

Graphical abstract
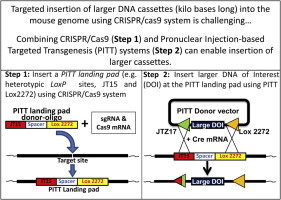
Abbreviations: Cas9, CRISPR-associated 9; Cre-PITT, Cre-Lox system-based PITT; CRISPR, clustered regularly interspaced short palindromic repeats; ES, embryonic stem cells; HDR, homology-directed repair; PI, pronuclear injection; PITT, pronuclear injection-based targeted transgenesis; RMCE, recombinase-mediated cassette exchange
Keywords: CRISPR/Cas9, ROSA26, Pronuclear, Targeted transgenesis, PITT/RMCE, Gene editing
Highlights
-
•
A Cas9 target that cleaves precisely at the ROSA26 original provirus integration site.
-
•
Combining CRISPR/Cas9 with the PITT system can enable insertion of larger DNAs.
-
•
Anomalous and mosaic sequence insertions can occur with the CRISPR/Cas9 system.
Abstract
Targeted transgenic mouse models, where an exogenous gene is inserted into a specified genomic locus to achieve its stable and reliable expression, have been widely used in biomedical research. However, the available methodologies for targeted insertion of sequences require many laborious steps that involve the use of embryonic stem (ES) cells. We recently developed Pronuclear Injection-based Targeted Transgenesis (PITT), a method that uses a recombinase-mediated cassette exchange (RMCE) to enable insertion of sequences at a predetermined genomic locus, such as ROSA26. The PITT technique uses fertilized eggs (instead of ES cells) collected from ‘seed mice’ that contain the RMCE landing pad. The PITT method can rapidly generate reliable targeted transgenic mice; it requires a seed mouse, which in our previous study was generated using ES cell targeting approaches. Here, we demonstrate that seed mice containing the RMCE landing pad can be developed rapidly by using the CRISPR/Cas9 system. One of the CRISPR targets tested in this study enabled the insertion of sequences precisely at the original ROSA26 provirus integration site. We anticipate that using a similar approach, PITT landing pad sequences can be rapidly and precisely inserted at other genomic loci to develop an array of PITT tools. This two-step strategy combines the best features of the two newer technologies—rapid creation of PITT landing pads using the CRISPR/Cas9 system and efficient and precise insertion of larger cassettes at the landing pads using PITT. This study also revealed that anomalous and mosaic sequence insertions can occur with the CRISPR/Cas9 system.
1. Introduction
To date, the transgenic mice generation process largely uses a method in which the DNA is injected into fertilized eggs (pronuclei). Even though the pronuclear injection (PI) method is quite efficient in generating multiple transgenic lines, the integration of the DNA at random places in the genome poses many problems—the transgene may not express if it gets integrated in an inactive chromatin region, multiple tandem copy integration can lead to subsequent inactivation of the transgene, and inadvertent integration of the transgene in a region that codes for important genes might disrupt their function. Because of such reasons, typically many independent transgenic lines are screened through strenuous and time taking steps of breeding them to identify the best suited line for further experiments.
To overcome the pitfalls of random transgenesis, certain labs have taken the embryonic stem (ES) cell approach to target a single copy of a transgene into well-studied genetic loci, such as ROSA26. However, this approach involves laborious and time-consuming steps, such as targeting vector construction, ES cell targeting, screening and expansion of correctly targeted clones, chimera generation and breeding them for germ line transmission. Despite the lengthy and expensive process that takes 7–8 months or longer, success is not guaranteed [3] and not surprisingly, this method is not routinely used.
Recently, we and others have developed a targeted transgenesis method through direct injection of DNA into pronuclei [7,6,5]. This method, termed pronuclear injection-based targeted transgenesis (PITT), involves prior development of a seed mouse that harbors landing pads for recombinase-mediated cassette exchange (RMCE) at a predetermined locus. In the Cre-Lox system-based PITT (Cre-PITT) that we developed the landing pad constituted heterotypic LoxP sites (JT15 and Lox2272) that allow uni-directional and precise exchange of the DNA cassettes from specially designed donor vectors containing compatible mutant LoxP sites for RMCE. Unlike the ES cell based approaches, the PITT approach takes about 3–4 months and provides considerable savings with respect to time and expense. However, this method uses a previously established PITT seed strain, which is usually generated through a traditional ES cell-based approach [6,7,10].
