

Highlights
-
•
Mutations in the DRY motif of CXCR1 abolish ligand binding and receptor activation.
-
•
Point mutations between TM6 and i3 loop result in constitutive activity of CXCR1.
-
•
Constitutive activity of mutant CXCR1 occurs via Gα15 signaling activation.
-
•
The highly conserved DRY motifs have distinct roles in CXCR1 and CXCR2.
Abbreviations: CXCR1, CXC receptor 1; GPCR, G protein-coupled receptor; DRY motif, Asp-Arg-Tyr motif; IL-8, interleukin 8; TMs, transmembrane domain; i2, intracellular loop 2; i3, intracellular loop 3; PLC, phospholipase C; WT, wild type; Kd, affinity constants; IP, inositol phosphate; PTX, pertussis toxin
Keywords: Chemokine receptor, CXCR1, G protein coupled receptor, Gα15, Gαi, Constitutive activity
Abstract
CXCR1, a receptor for interleukin-8 (IL-8), plays an important role in defending against pathogen invasion during neutrophil-mediated innate immune response. Human CXCR1 is a G protein-coupled receptor (GPCR) with its characteristic seven transmembrane domains (TMs). Functional and structural analyses of several GPCRs have revealed that conserved residues on TM3 (including the highly conserved Asp-Arg-Tyr (DRY) motif) and TM6 near intracellular loops contain domains critical for G protein coupling as well as GPCR activation. The objective of this study was to elucidate the role of critical amino acid residues on TM3 near intracellular loop 2 (i2) and TM6 near intracellular loop 3 (i3), including S1323.47 (Baldwin location), D1343.49, M2416.34, and F2516.44, in G protein coupling and CXCR1 activation. The results demonstrate that mutations of D1343.49 at DRY motif of CXCR1 (D134N and D134V) completely abolished the ligand binding and functional response of the receptor. Additionally, point mutations at positions 241 and 251 between TM6 and i3 loop generated mutant receptors with modest constitutive activity via Gα15 signaling activation. Our results show that D1343.49 on the highly conserved DRY motif has a distinct role for CXCR1 compared to its homologues (CXCR2 and KSHV-GPCR) in G protein coupling and receptor activation. In addition, M2416.34 and F2516.44 along with our previously identified V2476.40 on TM6 are spatially located in a “hot spot” likely essential for CXCR1 activation. Identification of these amino acid residues may be useful for elucidating mechanism of CXCR1 activation and designing specific antagonists for the treatment of CXCR1-mediated diseases.
1. Introduction
CXCR1 is an important member of the chemokine receptor family that mediates migration of neutrophils primarily, but is also important for acute and chronic inflammation, proliferation and development of lymphocytes [1,2]. Its most important ligand, IL-8, can not only bind to CXCR1, but also to its homologues CXCR2 and KSHV-GPCR [3]. CXCR1 and CXCR2 share 76% amino acid identity [1,2], are highly expressed on the surface of neutrophils and can trigger chemotactic signals in neutrophils in response to IL-8. Despite their high homology and similar co-expression on neutrophil that mediates chemotaxis, sequence differences between CXCR1 and CXCR2 lead to distinct activation patterns and subsequent regulation of physiological and pathophysiological processes involved in inflammation and cell proliferation. CXCR1 is specific for IL-8, whereas CXCR2 also binds to other CXC chemokines, and differential activation and regulation of CXCR1 and CXCR2 by IL-8 monomer and dimer have been demonstrated [4]. IL-8 promotes bacterial killing by neutrophils via CXCR1 but not CXCR2 at the site of inflammation [5], and impairment of CXCR1 increases susceptibility to bacterial infection [6]. Although both CXCR1 and CXCR2 promote tumor growth and angiogenesis, it is noteworthy that only CXCR2 plays an important role in regulating IL-8-mediated invasion and migration of human melanoma cells [7,8], and that CXCR1 blockade selectively depletes human breast cancer stem cells [9]. Interestingly, only CXCR2 can mediate the initial recruitment of neutrophils from the circulation [5] and from the bone marrow [10] as well as induce senescence in primary cells [11]. Efforts to elucidate the role of CXCR1 in vivo have been hampered for the following reasons: (1) the lack of specific CXCR1 agonists and antagonists, (2) human homologue of IL-8 does not exist in mice and (3) CXCR1 is not expressed in mouse neutrophils [12].
CXCR1 is a member of class A rhodopsin-like GPCRs, the largest class of integral membrane proteins responsible for cellular signal transduction as well as targeted as drug receptors [13,14]. In order to design specific antagonists for CXCR1 and CXCR2 that would have therapeutic benefits, it is essential to understand the structural and functional differences between the CXCR homologues. Despite recent progress on human CXCR1 structure determined by NMR spectroscopy [15], structural information on CXCR1 such as ligand specificity, selectivity, G protein coupling, and receptor activation is incomplete because the crystal structure of CXCR1, especially ligand-bound and/or G protein-bound forms, has not been available. However, recently published crystal structures of several other activated or inactive GPCRs have provided valuable information applicable for GPCR activation in general. For example, structural studies on CXCR4 and other GPCRs have indicated that TM3 and TM6 are critical regions for signaling mechanism [16–18]. Similar observations of agonist-bound structure of human A2A adenosine receptor and rhodopsin have shown that an outward tilt and rotation of the cytoplasmic half of TM6 and axial movement of TM3 toward the extracellular side are characteristic features of the active form of the receptor that is conformationally capable of coupling to G protein [19,20]. The movement of TM3 and TM6 at the cytoplasmic side of the membrane is a necessary step for GPCR activation [16–21] and constraint of the relative mobility of the cytoplasmic end of TM3 and TM6 maintains the receptor in the inactive state [16–21]. Due to their importance it is likely that disruption of these structures would likely affect G protein binding and activation. The movement of transmembrane helices due to ligand binding renders some amino acid residues exposed to G protein and results in receptor activation. The movement of TMs also induces changes in conformational structure of the intracellular loops (especially loops i2 and i3) that are crucial for G-protein interactions [22].
