Noelle RJ. Retinoic acid in the immune system. Ann NYAcad Sci. 2008;1143:170–87. doi: 10.1196/annals.1443.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.del Rio ML, Bernhardt G, Rodriguez-Barbosa JI, Forster R. Development and functional specialization of CD103+ dendritic cells. Immunol Rev. 2010;234(1):268–81. doi: 10.1111/j.0105-2896.2009.00874.x. Epub 2010/03/03. [DOI] [PubMed] [Google Scholar]
- 9.Gyongyosi A, et al. RDH10, RALDH2, and CRABP2 are required components of PPARgamma-directed ATRA synthesis and signaling in human dendritic cells. J Lipid Res. 2013;54(9):2458–74. doi: 10.1194/jlr.M038984. Epub 2013/07/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edele F, et al. Cutting edge: instructive role of peripheral tissue cells in the imprinting of T cell homing receptor patterns. J Immunol. 2008;181(6):3745–9. doi: 10.4049/jimmunol.181.6.3745. [DOI] [PubMed] [Google Scholar]
- 11.Ohoka Y, Yokota-Nakatsuma A, Maeda N, Takeuchi H, Iwata M. Retinoic acid and GM-CSF coordinately induce retinal dehydrogenase 2 (RALDH2) expression through cooperation between the RAR/RXR complex and Sp1 in dendritic cells. PLoS One. 2014;9(5):e96512. doi: 10.1371/journal.pone.0096512. Epub 2014/05/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsumoto M, Yokoyama H, Suzuki H, Shiraishi-Yokoyama H, Hibi T. Retinoic acid formation from retinol in the human gastric mucosa: role of class IV alcohol dehydrogenase and its relevance to morphological changes. AmJ Physiol Gastrointest Liver Physiol. 2005;289(3):G429–G33. doi: 10.1152/ajpgi.00502.2004. [DOI] [PubMed] [Google Scholar]
- 13.Jaensson-Gyllenback E, et al. Bile retinoids imprint intestinal CD103+ dendritic cells with the ability to generate gut-tropic T cells. Mucosal Immunol. 2011;4(4):438–47. doi: 10.1038/mi.2010.91. Epub 2011/02/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee SA, Belyaeva OV, Wu L, Kedishvili NY. Retinol dehydrogenase 10 but not retinol/sterol dehydrogenase(s) regulates the expression of retinoic acid-responsive genes in human transgenic skin raft culture. J Biol Chem. 2011;286(15):13550–60. doi: 10.1074/jbc.M110.181065. Epub 2011/02/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balmer JE, Blomhoff R. Gene expression regulation by retinoic acid. J Lipid Res. 2002;43(11):1773–808. doi: 10.1194/jlr.r100015-jlr200. Epub 2002/10/29. [DOI] [PubMed] [Google Scholar]
- 16.Yan ZH, Noonan S, Nagy L, Davies PJ, Stein JP. Retinoic acid induction of the tissue transglutaminase promoter is mediated by a novel response element. Mol Cell Endocrinol. 1996;120(2):203–12. doi: 10.1016/0303-7207(96)03826-9. Epub 1996/07/01. [DOI] [PubMed] [Google Scholar]
- 17.Chiocca EA, Davies PJ, Stein JP. The molecular basis of retinoic acid action. Transcriptional regulation of tissue transglutaminase gene expression in macrophages. J Biol Chem. 1988;263(23):11584–9. Epub 1988/08/15. [PubMed] [Google Scholar]
- 18.Gundemir S, Colak G, Tucholski J, Johnson GV. Transglutaminase 2: a molecular Swiss army knife. Biochim Biophys Acta. 2012;1823(2):406–19. doi: 10.1016/j.bbamcr.2011.09.012. Epub 2011/10/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McDonald KG, et al. Epithelial expression of the cytosolic retinoid chaperone cellular retinol binding protein II is essential for in vivo imprinting of local gut dendritic cells by lumenal retinoids. Am J Pathol. 2012;180(3):984–97. doi: 10.1016/j.ajpath.2011.11.009. Epub 2012/01/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Borel P, et al. Processing of vitamin A and E in the human gastrointestinal tract. Am J Physiol Gastrointest Liver Physiol. 2001;280(1):G95–G103. doi: 10.1152/ajpgi.2001.280.1.G95. [DOI] [PubMed] [Google Scholar]
- 21.Napoli JL. Biosynthesis and metabolism of retinoic acid: roles of CRBP and CRABP in retinoic acid: roles of CRBP and CRABP in retinoic acid homeostasis. J Nutr. 1993;123(2 Suppl):362–6. doi: 10.1093/jn/123.suppl_2.362. Epub 1993/02/01. [DOI] [PubMed] [Google Scholar]
- 22.O’Byrne SM, Blaner WS. Retinol and retinyl esters: biochemistry and physiology. J Lipid Res. 2013;54(7):1731–43. doi: 10.1194/jlr.R037648. Epub 2013/04/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iliev ID, et al. Human intestinal epithelial cells promote the differentiation of tolerogenic dendritic cells. Gut. 2009;58(11):1481–9. doi: 10.1136/gut.2008.175166. Epub 2009/07/03. [DOI] [PubMed] [Google Scholar]