In this work, we sought to explore options to generate a PITT seed mouse faster by directly inserting heterotypic LoxP sequences into the mouse ROSA26 locus using Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/CRISPR-associated 9 (Cas9) system. Our aim was to create a “simpler” seed mouse that would only contain essential elements for PITT and leave out nonessential elements such as a PGK-neomycin-polyA signal sequence that were necessary in the previous seed strain [7]. In addition, such a seed mouse would enable the insertion of larger cassettes through RMCE, in the next step. This two-step approach could be easily applied to other chromosomal locations that will circumvent the necessity to construct larger homology-arm containing plasmid vectors as required for ES cell based, or one-step CRISPR/Cas9-based approaches.
2. Materials and methods
2.1. Plasmids
The pBGK plasmid described in [2] was used as a template for Cas9 mRNA synthesis. pUC57-sgRNA expression vector (Addgene plasmid 51132; [8] was used as vector to clone sgRNA sequences that includes T7 promoter, convenient BsaI cloning sites for cloning of annealed sgRNA oligonucleotides and a DraI site for linearization.
2.2. Synthesis and purification of Cas9 mRNA and sgRNAs and donor oligos
The oligos corresponding to Cr2 and Cr4 sgRNAs were cloned into BsaI site of pUC57-sgRNA expression vector [8]. The positive clones were sequence confirmed and used for in vitro transcription of sgRNA. The Cas9 mRNA was transcribed from pBGK plasmid that was created by replacing iCre coding sequence with Cas9 in the pBBI vector [5] (Supplementary Fig. 1). The pBGK plasmid contains a stretch of 83 ‘A’s after the stop codon; this feature enables the direct synthesis of polyA containing mRNA and therefore the in vitro transcribed RNA does not require additional a poly-adenylation step. Linearized pBGK Cas9 by XbaI digestion was gel purified and used as the template for in vitro transcription using mMESSAGE mMACHINE T7 ULTRA kit (Ambion: AM 1345). The sgRNAs were synthesized using MEGAshortscript T7 kit (Ambion: AM 1354) from DraI linearized pUC57 vector templates. Both type of RNAs were purified using MEGAclear kit (Ambion: AM 1908) and eluted in RNase-free water. Single-stranded DNA Donors were purchased as Ultramer DNA oligos from Integrated DNA Technologies.
2.3. Pronuclear injection
B6/SJLF2 hybrids were used as embryo donors. Detailed description of CRISPR/Cas9-mediated mouse genome editing are described in [2]. Briefly, the injection mix contained 10 ng/ul of sgRNAs + 10 ng/ul of Cas9 mRNA. Donor oligo concentration included in some experiments was 20 ng/ul. We followed simultaneous cytoplasmic and nuclear injection method as described in [5]. The care, use, and disposition of animals used in this study were approved by the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center.
2.4. Genotyping of offspring and nucleotide sequencing
Genomic DNAs extracted from the offspring using Qiagen Gentra Puregene Tissue Kit were subjected to flanking primer PCR and internal (the donor oligo specific) and external primer PCR. Surveyor assay was performed as described by the manufacturer (Transgenomic). The primers used for amplifying the target sequence are given in the Supplementary Table 1. The assay products were analyzed using a 2.5% agarose gel. The bigger sized bands in flanking PCR genotyping assay of selected samples were gel purified and were subjected to direct sequencing. The upper bands of sample 6 (from internal + external PCR assay) and sample 23 (flanking PCR assay) were cloned into pCR 2.1 Topo cloning vector (Invitrogen: Cat # K4560) and the plasmids were sequenced using M13 Forward primer.
2.5. Off-target analysis
All potential off-target sites with homology to the 23 bases sequence (sgRNA target + PAM) were retrieved by a base-by-base scan of the whole genome, allowing for ungapped alignments with up to 5 mismatches in the sgRNA target sequence. The off-target sites were amplified using primers binding to flanking regions and were subjected to Surveyor assay.