Two important domains on TM3 and TM6 involved in interaction with G proteins have been identified based on crystal GPCR structures and mutagenesis data, the specific region on TM3 near the second intracellular loop (including the highly conserved DRY motif) and region on TM6 near the third intracellular loop [16–20]. In the current study, several potentially important amino acid residues on TM3 and TM6 near the intracellular loops that may be important for G protein coupling, including S1323.47and D1343.49 on TM3, and M2416.34 and F2516.44 on TM6, have been investigated. Previous data suggest that these specific residues on TM3 and TM6 are critically involved in the GPCRs coupling to G proteins. For example, substitution of S1203.47 with A (S120A) in M1 muscarinic acetylcholine receptor (S132A in CXCR1) leads to constitutive activation of the receptor [23]. D1343.49 is another residue for which the role of DRY in G protein coupling and receptor activation has been studied in two CXCR1 homologues, CXCR2 and KSHV-GPCR. Despite the high conservation of DRY motif at the junction of the third transmembrane domain to the second intracellular loop, the KSHV-GPCR contains a VRY sequence instead. The KSHV-GPCR exhibits constitutive signaling via activation of PLC in the absence of ligand [24–26] which is thought to play a role in the pathogenesis of Kaposi’s sarcoma [27,28]. Likewise, exchange of Asp1383.49 of the DRY sequence in CXCR2 with a Val (D138V), the corresponding amino acid in KSHV-GPCR, results in constitutive activity in inositol phosphate accumulation [25] and leads to altered activity similar to that seen in KSHV-GPCR [29]. In alpha1B-adrenergic receptor, mutation of A293, corresponding to M2416.34 on CXCR1, results in constitutive activity of the receptor [30]. Lastly, the phenylalanine at TM6.44 (corresponding to F2516.44 on CXCR1) is located in the central region of TM6 and highly conserved in 91% of the members of the rhodopsin family of GPCRs. TM6.44 (D633 on TSH receptor) [31,32] and D578 on LH receptor [33–35] are key switch residues involved in activation. Several naturally occurring mutations in TM6.44 (D578 in human luteinizing hormone receptors (LHR) such as D578H) are associated with constitutive activation of the receptor [34]. In addition, it has been suggested that the formation of a salt bridge between helices 3 (L457) and 6 (D578) is responsible for the constitutive activity of the naturally occurring L457R mutation of the human lutropin receptor [33]. Similarly, naturally occurred D633A, F, H mutation in TM6.44 of TSH receptor lead to high constitutive activation [31,32]. As these amino acid residues are relatively conserved in all GPCRs rhodopsin family, we sought to determine if these residues on TM3 and TM6 of human CXCR1 are involved in the interaction between receptor and G proteins. Our results show that mutant M241V and F251H are constitutively active mutants coupled to G proteins and lead to increase in IP accumulation in the absence of the ligand IL-8, suggesting an important role of these residues in G protein coupling of CXCR1.
2. Results
2.1. Construction of CXCR1 mutations by site-directed mutagenesis
CXCR1 is a member of rhodopsin-like GPCRs (Fig. 1). To elucidate the role of the highly conserved DRY motif in activation and function of CXCR1, we introduced a mutation in D1343.49 of DRY motif of CXCR1. Other amino acid residues on TM3 and TM6 potentially involved in G protein coupling and constitutive activity of GPCRs that were studied include S132 (TM3:47), M241 (TM6:34) and F251 (TM6:44). Among four amino acid residues chosen for our study, D1343.49 and F2516.44 are highly conserved whereas S1323.47 and M2416.34 are modestly conserved (Fig. 2). Selective mutations were made because of their critical roles in G protein coupling in several other GPCRs. These CXCR1 mutants were expressed in COS-7 cells or HEK239 cells and their expression, binding of IL-8, and Gα15- and Gαi coupling-induced IP production were determined.
Fig. 1.

Two-dimensional diagram of human CXCR1. Most rhodopsin-like GPCRs have a conserved Asp-Arg (DR) pair at the cytoplasmic end of TM3. CXCR1 consists of characteristic seven transmembrane helices (TM1–TM7) connected by three extracellular loops (e1∼e3) and three intracellular loops (i1∼i3). The positions of the residues (S1323.47, D1343.49, M2416.34 and F2516.44) that were targeted for mutagenesis in TM3 and TM6 of the CXCR1 are indicated by filled circles. The putative two disulphide bonds (gold), formed by Cys110/Cys187 are indicated by “S–S”.
Fig. 2.
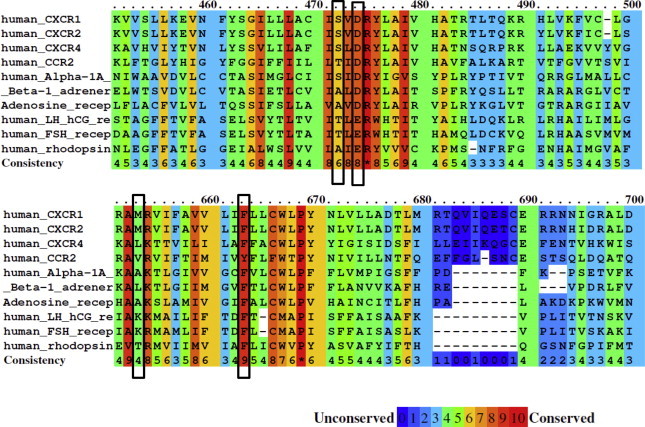
Color scheme showing alignment of amino acid conservation for CXCR1 and other rhodopsin family GPCRs. The conservation scoring is performed by PRALINE. The scoring scheme works from 0 for the least conserved alignment position up to 10 for the most conserved alignment position, as indicated by the color assignments. The four amino acid residues subjected to mutagenesis (S1323.47, D1343.49, M2416.34 and F2516.44) are highlighted with open bars.
2.2. Expression of CXCR1 and its mutants
The flow cytometry data demonstrated the expression of WT CXCR1 and mutants on transfected HEK293 cells (Fig. 3A and B) except F251A which was barely expressed. The surface expression of CXCR1 and its mutants were further supported by confocal imaging results (Fig. 4). Consistently, F251A mutant was poorly expressed, whereas other mutants, including S132A, D134N, D134V, M241V, F251H, and F251Y, were detectable on the cell surface (Figs. 3A, B, and 4).
Fig. 3.

Flow cytometry of CXCR1 WT and mutants. HEK 293 cells were transiently transfected with CXCR1 or its mutants. Cells were incubated with FITC-conjugated mouse anti-human CD181 (CXCR1). Specificity of signal was confirmed by staining the cells with mouse IgG1 isotype control. (A) Transfected HEK 293 cell population stained positive using the anti-CXCR1 antibody (M2 region). (B) Fluorescence of positively stained cells were quantified by FACS (n = 3, mean ± SEM, with CXCR1 WT set to 100%).
Fig. 4.

Confocal analysis of expression of CXCR1 and its mutants. Surface expression of CXCR1 and its mutants on COS-7 cells were measured as described. The green color represents the surface expression of CXCR1. The cells were counterstained with Dapi as shown in blue.
2.3. Ligand binding assay of CXCR1 and its mutants
Results of the ligand binding assay are summarized in Table 1 and Fig. 5. Mutant receptors S132A, F251H, and F251Y showed similar affinity constant to WT (4.15e–09 M, 4.86e–09 M, 3.48e–09 M vs. 5.55e–09 M of WT), whereas mutants F251A and M241V exhibited increased Kd values vs. WT (1.03e–09 M and 1.53e–09 M vs. 5.55e–09 M of WT). The antibody (anti-CD128a) utilized for both flow cytometry and confocal microscopy specifically recognizes NH2 terminus of CXCR1. Previously we demonstrated that conformational changes seen in the CXCR1 mutants can affect both their ability to bind to specific ligand (ligand binding assay) and their specific recognition by the same anti-CD128a CXCR1 antibody (flow cytometry and confocal microscopy) [36]. In agreement with this, although both D134N and D134V mutants were expressed on the surface of transfected cells (as determined by flow cytometry and confocal microscopy) (Figs. 3 and 4), they showed minimal specific binding to 125I-labeled IL-8 (Fig. 5). This suggests that replacement of the highly conserved D134 with N and V might impair the folding of the receptor that is necessary for ligand binding and subsequent signaling efficacy.