- 24.Wagner M, Han B, Jessell TM. Regional differences in retinoid release from embryonic neural tissue detected by an in vitro reporter assay. Development. 1992;116(1):55–66. doi: 10.1242/dev.116.1.55. Epub 1992/09/01. [DOI] [PubMed] [Google Scholar]
- 25.Uematsu S, et al. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nat Immunol. 2008;9(7):769–76. doi: 10.1038/ni.1622. [DOI] [PubMed] [Google Scholar]
- 26.Bimczok D, et al. Human primary gastric dendritic cells induce a Th1 response to H. pylori. Mucosal Immunol. 2010;3(3):260–9. doi: 10.1038/mi.2010.10. Epub 2010/03/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meredith MM, et al. Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. J Exp Med. 2012;209(6):1153–65. doi: 10.1084/jem.20112675. Epub 2012/05/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yokota A, et al. GM-CSF and IL-4 synergistically trigger dendritic cells to acquire retinoic acid-producing capacity. Int Immunol. 2009;21(4):361–77. doi: 10.1093/intimm/dxp003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. Retinoic acid imprints gut-homing specificity on T cells. Immunity. 2004;21(4):527–38. doi: 10.1016/j.immuni.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 30.Michetti M, Kelly CP, Kraehenbuhl JP, Bouzourene H, Michetti P. Gastric mucosal alpha(4)beta(7)-integrin-positive CD4 T lymphocytes and immune protection against helicobacter infection in mice. Gastroenterology. 2000;119(1):109–18. doi: 10.1053/gast.2000.8548. Epub 2000/07/13. [DOI] [PubMed] [Google Scholar]
- 31.Segal ED, Lange C, Covacci A, Tompkins LS, Falkow S. Induction of host signal transduction pathways by Helicobacter pylori. Proc Natl Acad Sci USA. 1997;94(14):7595–9. doi: 10.1073/pnas.94.14.7595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bamford KB, et al. Lymphocytes in the human gastric mucosa during Helicobacter pylori have a T helper cell 1 phenotype. Gastroenterology. 1998;114(3):482–92. doi: 10.1016/s0016-5085(98)70531-1. [DOI] [PubMed] [Google Scholar]
- 33.Harris PR, et al. Helicobacter pylori gastritis in children is associated with a regulatory T-cell response. Gastroenterology. 2008;134(2):491–9. doi: 10.1053/j.gastro.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 34.Robinson K, et al. Helicobacter pylori-induced peptic ulcer disease is associated with inadequate regulatory T cell responses. Gut. 2008;57(10):1375–85. doi: 10.1136/gut.2007.137539. [DOI] [PubMed] [Google Scholar]
- 35.Serrano C, et al. Downregulated Th17 responses are associated with reduced gastritis in Helicobacter pylori-infected children. [Epub 2013/01/10];Mucosal Immunol. 2013 doi: 10.1038/mi.2012.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lampen A, Meyer S, Arnhold T, Nau H. Metabolism of vitamin A and its active metabolite all-trans-retinoic acid in small intestinal enterocytes. J Pharmacol Exp Ther. 2000;295(3):979–85. Epub 2000/11/18. [PubMed] [Google Scholar]
- 37.McGrane MM. Vitamin A regulation of gene expression: molecular mechanism of a prototype gene. J Nutr Biochem. 2007;18(8):497–508. doi: 10.1016/j.jnutbio.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 38.Tang XH, Gudas LJ. Retinoids, retinoic acid receptors, and cancer. Annu Rev Pathol. 2011;6:345–64. doi: 10.1146/annurev-pathol-011110-130303. Epub 2010/11/16. [DOI] [PubMed] [Google Scholar]
- 39.Strate I, Min TH, Iliev D, Pera EM. Retinol dehydrogenase 10 is a feedback regulator of retinoic acid signalling during axis formation and patterning of the central nervous system. Development. 2009;136(3):461–72. doi: 10.1242/dev.024901. Epub 2009/01/15. [DOI] [PubMed] [Google Scholar]
- 40.Pinaire J, et al. Identification of a retinoid receptor response element in the human aldehyde dehydrogenase-2 promoter. Alcohol Clin Exp Res. 2003;27(12):1860–6. doi: 10.1097/01.ALC.0000100941.86227.4F. Epub 2003/12/24. [DOI] [PubMed] [Google Scholar]
- 41.Sun CM, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. 2007;204(8):1775–85. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coombes JL, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204(8):1757–64. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iliev ID, Mileti E, Matteoli G, Chieppa M, Rescigno M. Intestinal epithelial cells promote colitis-protective regulatory T-cell differentiation through dendritic cell conditioning. Mucosal Immunol. 2009 doi: 10.1038/mi.2009.13. [DOI] [PubMed] [Google Scholar]