3. Results
3.1. Designing of ROSA26-CRISPR target sequences
CRISPR target sequences in the first intron of mouse ROSA26 locus, the site where majority of the ROSA26 knock-in mutations have been created, were searched through UCSC browser http://www.genome-engineering.org/crispr/?page_id=41 (Fig. 1A). The target sequences were numbered ROSA-Cr 1 to 4. (ROSA-Cr1 is 270 base upstream of the XbaI site and ROSA-Cr2 to 4 are located 62 to 248 bases downstream of the XbaI site (Fig. 1B). We intended to test two independent CRISPR target sites: one that is immediately downstream of XbaI site; the most commonly used site for inserting transgenes and the second one close to the original pro-viral integration site. Among the ROSA-Cr1 to 4 target sites, Cr2 and Cr4 sites met these criteria and therefore were chosen for CRISPR/Cas9 mutagenesis experiments. The Cr4 site cleaves exactly at the original provirus integration site [9,1].
Fig. 1.

CRISPR target sequences in the ROSA26Sor locus. The CRISPR target sequences in the first intron of ROSA26 locus were searched through the UCSC genome browser available through genome-engineering.org/tools (Zhang lab). The results displayed 14 target sequences, of which 4 were within the vicinity of ∼270 bases from the XbaI site, the most commonly used site for targeted insertion of foreign sequences at this locus. These four target sites were named as ROSA Cr1, Cr2, Cr3 and Cr4 and only the Cr2 and Cr4 sites were tested in this study. The cut sites are indicated by a forward slash (/) and PAM sequences are underlined.
3.2. Targeted insertion of heterotypic LoxP sites at the ROSA26-CRISPR targets and genotyping of offspring
We designed experiments to insert short sequences at Cr2 and Cr4 target sites using oligonucleotide assisted homology directed repair process. These short single-stranded DNA donors contained 60 bases of homology on both the ends and two heterotypic LoxP sites (JT15 and lox2272) in the middle. The objective of choosing such an oligo design was to create a PITT seed mouse that can eventually be used for insertion of larger transgenes at the ROSA26 locus using Cre-PITT. Microinjection of CRISPR components were performed as described in Materials and Methods section and the microinjection data is compiled in Table 1.
Table 1.
Microinjection data.
| sgRNA | Embryos injected | Embryos transferred | # fosters used | # pregnant fosters | Animal id numbers (total) | Oligo insertion (%) |
|---|---|---|---|---|---|---|
| Cr2 | 108 | 98 | 5 | 3 | 1–5, 18–36, 47–55 (33) | 11 (33%) |
| Cr4 | 87 | 67 | 4 | 3 | 6–17, 37–46 (22) | 8 (36%) |
The target sequences from the genomic DNA of offspring were amplified using flanking primers and analyzed in a 2% agarose gel. As expected, some samples had slower migrating bands indicative of donor oligonucleotide insertion (Fig. 2A). While there were many founders that had the expected increase in size, some (founders 6, 9, 14, 15, 16, 20 and 23) had higher than the expected sized bands (Fig. 2A). The higher sized (or incorrect sized bands) may be the result of scenarios such as (i) addition of more than one copy of the donor-oligo, or (ii) duplication of some adjacent sequences near the target site before the donor-oligo gets inserted, or (iii) combination of varying lengths of deletion & addition of nucleotides followed by insertion of the donor-oligo, or (iv) due to unequal amplification of templates (higher sized bands being less favored) in the PCR reaction.
Fig. 2.