Table 1.
Summary of ligand binding assay of CXCR1 WT and mutants.1
| CXCR1 Residue | Location (Baldwin#) | CXCR1 and Mutants | Bmax (% of WT) | Kd (M) |
|---|---|---|---|---|
| WT | 100 ± 0 | 5.550e−09 (4.401e−009 to 7.000e−009) | ||
| Ser 132 | TM3, residue 47 | S132A | 179.6 ± 27.7 | 4.154e−09 (3.523e−009 to 4.897e−009) |
| Asp 134 | TM3, residue 49 | D134 N | 18.8 ± 2.0 | N.D. |
| D134V | 19.5 ± 0.5 | N.D. | ||
| Met 241 | TM6, residue 34 | M241V | 160.8 ± 13.3 | 1.531e−09 (1.068e−009 to 2.194e−009) |
| Phe 251 | TM6, residue 44 | F251A | 30.2 ± 5.2 | 1.026e−09 (8.500e−010 to 1.238e−009) |
| F251H | 256.9 ± 0.8 | 4.860e−09 (4.434e−009 to 5.328e−009) | ||
| F251Y | 213.0 ± 6.9 | 3.478e−09 (2.816e−009 to 4.294e−009) |
Bmax and Kd value were estimated from COS-7 cells ligand binding assay as described under “Section 4”. All assays were done in triplicates. Values are mean ± SEM from 3 experiments.
Fig. 5.

Binding of 125I-labeled IL-8 to COS-7 cells expressing WT CXCR1 and mutants. The CXCR1 mutants were transiently transfected in COS-7 cells. Maximal binding assay (A) and competition binding experiments (B) were performed as described under “Section 4”. (A) Maximal binding of 125I-IL-8 to CXCR1 WT and mutants. Mock was transfected with pSG5 plasmid. (B) Competitive binding studies were conducted using 125I-labeled IL-8 and various unlabeled ligands as described under “Section 4”. Nonspecific binding was determined by adding 250 nM unlabeled IL-8. The figure shows representative maximal binding and competitive binding experiments for the indicated CXCR1 mutants, performed in triplicate. Data are mean ± SEM of replicate wells from a representative experiments performed in triplicate. Curve fitting was done using GraphPad Prism data analysis program, and the affinity constants (Kd) for CXCR1 mutants are shown in Table 1.
2.4. Identification of CXCR1 mutants M241V and F251H possessing constitutive activity
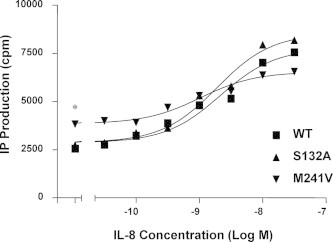
CXCR1 is coupled to both PTX-sensitive Gαi2 as well as PTX-resistant Gα15 in transfected COS-7 cells [37]. Mutation in D134 of DRY motif of CXCR1 did not retain G protein coupling or agonist-induced response (Fig. 6). Despite the high sequence identity between CXCR2 and KSHV-GPCR, both capable of binding IL-8 with high affinity, our results suggest that the conservative D on DRY motif plays a distinct role in G protein coupling from these two receptor homologues. This is probably responsible for the unique regulation and activation of effectors triggered by binding to IL-8. In contrast, among mutants detected compared with WT CXCR1, individual replacement of methionine (M241V) and phenylalanine (F251H) increased basal signal activity by 43.4% and 46.1%, respectively, in the absence of IL-8 (p < 0.001 vs. WT without IL-8 treatment), but not agonist-stimulated IP accumulation of the receptor. This suggests that substitution of M241V and F251H constitutively activates Gα15 protein (Fig. 6). In addition, M241V6.34 increased the IP accumulation basally as well as in response to IL-8 in a dose-dependent manner (Fig. 7). Moreover, in the absence of IL-8, IP accumulation was significantly higher in cells expressing M241V (p < 0.001) (Fig. 7). Two mutations on V247 (TM6:40), V247A and V247N, displayed constitutive activity as reported recently [36]. Although F251A mutant showed increased ligand affinity, it was nearly dysfunctional, which is likely attributed to its instability and its poor expression on cell surface (Figs. 3–5).
Fig. 6.
CXCR1 mutants coupled to Gα15. Basal and IL-8-stimulated inositol phosphate (IP) accumulation is shown in COS-7 cells transiently co-transfected with WT or mutant CXCR1 and pSG5 or Gα15. The release of inositol phosphate, induced by 40 nM IL-8, was measured 1 h after the treatment. Data for mutants are summarized from 3–7 experiments, each performed in triplicate, and are expressed as a percentage of WT CXCR1 baseline determined in parallel. The results are mean ± SEM. ∗∗P < 0.001, vs. WT + Gα15 (in the absence of IL-8).
Fig. 7.

Constitutive activation of CXCR1 mutant M241V. Basal and IL-8-stimulated inositol phosphate (IP) accumulation is shown in COS-7 cells transiently co-transfected with CXCR1 WT or mutant and Gα15. The cells were treated with various concentrations of IL-8. IP production was determined as described under “Section 4”. Data are mean ± SEM of replicate wells from a representative experiments performed in triplicate. Cells transfected with WT CXCR1 have the same basal IP production as cells transfected with vector alone. ∗P < 0.001, vs. WT + Gα15 (in the absence of IL-8).
2.5. Ability of CXCR1 WT and mutants to activate Gαi
To examine their coupling to Gαi protein, WT CXCR1 and mutants were co-transfected into COS-7 cells with Gαi2, Gβ1, Gγ2 and PLCβ2. This five-component co-transfection system (Gαi2-Gβ1-Gγ2-PLCβ2) has been successfully applied to investigate IL-8 signaling pathways in previous studies by our group and others [36,37]. This unique co-transfection system allows Gαi2 activation in response to IL-8, leading to the release of Gβγ subunits from Gαi2 and resulting in PLCβ2 activation and subsequent IP accumulation. Our results showed that mutants S132A, M241V, F251H and F251Y stimulated IP accumulation in response to IL-8, similar to CXCR1 WT (Fig. 8C). IP production in response to IL-8 was totally abolished by PTX, a specific inhibitor of Gαi, suggesting that CXCR1 and its mutants investigated here are coupled to Gαi2 (Fig. 8A and B). In contrast, IP production was mostly retained in response to IL-8 when Gα15 was expressed (data not shown). Thus, IP accumulation could be achieved via the PTX-insensitive Gα15 pathway or the PTX-sensitive Gαi2 pathway.
