

Abstract
Malignant gliomas are the most common malignant primary brain tumors and one of the most challenging forms of cancers to treat. Despite advances in conventional treatment, the outcome for patients remains almost universally fatal. This poor prognosis is due to therapeutic resistance and tumor recurrence after surgical removal. However, over the past decade, molecular targeted therapy has held the promise of transforming the care of malignant glioma patients. Significant progress in understanding the molecular pathology of gliomagenesis and maintenance of the malignant phenotypes will open opportunities to rationally develop new molecular targeted therapy options. Recently, therapeutic strategies have focused on targeting pro-growth signaling mediated by receptor tyrosine kinase/RAS/phosphatidylinositol 3-kinase pathway, proangiogenic pathways, and several other vital intracellular signaling networks, such as proteasome and histone deacetylase. However, several factors such as cross-talk between the altered pathways, intratumoral molecular heterogeneity, and therapeutic resistance of glioma stem cells (GSCs) have limited the activity of single agents. Efforts are ongoing to study in depth the complex molecular biology of glioma, develop novel regimens targeting GSCs, and identify biomarkers to stratify patients with the individualized molecular targeted therapy. Here, we review the molecular alterations relevant to the pathology of malignant glioma, review current advances in clinical targeted trials, and discuss the challenges, controversies, and future directions of molecular targeted therapy.
Abbreviations: BBB, blood-brain barrier; CDK4, cyclin-dependent kinase 4; CDKN2A, cyclin-dependent kinase inhibitor 2A; c-KIT, v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog; EGFR, epidermal growth factor receptor; FT, farnesyl transferase; FTI, FT inhibitor; GBMs, glioblastomas; GSCs, glioma stem cells; HDAC, histone deacetylase; IDH, isocitrate dehydrogenase; MAPK, mitogen-activated protein kinase; MGMT, O6-methylguanine DNA methyltransferase; miRNAs, microRNAs; mTOR, mammalian target of rapamycin; OS, overall survival; PDGFR, PDGF receptor; PFS, progression-free survival; PI3K, phosphatidylinositol 3-kinase; PTEN, phosphatase and tensin homolog; RB1, retinoblastoma susceptibility protein 1; RTK, receptor tyrosine kinase; SHH, sonic hedgehog; TCGA, The Cancer Genome Atlas; TKIs, receptor tyrosine kinase inhibitors; TMZ, temozolomide; VEGF, vascular endothelial growth factor
Introduction
Gliomas account for about 80% of primary malignant tumors in the central nervous system, and World Health Organization (WHO) classification divides gliomas into four grades with increasing degree of malignancy [1]. Each subgroup has a relatively specific prognosis that guides the clinical management; unfortunately, anaplastic gliomas (WHO III) and glioblastomas (GBMs, WHO IV) constitute the majority of gliomas and are essentially incurable.
Currently, only surgical resection and adjuvant chemotherapy with temozolomide (TMZ) combined with radiotherapy are standard-of-care treatment strategies for this disease. However, the malignant behavior of these cancers with resistance to chemotherapy and radiation results in a high recurrence rate, and thus, patients with malignant glioma derive little benefit from standard treatments [2]. The disease ultimately follows a fatal course with the median survival of 12 to 15 months and 2 to 5 years for patients with GBM and anaplastic glioma, respectively [3].
To break through these challenges for malignant glioma therapy posed by limitations in the current therapeutic strategies, novel therapies such as molecular targeted therapy, immunotherapy, gene therapy, stem cell–based therapies, and nanotechnology have emerged from the interface between preclinical and clinical research [4]. Due to the success of molecular targeted therapy in several other cancer types such as non–small cell lung cancer [5], melanoma [6], and chronic myelogenous leukemia [7], this therapeutic strategy holds significant promise for the treatment of malignant glioma and has greatly advanced over the past decade, with such molecularly targeted therapeutics as bevacizumab, a monoclonal antibody against vascular endothelial growth factor (VEGF), being granted approval by the US Food and Drug Administration for treating recurrent GBM in 2009 [8], [9], [10].
However, despite increasing radiographic response and progression-free survival (PFS), bevacizumab does not benefit overall survival (OS) in either recurrent GBM or newly diagnosed GBM [11], [12], [13], [14]. Hence, with an increasing understanding of the molecular pathology of malignant glioma, novel signaling pathways driving gliomagenesis and progression that are candidates to become therapeutic targets and novel agents that may target relevant pathways more effectively are urgently needed. Herein, we set forth the rationales for targeting molecular pathways in malignant glioma, review current clinical trials for these tumors, and discuss the challenges, controversies, and future directions of molecular targeted therapy.
Multiple Core Signaling Pathways in Malignant Glioma
With high genetic and pathologic heterogeneity even in the same tumor sample, and low prevalence of each molecular abnormality, malignant gliomas are usually not defined by a single genetic mutation or molecular alteration. Thus, a “single gene–based” process of target identification and targeted therapy development is prohibitively difficult. It will be necessary to understand the pathways within which different genetic alterations function to drive gliomagenesis, progression, and treatment resistance and then focus our efforts in the development of a biologically meaningful classification scheme for treating these tumors.