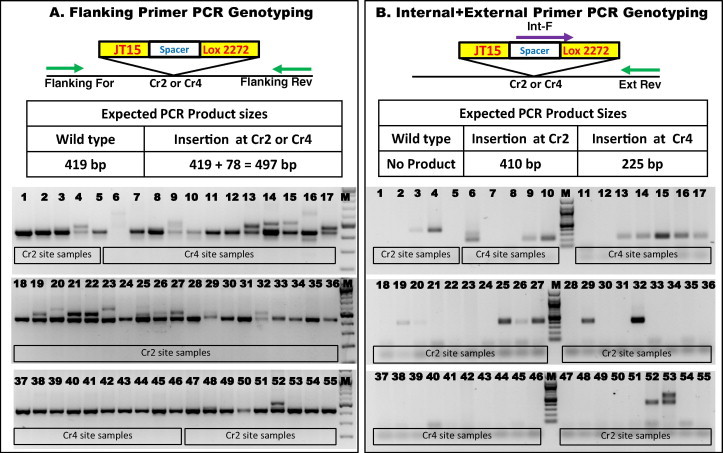
Genotyping of offspring. (A) Schematic of Flanking Primer PCR assay. The primers bind on the flanking sequences. Insertion of the JT15-Spacer-Lox2272 sites adds 78 bases at the target site that increases the PCR band size from 419 bp (wild type) to 497 bp (mutant). Some samples show incorrect sized mutant band (see text for details). (B) Internal + External primer PCR. Genotyping PCR with a forward primer in the repair oligo region and a reverse primer downstream of Cr4 site. Insertion at Cr2 and Cr4 sites will yield bands of 410 and 225 base pairs respectively. See text for discussion on results for samples 6 and 53. M: 100 base pair Marker.
We further analyzed the samples by a PCR using an internal primer that binds to the donor-oligo sequence and an external primer that binds outside the homology arms. In this ‘internal + external primer PCR’ assay, a band is detected only if there is an insertion of the donor-oligo at the cleavage site. As expected, most of the samples that showed higher migrating bands in the ‘flanking primer PCR’ assay were positive for PCR using this primer set (Fig. 2B). Collective results from the two different types of genotyping assays showed that about 33% (11/33) and 36% (8/22) of offspring had targeted insertion of donor-oligo at Cr2 and Cr4 sites respectively. Interestingly, two samples (# 6 and # 53) showed two bands (instead of expected one band) in this PCR; these results are discussed further (see below).
Higher migrating sharper bands from flanking PCR assay for a few samples were purified and sequenced. While some samples showed clean sequence (Fig. 3A) with precise insertion of the donor-oligo sequence, others contained mixtures of sequences indicative of mosaicism (data not shown), possibly due to insertions occurring after the 2 cell stage [4]. Since we obtained more than sufficient number of founders with the precise oligo sequence insertion, we only bred two founders each (for Cr2 or Cr4 insertions) from these and we did not pursue breeding of the other founders. Out of curiosity, we further analyzed the nature of mutations in a few samples that showed atypical banding pattern in the genotyping PCRs. The results uncovered some anomalous mutations in those samples that are presented below.
Fig. 3.
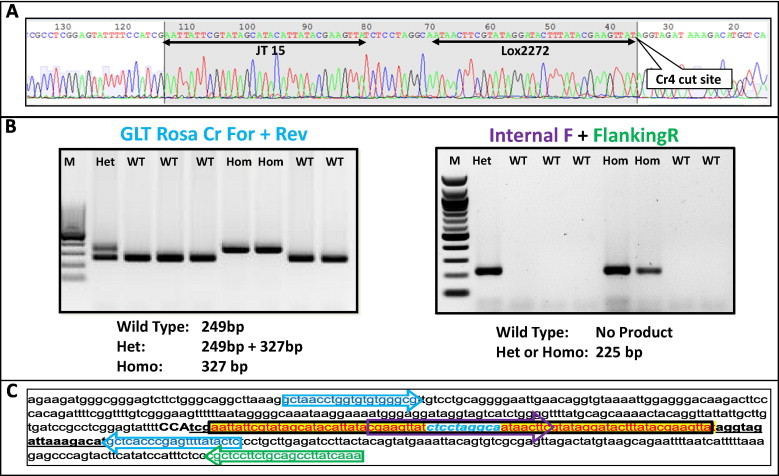
Germ line transmission and establishment of a homozygous Cr4 insertion mutation. (A) A sequencing reaction chromatogram from a founder mouse showing a correct donor-oligo insertion at the Cr4 target site. (B) The sequence confirmed mutant was bred with a wild type mouse and germ line transmission was established. Genotyping of F2 generation litter with flanking primers PCR (left) and with Internal + External primers PCR (right). The expected product sizes and the genotypes are given below the gel images. M: 100 base pair Marker. (C) Nucleotide sequence of the locus containing the correct insertion sequence. The locations of primers are shown with the corresponding color coded arrows. Cr4 sequence is underlined and PAM sequence is shown in uppercase.