Fig. 8.

CXCR1 and mutants coupled to Gαi2. (A) COS-7 cells were co-transfected with equal amounts of cDNA (0.1 μg per well per component) encoding Gαi2, Gβ1, Gγ2, PLCβ2, as well as WT CXCR1 or its mutants. (A and B) The influence of PTX on the release of inositol phosphates. The cells were treated with or without PTX (100 ng/ml) for 18 h as indicated. The release of inositol phosphates was measured 1 h after the treatment in the absence (A) or presence (B) of 40 nM IL-8. “M” denotes cells co-transfected with cDNA components encoding Gαi2, Gβ1, Gγ2, and PLCβ2. (C) COS-7 cells were co-transfected with 0.3 μg of cDNA encoding Gαqi5 and the WT CXCR1 or its mutants. The release of inositol phosphates, induced by 40 nM IL-8, was measured 1 h after the treatment as described in detail under “Section 4”. Data are mean ± SEM of replicate wells from a representative experiment.
The low transfection rate in cells co-transfected with five different components is probably caused by inherent difficulty in simultaneous transfection with five vectors, which may account for the low reading of IP accumulation. The coupling of CXCR1 and mutants to Gαi was further demonstrated by introducing the chimeric G protein, Gαqi5, which contains the main structure of Gαq with the last five C-terminal amino acids of Gαq replaced with the corresponding amino acids from Gαi2. Gαqi5 allows Gαi protein-coupled receptors to participate in PLC-mediated signal pathways, and such Gαqi5-mediated IP production has been used to assess Gαi-mediated signaling [36,38,39]. As shown in Fig. 8C, IP production by CXCR1 WT, S132A, M241V, F251H and F251Y was stimulated in response to IL-8 in Gαqi5-expressing cells, suggesting that CXCR1 WT and these mutants are indeed coupled to Gαi protein. M241V and F251H constitutively activate Gα15 protein (Fig. 5). We observed a slight increase in basal IP accumulation in Gαqi5-co-transfected cells for M241V (p < 0.001 vs. WT without IL-8 treatment) and for F251H (p < 0.001 vs. WT without IL-8 treatment) (Fig. 8B). Based on the results we draw the conclusion that M241V and F251H are constitutively active mutants coupled to both Gα15 and PTX-sensitive Gαi protein.
3. Discussion
Studies of GPCR crystal structures have revealed the role of DRY motif in stabilizing GPCR conformation through formation of hydrogen and ionic bonds with other amino acid residues that impact GPCR activation and G protein coupling [17,18,40–43]. Substitution mutations of Asp1343.49 to asparagine (D134N) or valine (D134V) resulted in nonfunctional receptors that were devoid of ligand binding. Similar findings have been reported in TM 3.49 of GPCRs such as rat angiotensin II (Ang II) receptor type 1A (AT1A) [44], vasopressin V1a receptor (V1aR) [45], muscarinic receptor [46], and cannabinoid 2 (CB2R) [47,48]. Although D134 mutants are expressed on the surface of transfected cells, they barely bind to the ligand IL-8, suggesting that inactivity of D134 mutants could be due to impaired receptor folding. Recent data indicates that CXCR1 activity involves binding of IL-8 at two distinct receptor sites (N-terminal residues, and extracellular loop/transmembrane residues of receptor) and that coupling between these sites is essential for CXCR1 activation [49]. Considering mutations such as D to N result in loss of specific electrostatic interactions, it is possible that D134 mediates coupling between these two sites that is disrupted in the mutants.
Given that CXCR1 and CXCR2 share 77% amino acid identity and can both bind to IL-8 and initiate chemotaxis and innate immune response, it is unusual that the conserved D134 of CXCR1 plays a distinct role in ligand binding and G protein coupling. This is because CXCR1 is a close homologue to KSHV-GPCR and CXCR2. Importantly, a mutation (D142V) within its DRY motif at the junction of TM3 and i2 loop renders KSHV-GPCR constitutively active, which is responsible for Kaposi’s sarcoma development [26]. It is known that the exchange of Asp138 of the DRY sequence in the CXCR2 with a Val (D138V) (the corresponding amino acid in KSHV-GPCR) results in constitutive activity of CXCR2 and high levels of inositol phosphate accumulation [25]. Our data suggest that the conserved aspartic acid residue within the DRY motif of CXCR1 plays a role in G protein coupling and receptor activation that is distinct from that of KSHV-GPCR and CXCR2. These insights will have important implications in identifying the critical amino acid residues or motifs that are responsible for the selective activation of CXCR1 and CXCR2, and to elucidate distinct functional mechanisms based on structural differences between CXCR1 and CXCR2. Interestingly, an earlier study suggested that KSHV-GPCR originated from CXCR1 or CXCR2 [50] during viral evolution. The similarity in the signaling of the CXCR2 mutant D138V and the KSHV-GPCR supports the hypothesis that KSHV-GPCR and CXCR2 share the same genetic origin [25]. Our results show a distinct role of the conserved D1343.49 on DRY motif in receptor activation between CXCR1 and its homologues (CXCR2 and KSHV-GPCR), another evidence that KSHV likely originated from CXCR2 rather than CXCR1.
Our recent study highlighted the important role of amino acid residues on TM3 and TM6, especially those near intracellular loop of CXCR1, in G protein coupling and receptor activation [36]. Other recent studies on agonist-bound adenosine A2A receptor and rhodopsin structures revealed that the cytoplasmic end of TM6 partially occludes the G-protein-binding site [19,51], suggesting an essential role of the cytoplasmic end of TM6 in G protein coupling and receptor activation. The comparison of agonist-bound, active-state crystal structure and inactive structure of the human beta2 adrenergic receptor and human A2A adenosine receptor demonstrated an outward movement of the cytoplasmic end of transmembrane segment 6 in the active form of the receptor that are remarkably similar to those observed between active rhodopsin and inactive opsin [17,19,41,42,52]. In fact, many constitutively active mutations of GPCRs are found in the bent region at the cytoplasmic end of TM6, particularly in position 6.34, suggesting that this region plays an important role in receptor activation. Mutations on 6.34 site of M5 receptor [53], rat μ-opioid receptor [54], luteinizing hormone receptor [55], thyrotropin receptor [56], and β(1)-adrenergic receptor [20] may generate different degrees of constitutive activity. In addition, highly conserved F2516.44 on TM6 is a key switch residue involved in activation, and mutation on this site in TSH receptor and in LH receptor caused constitutive activation [32,57]. In accordance, our findings indicate that substitution of M2416.34 with Val and F2516.44 with His caused constitutive activity of the receptor, indicating that these conserved amino acids are critical for maintaining the inactive conformation structure of CXCR1 and that mutations on M2416.34 and F2516.44 may disrupt the constrained inactive conformation and result in the constitutive activation of CXCR1.