Recently, The Cancer Genome Atlas (TCGA) research network identified three core signaling pathways underlying malignant glioma pathogenesis: receptor tyrosine kinase (RTK)/RAS/phosphatidylinositol 3-kinase (PI3K), p53, and retinoblastoma protein (RB) signaling pathways [15] (as shown in Figure 1). In addition, other canonical signaling pathways like proangiogenic pathway are important for gliomagenesis and maintenance of glioma phenotypes.
Figure 1.
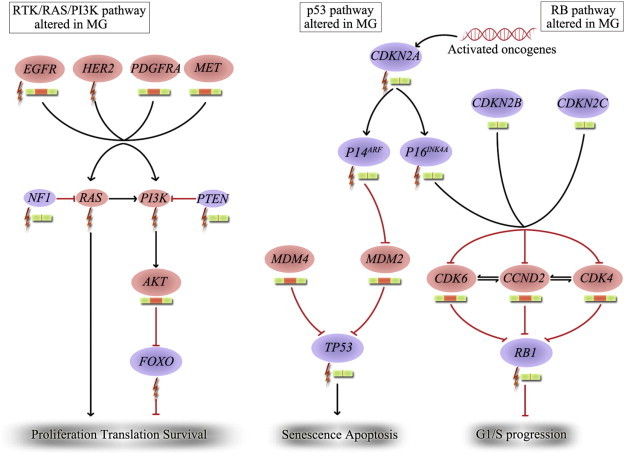
Three core signaling pathways altered in malignant gliomas. DNA alterations and copy number changes in the RTK/RAS/PI3K, RB, and p53 are shown. Moreover, activating genetic alterations are indicated in red, and inactivating genetic alterations are indicated in purple. In each pathway, the altered components and the type of alteration are indicated. The types of alteration are represented by different patterns as follows:  represents mutation,
represents mutation,  represents amplification and
represents amplification and 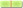 represents homozygous deletion, while
represents homozygous deletion, while  represents gene with normal copy number. MG indicates malignant glioma; HER, human epithelial receptor; MET, mesenchymal epithelial transition factor.
represents gene with normal copy number. MG indicates malignant glioma; HER, human epithelial receptor; MET, mesenchymal epithelial transition factor.
RTK/RAS/PI3K Pathway
Growth factor receptors [e.g., epidermal growth factor receptor (EGFR) and PDGF receptor A (PDGFRA)] are RTKs that are plasma membrane–spanning proteins consisting of extracellular domains that can be bound by the receptors' respective ligands (e.g., EGF and PDGF) and intracellular domains associated with tyrosine kinase activity. Activation of several RTKs is frequently found in malignant glioma, such as EGFR gene amplification that occurs in approximately 40% of patients with GBM and PDGFRA gene amplification that occurs in up to 16% of GBM [16], [17]. Usually, RTKs are activated through the interaction of growth factors and RTKs, but a unique EGFR variant (EGFRvIII) shows ligand-independent constitutive activation of the receptor. This deletion mutant is observed in approximately 30% to 50% of EGFR-amplified gliomas [16]. Mutated RTKs will contribute to recruitment of PI3K, RAS, and so on, to the cell membrane to trigger signal transduction cascades. It is thought that PI3K then initiates activation of downstream effectors such as AKT and mammalian target of rapamycin (mTOR) that function as central regulators of cellular metabolism, proliferation, cell cycle control, differentiation, and angiogenesis [18]. This activation pathway is held in check by the phosphatase and tensin homolog (PTEN) protein inhibiting PI3K activity. However, biallelic inactivation of the PTEN gene located on chromosome 10q occurs in up to 40% of malignant gliomas, which makes the PI3K/AKT pathway active constitutively [18], [19]. Mutation of RAS increases the activity of the RAS–RAF–mitogen-activated protein kinase (RAS/RAF/MAPK) pathway and results in uncontrolled cell growth and proliferation; however, RAS mutation is a fairly rare occurrence in malignant glioma. Mutation or amplification of upstream RTKs and mutation or deletion of the neurofibromin 1 gene that encodes neurofibromin functioning as a negative regulator of RAS seem to accomplish the result of a permanent activation of RAS, leading to proliferation, motility, and survival [15], [20]. In sum, all genetic alterations of the RTK/RAS/PI3K pathway in GBM were confirmed by TCGA with a total percentage up to 88% of tumors [15].