3.3. Occurrence of anomalous and/or mosaic insertions of donor-oligo in CRISPR/Cas9-mediated genome editing
In the ‘internal + external primer PCR assay’, the sample 6 showed one expected sized band and another unexpected sized (larger) band. Notably, this sample did not amplify a wild type band in the flanking PCR but there were two faint and diffused larger sized bands (Fig. 2B). The lack of wild type sized band indicates that both alleles may have got mutated with a possibility of loss of sequences near the forward primer binding site. We cloned and sequenced the larger sized band (from internal + external primer PCR) which revealed an anomalous insertion event—there was a stretch of 87 extra nucleotides inserted upstream of the correctly inserted donor cassette of 78 nucleotides. This 87 nucleotide extra sequence constituted (i) duplication of 59 nucleotides immediately upstream of the cleavage site followed by (ii) a 26 base long inverted sequence originated from the 7 to 32 nucleotides downstream of the cut site and (iii) two nucleotides (insertion) of unknown origin (Fig. 4C and D). It is difficult to ascertain how such a donor event must have occurred, but it clearly indicates that anomalous insertions of donor-oligos can occur in CRISPR/Cas9-mediated genome editing.
Fig. 4.
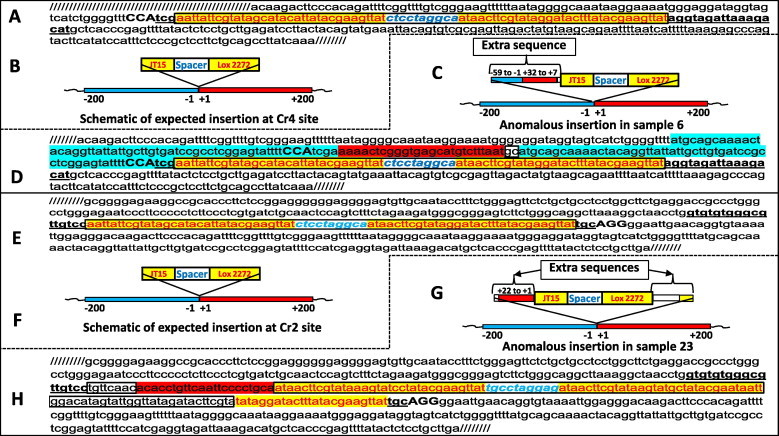
Examples of anomalous sequence insertions occurring during HDR mediated CRISPR/Cas9 genome editing events. Nucleotide sequence after the expected donor-oligo insertion at the ROSA-Cr4 site (A), and its schematic (B). Schematic of anomalous insertion in the sample #6 (C), and its nucleotide sequence (D). Nucleotide sequence after the expected donor-oligo insertion at the ROSA-Cr2 site (E), and its schematic (F). Schematic of anomalous insertion in the sample # 23 (G) and its nucleotide sequence (H). The schematics include blue (left) or red (right) bars and the nucleotide numbering include “−” (left) or “ + ” (right) signs that demarcate the Cas9 cut sites. The stretches of extra sequences (in D and H) are highlighted with corresponding (blue or red) color to indicate their origin. All LoxP sequences are highlighted in yellow and boxed except the partial LoxP site (in H), which is only highlighted and not boxed. The sequences originating from unknown origin are boxed and are not highlighted with any color (in D and H).