We recently reported that V2476.40 is critical in CXCR1 activation and that mutations on V2476.40 result in constitutive activation of CXCR1 [36]. In comparison, the two mutants from the current study, M241V and F251H, showed modest constitutive activity. V247A and V247N are potent mutants with constitutive activity which caused the basal IP production to be increased by 78% and 130%, respectively, compared with CXCR1 WT. On the other hand, IP production was increased by 43% and 46% for M241V and F251H, respectively, compared with CXCR1 in the absence of IL-8. M2416.34, V2476.40, and F2516.44 share spatial proximity and are all located at the junction of TM6 and i3 loop of CXCR1. The intracellular loops of GPCRs are crucial for G-protein interactions [22]. The i3 loop of CXCR1, extending from Thr 228 to Gln 236, protrudes into the cytoplasm where it is available for G-protein binding [15]. The fact that different degrees of constitutive activity of CXCR1 can result from mutations of either M241, V247, or F251 on TM6 support the concept that an agonist needs to disrupt just one key intramolecular interaction to stabilize the inactive state of CXCR1, and trigger interaction with G proteins. Our results suggest that regions containing M241, V247, F251 (and probably other surrounding amino acid residues) on TM6, especially those near i3 loop are “hot spots” for stabilizing the inactive configuration of CXCR1 critical for G protein activation and receptor function.
Despite recent demonstration of the structure of resting CXCR1 by NMR [15], the critical structural information of CXCR1, especially ligand-bound as well as G protein-bound inactive and active forms of CXCR1, are not available. We identified a critical amino acid residue (D1343.49) on conserved glutamic acid/aspartic acid–arginine–tyrosine (i.e., the E/DRY) triplet motif at the boundary between TM3 and intracellular loop 2 of class A GPCRs (rhodopsin family) that demonstrated distinct roles in G protein coupling and receptor activation between CXCR1 and its homologues (CXCR2 and KSHV-GPCR). The D1343.49 within the evolutionarily conserved E/DRY motif is important for protein stabilization and/or G protein activation [21,22,58,59]. We showed that, unlike CXCR2 and KSHV-GPCR, substituting the D1343.49 of DRY motif of CXCR1 with N and V results in an almost silent receptor devoid of G protein coupling that was strongly impaired in its ability to bind IL-8. In contrast, mutations on M2416.34 (M241V) and F2516.44 (F251H) led to modest constitutive activation of CXCR1, suggesting that M2416.34 and F2516.44 play a role in stabilizing CXCR1 in the ground (inactive) state and that disruption of intramolecular constrains caused by introducing a residue may lead to constitutive activation of CXCR1. The critical amino acid residues identified in this study together the important residues that we previously identified on CXCR1, provide new insights into ligand binding, G protein coupling, as well as receptor activation of CXCR. This knowledge will hopefully lead to better understanding of the receptor structure and function so that novel and selective agonists/antagonists could be designed for the treatment of COPD or other inflammatory diseases caused by the excessive neutrophil activation as well as for the disruption of breast cancer stem cells.
4. Materials and methods
4.1. Construction of expression vectors and site-directed mutagenesis
Wild type (WT) human CXCR1 cloned into pSFFV·neo vector was provided as a gift from Professor Ingrid Schraufstatter at the La Jolla Institute for Experimental Medicine. After digestion with EcoRI, WT human CXCR1 fragment was subcloned into the pSG5 vector. Oligonucleotides for site-directed mutagenesis were designed to yield several different amino acid substitutions and were synthesized by Invitrogen (Carlsbad, CA). CXCR1 mutations were generated with the Transformer mutagenesis kit (Clontech, Palo Alto, CA) and the final constructs were confirmed by Big Dye Terminator Cycle Sequencing (Perkin Elmer). Plasmid DNA for transient transfection was purified with the EndoFree Plasmid Maxi Kit from Qiagen.
4.2. Transient transfection
COS-7 cells and human embryonic kidney 293 cells (HEK293 cells) from the American Type Culture Collection (ATCC, Rockville, MD) were maintained at 37 °C in humidified air containing 5% CO2 in DMEM with 10% FBS. Cells were grown to 60∼80% confluency prior to transient transfection. Transfection was performed using LipofectAMINE reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instruction. COS-7 cells were incubated with the transfection complex for 5 h at 37 °C. After removal of the transfection medium, the cells were incubated in DMEM with 10% FCS overnight.
4.3. Flow cytometry
HEK 293 cells were transfected with either pSG5 plasmid, or CXCR1 WT or mutants. After 48 h, cells were dissociated and fixed with 4% paraformaldehyde. Cells were then washed three times with cold stain buffer before incubation on ice for 20 min with mouse anti human CD128 FITC (CXCR1) from BD Biosciences (Cat# 555939) (San Diego, CA). Cells were washed three times and resuspended in 0.5 ml of stain buffer. Stained cell samples were analyzed by flow cytometry [36].
4.4. Confocal microscopy
COS-7 cells were transfected with either pSG5 plasmid, or CXCR1 WT or mutants. After 24 h, cells were fixed with 4% paraformaldehyde followed by blocking with 1% BSA in HBSS. Cells were incubated with primary antibody (mouse anti human CD128a (CXCR1)) from BD Pharmingen (San Diego, CA) at 4 °C overnight. After washing three times, the cells were incubated with the secondary antibody (Alexa Fluor 488, goat anti-mouse IgG(H + L) from Invitrogen, CA) at RT for 1 h and counterstained with Dapi. Expression of CXCR1 or the mutants was observed using confocal microscope.
4.5. Inositol Phosphate (IP) Assays
Inositol Phosphate (IP) Assays was conducted as we previously described [36]. Briefly, COS-7 cells were plated in 24-well plates with a density of 1 × 105 cells per well the day before transfection. The cells were co-transfected with 0.3 μg CXCR1 plasmid DNA (or its mutants) and 0.3 μg Gα15 (or Gαqi5) plus 1.5 μl LipofectAMINE reagent and 0.5 μl Plus reagent. In experiments of co-transfection with WT CXCR1 or its mutants and plasmids encoding Gαi2, Gβ1, Gγ2 and phospholipase C (PLC)β2, COS-7 cells were co-transfected with equal amounts of cDNA (0.1 μg per well per component). 24 h after transfection, cells were incubated with inositol-free medium containing 2 μl/ml myo-[2-3H] inositol (Dupont-NEN, Boston, MA) in the absence or presence of 100 ng/ml pertussis toxin (PTX) as indicated for 18 h. At 48 h from the start of transfection, cells were washed with assay buffer [Hank’s balanced salt solution, 0.5% (w/v) crystalline bovine serum albumin, 20 mM HEPES-NaOH, pH 7.4]. IP production was measured as described before [36]. All assays were performed in triplicate, on at least 3 separate occasions with different batches of cells, and always included control cells transfected with WT CXCR1. Data were analyzed using GraphPad Prism (San Diego, CA) and expressed as fold-increase over basal conditions in cells co-transfected with WT CXCR1 and Gα15 plasmid.