- 44.Manicassamy S, et al. Toll-like receptor 2-dependent induction of vitamin A-metabolizing enzymes in dendritic cells promotes T regulatory responses and inhibits autoimmunity. Nat Med. 2009;15(4):401–9. doi: 10.1038/nm.1925. Epub 2009/03/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Szatmari I, et al. PPARgamma controls CD1d expression by turning on retinoic acid synthesis in developing human dendritic cells. J Exp Med. 2006;203(10):2351–62. doi: 10.1084/jem.20060141. Epub 2006/09/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nagy TA, et al. Helicobacter pylori induction of eosinophil migration is mediated by the cag pathogenicity island via microbial-epithelial interactions. Am J Pathol. 2011;178(4):1448–52. doi: 10.1016/j.ajpath.2010.12.018. Epub 2011/03/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harrison EH. Mechanisms of digestion and absorption of dietary vitamin A. Annu Rev Nutr. 2005;25:87–103. doi: 10.1146/annurev.nutr.25.050304.092614. [DOI] [PubMed] [Google Scholar]
- 48.Cassani B, Villablanca EJ, De Calisto J, Wang S, Mora JR. Vitamin A and immune regulation: role of retinoic acid in gut-associated dendritic cell education, immune protection and tolerance. Mol Aspects Med. 2012;33(1):63–76. doi: 10.1016/j.mam.2011.11.001. Epub 2011/11/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Watchmaker PB, et al. Comparative transcriptional and functional profiling defines conserved programs of intestinal DC differentiation in humans and mice. Nat Immunol. 2014;15(1):98–108. doi: 10.1038/ni.2768. Epub 2013/12/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robinson PW, Green SJ, Carter C, Coadwell J, Kilshaw PJ. Studies on transcriptional regulation of the mucosal T-cell integrin alphaEbeta7 (CD103) Immunology. 2001;103(2):146–54. doi: 10.1046/j.1365-2567.2001.01232.x. Epub 2001/06/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guilliams M, et al. Skin-draining lymph nodes contain dermis-derived CD103(−) dendritic cells that constitutively produce retinoic acid and induce Foxp3(+) regulatory T cells. Blood. 2010;115(10):1958–68. doi: 10.1182/blood-2009-09-245274. Epub 2010/01/14. [DOI] [PubMed] [Google Scholar]
- 52.Epplein M, et al. Helicobacter pylori prevalence and circulating micronutrient levels in a low-income United States population. Cancer Prev Res (Phila) 2011;4(6):871–8. doi: 10.1158/1940-6207.CAPR-10-0398. Epub 2011/03/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ozturk Y, Buyukgebiz B, Arslan N, Ozer E, Lebe B. Serum vitamin A and total carotene concentrations in well-nourished children with Helicobacter pylori infection. J Pediatr Gastroenterol Nutr. 2003;36(4):502–4. doi: 10.1097/00005176-200304000-00018. Epub 2003/03/27. [DOI] [PubMed] [Google Scholar]
- 54.Hu KW, Chen FH, Ge JF, Cao LY, Li H. Retinoid receptors in gastric cancer: expression and influence on prognosis. Asian Pac J Cancer Prev. 2012;13(5):1809–17. doi: 10.7314/apjcp.2012.13.5.1809. Epub 2012/08/21. [DOI] [PubMed] [Google Scholar]
- 55.Kim JH, Choi YK, Kwon HJ, Yang HK, Choi JH, Kim DY. Downregulation of gelsolin and retinoic acid receptor beta expression in gastric cancer tissues through histone deacetylase 1. J Gastroenterol Hepatol. 2004;19(2):218–24. doi: 10.1111/j.1440-1746.2004.03336.x. Epub 2004/01/21. [DOI] [PubMed] [Google Scholar]
- 56.Smoot DT, Sewchand J, Young K, Desbordes BC, Allen CR, Naab T. A method for establishing primary cultures of human gastric epithelial cells. Methods Cell Sci. 2000;22(2–3):133–6. doi: 10.1023/a:1009846624044. [DOI] [PubMed] [Google Scholar]
- 57.Bimczok D, et al. Helicobacter pylori infection inhibits phagocyte clearance of apoptotic gastric epithelial cells. J Immunol. 2013;190(12):6626–34. doi: 10.4049/jimmunol.1203330. Epub 2013/05/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun X, et al. TLR2 mediates Helicobacter pylori-induced tolerogenic immune response in mice. PLoS One. 2013;8(9):e74595. doi: 10.1371/journal.pone.0074595. Epub 2013/09/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Napoli JL, Horst RL. Quantitative analyses of naturally occurring retinoids. Methods Mol Biol. 1998;89:29–40. doi: 10.1385/0-89603-438-0:29. Epub 1998/07/17. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.