The mutant amplicon (upper band) in the flanking primer PCR assay of the sample 23 was higher size than the anticipated increase of 78 bases (size of the donor template) from the wild type. We cloned and sequenced the upper band, which revealed an insertion of a stretch of extra 84 nucleotides of a random mix of various short fragments—8 nucleotides of unknown origin, 22 nucleotides of inverted sequence originated from immediately-downstream of the Cr2 cut site followed by desired insertion cassette with one nucleotide mismatch and one nucleotide deletion, 30 nucleotides of unknown origin and partial Lox2272 sequence of 25 nucleotide long (Fig. 4G and H).
Even though the sample 53 showed two bands in the internal + external primer PCR, it did not show a higher migrating band in the flanking primer PCR assay (Fig. 2A). This indicates the possibility that this founder constitutes at least three alleles (a wild type allele detected in flanking PCR assay and two different mutant alleles detected by internal + external primer PCR assay) and therefore should contain mosaic mutations. It is also likely that the wild type template would have preferentially been amplified (in the flanking PCR assay) because of its relative abundance in the mosaic genomic DNA and the mutant bands might not have got amplified. Another reason for the absence of mutant bands (larger sized bands in flanking PCR assay) could be that the forward primer binding site may have been lost in the mutant alleles of this sample. We sequenced the upper band from the internal + external PCR reaction. Although, the direct sequencing of the gel purified PCR product did not yield a quality sequencing reaction to clearly identify the extra sequence and its exact length, the results were indicative of anomalous insertion event (data not shown). Another sample indicative of mosaic mutant pattern is #16 that contains one prominent wild type, and two minor bands (one band much larger than- and the other smaller than- the wild type).
Taken together, the results from the analysis of the extra bands in the ‘internal + external primer PCRs’ and atypical bands in ‘flanking primer PCRs’ revealed that anomalous insertions and mosaic mutations occur in CRISPR/Cas9-mediated genome editing experiments that include donor-oligo insertion designs.
Two founders each harboring insertions at Cr2 or Cr4 sites, that were sequence-confirmed, were mated to achieve germ line transmission of the mutation. The schematic of genotyping assays and a representative gel images showing the germ line transmission for a litter from Cr4 insertion site are shown in Fig. 3B. Since the Cr4 site is located precisely at the original pro-viral integration site, we reasoned that Cr4 site mutants would be preferred over the Cr2 site to use them as PITT seed strain for future studies and therefore, we verified if there were any off-target cleavages in any of the eight Cr4 mutant animals. Seven potential off-target sites of Cr4 target sequence were identified in the mouse genome and a surveyor assay was performed at all these off-target sites in all of the eight Cr4 positive animals. We did not observe off-target Cas9 activity in any animals at any of the 7 potential off-target sites, as indicated by lack of cleavage products in the surveyor assay (Supplementary Fig. 2).
4. Discussion
Traditionally, insertion of transgenes at a predetermined site in the mouse genome has been achieved through laborious and time-consuming steps such as targeting vector construction, ES cell targeting, screening and expansion of correctly targeted clones, chimera generation and breeding them for germ line transmission. Using such approaches several transgenes have been targeted to the ROSA26 locus in the mouse genome. During the recent 3–4 years, we and others have shown that all of the steps involved in traditional targeted transgenesis method can be bypassed using the newly developed PITT system [7,10,6,5]. PITT involves prior development of a seed mouse that has heterotypic LoxP sites called landing pads at the genomic locus of interest. In the second step, viz. PITT, the fertilized eggs from the seed mouse are used for injecting a donor plasmid containing the transgenic cassette of interest along with Cre mRNA which results in precise insertion of the transgenic cassette at the landing pad. In this report, we tested if a PITT seed mouse can be created at the ROSA26 locus by co-injecting ROSA sgRNA (either ROSA-Cr2 or ROSA-Cr4 sites), Cas9 mRNA and a donor-oligo into fertilized eggs. We reasoned that by using CRISPRs, just the LoxP sites required for the PITT seed mice can be inserted, without any additional sequences such as a neomycin cassette as needed in the traditional ES cell approach. The mutant mouse so created would serve as a seed mouse for future PITT experiments to insert larger sequences using the Cre-Lox-based Recombinase-Mediated Cassette Exchange (RMCE) mechanism [7,6]. Upon publication, the new seed mouse strain will be made available to the research community which could be used for precise insertion of desired transgenes through PITT.