4.6. IL-8 binding assay
IL-8 binding assay was conducted as described [36]. COS-7 cells were plated in 12-well plates at a density of 1.6 × 105 cells/ml of DMEM/10% FCS and incubated overnight. The cells were transfected with 0.7 μg DNA plus 2.5 μl of LipofectAMINE reagent and 5 μl Plus Reagent (total transfection volume was 0.5 ml/well). After 48 h from the start of transfection, transfected cells were washed in the binding buffer (HBSS medium containing 0.5% BSA and 25 mM HEPES buffer) and then incubated for 2 h at 4 °C. The final concentration of 125I-IL-8 in the 0.5 ml/well (for 12-well plate) of medium was 0.07 nM (0.05 μCi). The range of unlabeled IL-8 concentrations in the binding assays was 0.1–300 nM [0, 0.1, 0.3, 1, 3, 10, 30, 100, 300 nM]. Non-specific 125I-IL-8 binding was determined by incubating the cells with 125I-IL-8 in the presence of 250 nM of cold IL-8. For maximal binding of 125I-IL-8 to CXCR1 WT and mutants, unlabelled IL-8 was not added. After washing three times with 2 ml of ice-cold binding buffer, the cells were lysed in 0.5 ml of 1 N NaOH and the lysates were counted using a scintillation counter. The data were curve fitted and affinity constant (Kd) and maximum total binding (Bmax) values were calculated using GraphPad Prism (San Diego, CA). All experiments were carried out in triplicate.
4.7. Statistical analysis
Results were expressed as mean ± standard error of the mean (SEM) from n determinations. Statistical analyses were performed with GraphPad Prism 5.1 software. The significance of differences between the means was determined with the Student’s two tailed t test. Values of p < 0.05 were considered as statistically significant and are denoted with an asterisk (*): ∗p < 0.05; ∗∗p < 0.01. All the experiments were repeated at least three times and each point was tested in triplicate.
Author contributions
XH and YF designed the study, performed experiments, analyzed data, and wrote the manuscript. XC and CG acquired data and interpreted the results. WAB designed the study, provided funding and wrote the manuscript.
Acknowledgments
We are grateful to Dr. A. Shenker for critical reading and comments on the manuscript. We thank Z. Stender, Dr. G. Liu, and J. Wu for their technical help with inositol phosphate assay. This project was funded by NIH grants R01HL075677 and R01HL081863 to WAB. This work was performed within the Russian Government Program of Competitive Growth of Kazan Federal University.
Contributor Information
Xinbing Han, Email: XinbingHan12@gmail.com.
William A. Boisvert, Email: wab@hawaii.edu.
References
- 1.Stillie R., Farooq S.M., Gordon J.R., Stadnyk A.W. The functional significance behind expressing two IL-8 receptor types on PMN. J. Leukoc. Biol. 2009;86:529–543. doi: 10.1189/jlb.0208125. [DOI] [PubMed] [Google Scholar]
- 2.Bizzarri C., Beccari A.R., Bertini R., Cavicchia M.R., Giorgini S., Allegretti M. ELR+ CXC chemokines and their receptors (CXC chemokine receptor 1 and CXC chemokine receptor 2) as new therapeutic targets. Pharmacol. Ther. 2006;112:139–149. doi: 10.1016/j.pharmthera.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 3.Cannon M. The KSHV and other human herpesviral G protein-coupled receptors. Curr. Top. Microbiol. Immunol. 2007;312:137–156. doi: 10.1007/978-3-540-34344-8_5. [DOI] [PubMed] [Google Scholar]
- 4.Nasser M.W., Raghuwanshi S.K., Grant D.J., Jala V.R., Rajarathnam K., Richardson R.M. Differential activation and regulation of CXCR1 and CXCR2 by CXCL8 monomer and dimer. J. Immunol. 2009;183:3425–3432. doi: 10.4049/jimmunol.0900305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chuntharapai A., Kim K.J. Regulation of the expression of IL-8 receptor A/B by IL-8: possible functions of each receptor. J. Immunol. 1995;155:2587–2594. [PubMed] [Google Scholar]
- 6.Hartl D., Latzin P., Hordijk P., Marcos V., Rudolph C., Woischnik M., Krauss-Etschmann S., Koller B., Reinhardt D., Roscher A.A., Roos D., Griese M. Cleavage of CXCR1 on neutrophils disables bacterial killing in cystic fibrosis lung disease. Nat. Med. 2007;13:1423–1430. doi: 10.1038/nm1690. [DOI] [PubMed] [Google Scholar]
- 7.Shamaladevi N., Lyn D.A., Escudero D.O., Lokeshwar B.L. CXC receptor-1 silencing inhibits androgen-independent prostate cancer. Cancer Res. 2009;69:8265–8274. doi: 10.1158/0008-5472.CAN-09-0374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gabellini C., Trisciuoglio D., Desideri M., Candiloro A., Ragazzoni Y., Orlandi A., Zupi G., Del Bufalo D. Functional activity of CXCL8 receptors, CXCR1 and CXCR2, on human malignant melanoma progression. Eur. J. Cancer. 2009;45:2618–2627. doi: 10.1016/j.ejca.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 9.Ginestier C., Liu S., Diebel M.E., Korkaya H., Luo M., Brown M., Wicinski J., Cabaud O., Charafe-Jauffret E., Birnbaum D., Guan J.L., Dontu G., Wicha M.S. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J. Clin. Invest. 2010;120:485–497. doi: 10.1172/JCI39397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eash K.J., Greenbaum A.M., Gopalan P.K., Link D.C. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J. Clin. Invest. 2010;120:2423–2431. doi: 10.1172/JCI41649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Acosta J.C., O’Loghlen A., Banito A., Guijarro M.V., Augert A., Raguz S., Fumagalli M., Da Costa M., Brown C., Popov N., Takatsu Y., Melamed J., d’Adda di Fagagna F., Bernard D., Hernando E., Gil J. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–1018. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 12.Fan X., Patera A.C., Pong-Kennedy A., Deno G., Gonsiorek W., Manfra D.J., Vassileva G., Zeng M., Jackson C., Sullivan L., Sharif-Rodriguez W., Opdenakker G., Van Damme J., Hedrick J.A., Lundell D., Lira S.A., Hipkin R.W. Murine CXCR1 is a functional receptor for GCP-2/CXCL6 and interleukin-8/CXCL8. J. Biol. Chem. 2007;282:11658–11666. doi: 10.1074/jbc.M607705200. [DOI] [PubMed] [Google Scholar]