During our search for CRISPR target sequences in the ROSA26 locus, we noted that the ROSA-Cr4 site is exactly where the initial provirus mediated LacZ insertion was characterized [9,1]. We reasoned that if the Cr4 target site can be validated to insert sequences using the CRISPR/Cas9 system, it may be a better targeting approach, unlike the available targeting vector tools that are designed to insert sequences at the XbaI site which is 248 bases upstream of the provirus integration (/Cr4) site. As demonstrated in this work, exogenous sequences can be inserted precisely at the original provirus insertion site using the CRISPR/Cas9 system (Fig. 2C). The short stretch of sequences (mutant LoxP sites) inserted in this work serves as a landing pad for PITT method using which larger cassettes can be inserted into the landing pad. The PITT technique can typically insert 8–10 kb long cassettes and the largest DNA piece inserted so far is 14.4 kb [5].
Mosaicism and allele complexities have been reported in gene disruption experiments using CRISPR/Cas9 system [4,11]. Li et al., in their ‘Th’ locus disruption experiment, reported that as many as six different mutations were detected in the offspring when a founder was bred even when they could only identify two mutant alleles in the founder [4]. Yen et al., readily detected somatic mosaicism and allele complexities in their experiment which targeted the tyrosinase (Tyr) gene in C57BL/6 mice that resulted in albino as well as patchy coat colored offspring and the co-existing mutations were segregated when the founders were bred [11]. While these reports have observed mosaicism and allele complexities in Non-Homologous End Joining (NHEJ)-mediated CRISPR/Cas9 genome editing experiments, our report presents evidence that anomalous insertions and mosaicism can occur in Homology Directed Repair (HDR)-mediated CRISPR/Cas9 genome editing experiments.
As noted in the results section, we only sequenced the alleles of a few founders that appeared to contain atypical genotyping pattern. A comprehensive analysis of multitude of mutant alleles among the founders in a CRISPR/Cas9 mutagenesis experiment may be of scientific interest to understand the nature of HDR mechanism that occur during Cas9 cleavage. Such analysis can be done by systematically breeding many F0 founders to segregate multiple alleles and analyzing multiple different mutations among a library of offspring from each founder.
In summary, this work validates the CRISPR target sequences at ROSA26 locus, the most commonly used genetic locus in mouse for targeted knocking-in of exogenous sequences. Furthermore, the model created using the CRISPR/Cas9 system will serve as a simplified PITT seed mouse that can be used to insert larger cassettes through RMCE, similar to the previously generated seed mice. The two-step strategy—introducing heterotypic LoxP sites using CRISPR/Cas9 system and then inserting larger cassettes through PITT approach offers two distinct advantages; i) it overcomes the current limitations of the two techniques—the lengthy and expensive steps of creating a seed mouse needed for the PITT technique and relative inefficiency of CRISPR/Cas9 system to readily and precisely insert larger cassettes and ii) it combines the best features of the two new techniques—CRISPR/Cas9 system assisted rapid creation of PITT seed mice by targeted insertion of a landing pad at a desired locus and the capability of PITT system to precisely insert larger DNA cassettes into the landing pad. The two-step strategy (introducing heterotypic LoxP sites using CRISPR/Cas9 system and then inserting larger cassettes), can also be applied to other chromosomal locations and may circumvent the necessity to construct larger homology containing plasmid vectors as required for ES cell-based or one-step CRISPR/Cas9-based strategies.
Authors’ contributions
CBG conceived the study. CBG and MO designed the study. RMQ and DWH performed pronuclear injections, mouse breeding experiments and acquired the data. RMQ and CBG performed genotyping assays. CBG and MO analyzed and interpreted data and wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This work was partially supported by an Institutional Development Award (IDeA) to CBG (PI: Shelley Smith) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103471 to CBG, by Grant-in-Aid for Scientific Research (25290035) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), and 2014 Tokai University School of Medicine Research Aid to MO. We thank Bin Shen and Xingxu Huang for providing pUC57-sgRNA vector and for valuable suggestions on the manuscript. We thank Manju George for critical reading of the manuscript and for English corrections. We thank Ronald Redder and Gregory Kubik, UNMC DNA sequencing core facility for their help with sequencing. The DNA Sequencing Core receives partial support from the NCRR (1S10RR027754-01, 5P20RR016469, RR018788-08) and the National Institute for General Medical Science (NIGMS) (8P20GM103427, P20GM103471). RMQ, DWH and CBG acknowledge the Nebraska Research Initiative and UNMC Vice Chancellor for Research Office for supporting the mouse genome engineering core facility. MO acknowledges the people in Support Center for Medical Research and Education in Tokai University for technical assistance.
Appendix A. Supplementary data
Supplementary material.
References
- 1.Friedrich G., Soriano P. Promoter traps in embryonic stem cells: a genetic screen to identify and mutate developmental genes in mice. Genes Dev. 1991;5:1513–1523. doi: 10.1101/gad.5.9.1513. [DOI] [PubMed] [Google Scholar]
- 2.D.W. Harms, R.M. Quadros, D. Seruggia, M. Ohtsuka, G. Takahashi, L. Montoliu, C.B. Gurumurthy, 2014. Mouse Genome Editing Using the CRISPR/Cas System. Current Protocols in Human Genetics/Editorial Board, Jonathan L. Haines, [et al.] 83:15.7.1–15.7.27. [DOI] [PMC free article] [PubMed]
- 3.Lee A.Y., Lloyd K.C.K. Conditional targeting of Ispd using paired Cas9 nickase and a single DNA template in mice. FEBS Open Biol. 2014;4:637–642. doi: 10.1016/j.fob.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li D., Qiu Z., Shao Y., Chen Y., Guan Y., Liu M., Li Y., Gao N., Wang L., Lu X. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat. Biotechnol. 2013;31:681–683. doi: 10.1038/nbt.2661. [DOI] [PubMed] [Google Scholar]
- 5.Ohtsuka M., Miura H., Hayashi H., Nakaoka H., Kimura M., Sato M., Gurumurthy C.B., Inoko H. Improvement of pronuclear injection-based targeted transgenesis (PITT) by iCre mRNA-mediated site-specific recombination. Trans. Res. 2013;22:873–875. doi: 10.1007/s11248-013-9703-x. [DOI] [PubMed] [Google Scholar]
- 6.Ohtsuka M., Miura H., Sato M., Kimura M., Inoko H., Gurumurthy C.B. PITT: pronuclear injection-based targeted transgenesis, a reliable transgene expression method in mice. Exp. Anim./Jpn. Assoc. Lab. Anim. Sci. 2012;61:489–502. doi: 10.1538/expanim.61.489. [DOI] [PubMed] [Google Scholar]
- 7.Ohtsuka M., Ogiwara S., Miura H., Mizutani A., Warita T., Sato M., Imai K., Hozumi K., Sato T., Tanaka M. Pronuclear injection-based mouse targeted transgenesis for reproducible and highly efficient transgene expression. Nucl. Acids Res. 2010;38:e198. doi: 10.1093/nar/gkq860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen B., Zhang W., Zhang J., Zhou J., Wang J., Chen L., Wang L., Hodgkins A., Iyer V., Huang X. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Methods. 2014;11:399–402. doi: 10.1038/nmeth.2857. [DOI] [PubMed] [Google Scholar]
- 9.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Gene. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 10.Tasic B., Hippenmeyer S., Wang C., Gamboa M., Zong H., Chen-Tsai Y., Luo L. Site-specific integrase-mediated transgenesis in mice via pronuclear injection. Proc. Natl. Acad. Sci. U.S.A. 2011;108:7902–7907. doi: 10.1073/pnas.1019507108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yen S.-T., Zhang M., Deng J.M., Usman S.J., Smith C.N., Parker-Thornburg J., Swinton P.G., Martin J.F., Behringer R.R. Somatic mosaicism and allele complexity induced by CRISPR/Cas9 RNA injections in mouse zygotes. Dev. Biol. 2014;393:3–9. doi: 10.1016/j.ydbio.2014.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material.