- 13.Rajagopal S., Rajagopal K., Lefkowitz R.J. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat. Rev. Drug Discov. 2010;9:373–386. doi: 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katritch V., Cherezov V., Stevens R.C. Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol. Sci. 2012;33:17–27. doi: 10.1016/j.tips.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park S.H., Das B.B., Casagrande F., Tian Y., Nothnagel H.J., Chu M., Kiefer H., Maier K., De Angelis A.A., Marassi F.M., Opella S.J. Structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature. 2012;491:779–783. doi: 10.1038/nature11580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu B., Chien E.Y., Mol C.D., Fenalti G., Liu W., Katritch V., Abagyan R., Brooun A., Wells P., Bi F.C., Hamel D.J., Kuhn P., Handel T.M., Cherezov V., Stevens R.C. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scheerer P., Park J.H., Hildebrand P.W., Kim Y.J., Krauss N., Choe H.W., Hofmann K.P., Ernst O.P. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 18.Hofmann K.P., Scheerer P., Hildebrand P.W., Choe H.W., Park J.H., Heck M., Ernst O.P. A G protein-coupled receptor at work: the rhodopsin model. Trends Biochem. Sci. 2009;34:540–552. doi: 10.1016/j.tibs.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 19.Xu F., Wu H., Katritch V., Han G.W., Jacobson K.A., Gao Z.G., Cherezov V., Stevens R.C. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moukhametzianov R., Warne T., Edwards P.C., Serrano-Vega M.J., Leslie A.G., Tate C.G., Schertler G.F. Two distinct conformations of helix 6 observed in antagonist-bound structures of a beta1-adrenergic receptor. Proc. Natl. Acad. Sci. U.S.A. 2011;108:8228–8232. doi: 10.1073/pnas.1100185108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ballesteros J.A., Jensen A.D., Liapakis G., Rasmussen S.G., Shi L., Gether U., Javitch J.A. Activation of the beta 2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J. Biol. Chem. 2001;276:29171–29177. doi: 10.1074/jbc.M103747200. [DOI] [PubMed] [Google Scholar]
- 22.Oldham W.M., Hamm H.E. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 2008;9:60–71. doi: 10.1038/nrm2299. [DOI] [PubMed] [Google Scholar]
- 23.Lu Z.L., Hulme E.C. The functional topography of transmembrane domain 3 of the M1 muscarinic acetylcholine receptor, revealed by scanning mutagenesis. J. Biol. Chem. 1999;274:7309–7315. doi: 10.1074/jbc.274.11.7309. [DOI] [PubMed] [Google Scholar]
- 24.Arvanitakis L., Geras-Raaka E., Varma A., Gershengorn M.C., Cesarman E. Human herpesvirus KSHV encodes a constitutively active G-protein-coupled receptor linked to cell proliferation. Nature. 1997;385:347–350. doi: 10.1038/385347a0. [DOI] [PubMed] [Google Scholar]
- 25.Burger M., Burger J.A., Hoch R.C., Oades Z., Takamori H., Schraufstatter I.U. Point mutation causing constitutive signaling of CXCR2 leads to transforming activity similar to Kaposi’s sarcoma herpesvirus-G protein-coupled receptor. J. Immunol. 1999;163:2017–2022. [PubMed] [Google Scholar]
- 26.O’Hayre M., Vazquez-Prado J., Kufareva I., Stawiski E.W., Handel T.M., Seshagiri S., Gutkind J.S. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat. Rev. Cancer. 2013;13:412–424. doi: 10.1038/nrc3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yarchoan R. Key role for a viral lytic gene in Kaposi’s sarcoma. N. Engl. J. Med. 2006;355:1383–1385. doi: 10.1056/NEJMcibr063911. [DOI] [PubMed] [Google Scholar]
- 28.Philpott N., Bakken T., Pennell C., Chen L., Wu J., Cannon M. The Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor contains an immunoreceptor tyrosine-based inhibitory motif that activates Shp2. J. Virol. 2011;85:1140–1144. doi: 10.1128/JVI.01362-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burger M., Hartmann T., Burger J.A., Schraufstatter I. KSHV-GPCR and CXCR2 transforming capacity and angiogenic responses are mediated through a JAK2-STAT3-dependent pathway. Oncogene. 2005;24:2067–2075. doi: 10.1038/sj.onc.1208442. [DOI] [PubMed] [Google Scholar]
- 30.Chen S., Xu M., Lin F., Lee D., Riek P., Graham R.M. Phe310 in transmembrane VI of the alpha1B-adrenergic receptor is a key switch residue involved in activation and catecholamine ring aromatic bonding. J. Biol. Chem. 1999;274:16320–16330. doi: 10.1074/jbc.274.23.16320. [DOI] [PubMed] [Google Scholar]
- 31.Govaerts C., Lefort A., Costagliola S., Wodak S.J., Ballesteros J.A., Van Sande J., Pardo L., Vassart G. A conserved Asn in transmembrane helix 7 is an on/off switch in the activation of the thyrotropin receptor. J. Biol. Chem. 2001;276:22991–22999. doi: 10.1074/jbc.M102244200. [DOI] [PubMed] [Google Scholar]
- 32.Neumann S., Krause G., Chey S., Paschke R. A free carboxylate oxygen in the side chain of position 674 in transmembrane domain 7 is necessary for TSH receptor activation. Mol. Endocrinol. 2001;15:1294–1305. doi: 10.1210/mend.15.8.0672. [DOI] [PubMed] [Google Scholar]
- 33.Zhang M., Mizrachi D., Fanelli F., Segaloff D.L. The formation of a salt bridge between helices 3 and 6 is responsible for the constitutive activity and lack of hormone responsiveness of the naturally occurring L457R mutation of the human lutropin receptor. J. Biol. Chem. 2005;280:26169–26176. doi: 10.1074/jbc.M502102200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lei Y., Hagen G.M., Smith S.M., Liu J., Barisas G., Roess D.A. Constitutively-active human LH receptors are self-associated and located in rafts. Mol. Cell Endocrinol. 2007;260–262:65–72. doi: 10.1016/j.mce.2005.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu S.M., Leschek E.W., Rennert O.M., Chan W.Y. Luteinizing hormone receptor mutations in disorders of sexual development and cancer. Front. Biosci. 2000;5:D343–352. doi: 10.2741/wu. [DOI] [PubMed] [Google Scholar]
- 36.Han X., Tachado S.D., Koziel H., Boisvert W.A. Leu128(3.43) (l128) and Val247(6.40) (V247) of CXCR1 are critical amino acid residues for g protein coupling and receptor activation. PLoS One. 2012;7:e42765. doi: 10.1371/journal.pone.0042765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu D., LaRosa G.J., Simon M.I. G protein-coupled signal transduction pathways for interleukin-8. Science. 1993;261:101–103. doi: 10.1126/science.8316840. [DOI] [PubMed] [Google Scholar]
- 38.White C.D., Coetsee M., Morgan K., Flanagan C.A., Millar R.P., Lu Z.L. A crucial role for Galphaq/11, but not Galphai/o or Galphas, in gonadotropin-releasing hormone receptor-mediated cell growth inhibition. Mol. Endocrinol. 2008;22:2520–2530. doi: 10.1210/me.2008-0122. [DOI] [PubMed] [Google Scholar]
- 39.Conklin B.R., Farfel Z., Lustig K.D., Julius D., Bourne H.R. Substitution of three amino acids switches receptor specificity of Gq alpha to that of Gi alpha. Nature. 1993;363:274–276. doi: 10.1038/363274a0. [DOI] [PubMed] [Google Scholar]
- 40.Jaakola V.P., Griffith M.T., Hanson M.A., Cherezov V., Chien E.Y., Lane J.R., Ijzerman A.P., Stevens R.C. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rasmussen S.G., Choi H.J., Fung J.J., Pardon E., Casarosa P., Chae P.S., Devree B.T., Rosenbaum D.M., Thian F.S., Kobilka T.S., Schnapp A., Konetzki I., Sunahara R.K., Gellman S.H., Pautsch A., Steyaert J., Weis W.I., Kobilka B.K. Structure of a nanobody-stabilized active state of the β(2) adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park J.H., Scheerer P., Hofmann K.P., Choe H.W., Ernst O.P. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature. 2008;454:183–187. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- 43.Rasmussen S.G., Choi H.J., Rosenbaum D.M., Kobilka T.S., Thian F.S., Edwards P.C., Burghammer M., Ratnala V.R., Sanishvili R., Fischetti R.F., Schertler G.F., Weis W.I., Kobilka B.K. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 44.Ohyama K., Yamano Y., Sano T., Nakagomi Y., Wada M., Inagami T. Role of the conserved DRY motif on G protein activation of rat angiotensin II receptor type 1A. Biochem. Biophys. Res. Commun. 2002;292:362–367. doi: 10.1006/bbrc.2002.6670. [DOI] [PubMed] [Google Scholar]
- 45.Hawtin S.R. Charged residues of the conserved DRY triplet of the vasopressin V1a receptor provide molecular determinants for cell surface delivery and internalization. Mol. Pharmacol. 2005;68:1172–1182. doi: 10.1124/mol.105.013359. [DOI] [PubMed] [Google Scholar]
- 46.Lu Z.L., Curtis C.A., Jones P.G., Pavia J., Hulme E.C. The role of the aspartate–arginine–tyrosine triad in the m1 muscarinic receptor: mutations of aspartate 122 and tyrosine 124 decrease receptor expression but do not abolish signaling. Mol. Pharmacol. 1997;51:234–241. doi: 10.1124/mol.51.2.234. [DOI] [PubMed] [Google Scholar]
- 47.Rhee M.H., Nevo I., Levy R., Vogel Z. Role of the highly conserved Asp-Arg-Tyr motif in signal transduction of the CB2 cannabinoid receptor. FEBS Lett. 2000;466:300–304. doi: 10.1016/s0014-5793(00)01094-2. [DOI] [PubMed] [Google Scholar]
- 48.Feng W., Song Z.H. Effects of D3.49A, R3.50A, and A6.34E mutations on ligand binding and activation of the cannabinoid-2 (CB2) receptor. Biochem. Pharmacol. 2003;65:1077–1085. doi: 10.1016/s0006-2952(03)00005-4. [DOI] [PubMed] [Google Scholar]
- 49.Joseph P.R., Sawant K.V., Isley A., Pedroza M., Garofalo R.P., Richardson R.M., Rajarathnam K. Dynamic conformational switching in the chemokine ligand is essential for G-protein-coupled receptor activation. Biochem. J. 2013;456:241–251. doi: 10.1042/BJ20130148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gershengorn M.C., Geras-Raaka E., Varma A., Clark-Lewis I. Chemokines activate Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor in mammalian cells in culture. J. Clin. Invest. 1998;102:1469–1472. doi: 10.1172/JCI4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lebon G., Warne T., Edwards P.C., Bennett K., Langmead C.J., Leslie A.G., Tate C.G. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011;474:521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rosenbaum D.M., Zhang C., Lyons J.A., Holl R., Aragao D., Arlow D.H., Rasmussen S.G., Choi H.J., Devree B.T., Sunahara R.K., Chae P.S., Gellman S.H., Dror R.O., Shaw D.E., Weis W.I., Caffrey M., Gmeiner P., Kobilka B.K. Structure and function of an irreversible agonist-β(2) adrenoceptor complex. Nature. 2011;469:236–240. doi: 10.1038/nature09665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kjelsberg M.A., Cotecchia S., Ostrowski J., Caron M.G., Lefkowitz R.J. Constitutive activation of the alpha 1B-adrenergic receptor by all amino acid substitutions at a single site. Evidence for a region which constrains receptor activation. J. Biol. Chem. 1992;267:1430–1433. [PubMed] [Google Scholar]
- 54.Huang P., Li J., Chen C., Visiers I., Weinstein H., Liu-Chen L.Y. Functional role of a conserved motif in TM6 of the rat mu opioid receptor: constitutively active and inactive receptors result from substitutions of Thr6.34(279) with Lys and Asp. Biochemistry. 2001;40:13501–13509. doi: 10.1021/bi010917q. [DOI] [PubMed] [Google Scholar]
- 55.Schulz A., Schoneberg T., Paschke R., Schultz G., Gudermann T. Role of the third intracellular loop for the activation of gonadotropin receptors. Mol. Endocrinol. 1999;13:181–190. doi: 10.1210/mend.13.2.0233. [DOI] [PubMed] [Google Scholar]
- 56.Parma J., Duprez L., Van Sande J., Cochaux P., Gervy C., Mockel J., Dumont J., Vassart G. Somatic mutations in the thyrotropin receptor gene cause hyperfunctioning thyroid adenomas. Nature. 1993;365:649–651. doi: 10.1038/365649a0. [DOI] [PubMed] [Google Scholar]
- 57.Latronico A.C., Abell A.N., Arnhold I.J., Liu X., Lins T.S., Brito V.N., Billerbeck A.E., Segaloff D.L., Mendonca B.B. A unique constitutively activating mutation in third transmembrane helix of luteinizing hormone receptor causes sporadic male gonadotropin-independent precocious puberty. J. Clin. Endocrinol. Metab. 1998;83:2435–2440. doi: 10.1210/jcem.83.7.4968. [DOI] [PubMed] [Google Scholar]
- 58.Rovati G.E., Capra V., Neubig R.R. The highly conserved DRY motif of class A G protein-coupled receptors: beyond the ground state. Mol. Pharmacol. 2007;71:959–964. doi: 10.1124/mol.106.029470. [DOI] [PubMed] [Google Scholar]
- 59.Kamps A.R., Pruitt M.M., Herriges J.C., Coffman C.R. An evolutionarily conserved arginine is essential for Tre1 G protein-coupled receptor function during germ cell migration in Drosophila melanogaster. PLoS One. 2010;5:e11839. doi: 10.1371/journal.pone.0011839. [DOI] [PMC free article] [PubMed] [Google Scholar]