P53 Pathway
The Tumor Protein p53 (TP53) tumor suppressor gene encodes a protein that regulates several cellular programs including the cell cycle arrest, response of cells to DNA damage, senescence, apoptosis, and differentiation [21], [22]. When cells are under genotoxic and cytotoxic stress, p53 functions as a transcription factor to regulate expression of downstream effector genes to determine cell fate [23], [24]. Loss of normal TP53 function resulting from TP53/mouse double-minute 2 (MDM2)/MDM4/p14ARF alterations has been linked to clonal expansion of glioma cells [25]. The human homolog of MDM2 inhibits p53 function, MDM4 regulates p53 activity, and p14ARF is negatively regulated by p53 [21], [24]. Of these classic pathway targets, TCGA has demonstrated altered constituents of the pathway including TP53 mutation or deletion (35%), MDM2 amplification (14%), MDM4 amplification (7%), and p14ARF mutation or deletion (49%) in GBM [15]. Notably, MDM2 amplification and TP53 mutation are alterations found in a mutually exclusive fashion, as well as p14ARF alteration and TP53 mutation [21]. However, perhaps as a result of its near-ubiquitous pathway inactivation, TP53 status has not been found to display any clear relationship with treatment and outcome in malignant glioma [22].
RB Pathway
Like TP53, RB is a tumor suppressor gene encoding the retinoblastoma susceptibility protein 1 (RB1) that inhibits entry of cells through G1 into the S-phase of the cell cycle [21]. When phosphorylated by cyclin D, cyclin-dependent kinase 4 (CDK4), and CDK6, RB1 will be inactive, thereby disinhibiting progression through the cell cycle [25]. Thus, aberration of associated cell-cycle regulators from genetic alteration of p16INK4a/CDK4/RB1 pathway components leads to glioma proliferation [23]. RB1 mutation or deletion and CDK4 amplification account for inactivation of RB1, and cyclin-dependent kinase inhibitor 2A (CDKN2A) gene mutation or homozygous deletion also results in loss of normal RB1 function [15], [21]. Overall, the frequency of genetic alterations in this pathway amounts to 78% of GBMs, with CDKN2B deletion (47%), CDKN2C deletion (2%), cyclin D2 (CCND2) amplification (2%), CDK6 amplification (1%), RB1 mutation or deletion (11%), CDK4 amplification (18%), and CDKN2A(p16INK4a) mutation or homozygous deletion (52%), as reported by TCGA [15]. Among them, CDKN2A mutation or homozygous deletion leads to loss of p16INK4a, which is an inhibitor of CDK4, and the CDKN2A gene encodes p16INK4a and p14ARF that exert respective functions in the RB and p53 pathways, therefore revealing the critical importance of the single genetic inactivation of CDKN2A for these two core pathways in the growth of glioma [25].
Proangiogenic Pathway
For angiogenesis, several signaling pathways are theorized to contribute to the process of vasculature development. In one model of step-wise progression, the first step of glioma achieving its vasculature is vascular co-option, a process by which several proangiogenic factors such as angiopoietin-2 (ANG-2) and its receptor tyrosine kinase with immunoglobulin-like and epidermal growth factor homology domains 2 (TIE-2) are upregulated in endothelial and tumor cells that promote vessel disruption, and then VEGF binding to VEGF receptor (VEGFR) activates intracellular signaling cascades transduced by RAS/MAPK and PI3K/AKT pathways to promote migration and proliferation of endothelial cells and stimulate formation of new blood vessels and also induces endothelium to express integrin that mediates largely the final stages of angiogenesis [26], [27], [28], [29]. Activated endothelial cells also secrete PDGF to recruit pericytes to the new vessels, stabilizing them in a process mediated by the angiopoietin/TIE pathway [30], [31]. Furthermore, several other pathways have been proposed to contribute to the process of angiogenesis, such as erythropoietin and its receptor, Delta-like 4 and its receptor Notch, hypoxia-induced factor-1α, basic fibroblast growth factor, neuropilin, and stromal-derived factor 1 [25], [28], [32], [33]. In addition, endogenous angiogenesis inhibitors such as the soluble form of the VEGFR 1, thrombospondin-1, angiostatin, vasculostatin, and endostatin can play important roles in the delicate balance of angiogenic potential in tumors [29], [32].
Current Standard Therapeutic Modalities in Malignant Glioma
Radiotherapy + TMZ
Of all adjuvant therapies, radiotherapy offers relatively greater magnitude of survival benefit, so that almost all patients with malignant gliomas receive it right after surgery [34]. More recently, radiotherapy plus the chemotherapy agent TMZ, a new-generation alkylating agent, for GBM patients has been evaluated in a randomized phase III trial. This regimen, which has minimal additional toxicity, was demonstrated to increase the median OS to 14.6 months and the 2-year OS to 27.2%, compared to 12.1 months and 10.9%, respectively, in the radiotherapy alone group [35], [36]. Moreover, patients with epigenetic silencing of O6-methylguanine DNA methyltransferase (MGMT) benefitted more from TMZ treatment [37]. On the basis of these findings, TMZ has been incorporated into multimodal treatment strategies for malignant gliomas.
Antiangiogenesis
Contemporary clinical studies also focus on the role of angiogenesis, which is a hallmark of GBM. Currently, antiangiogenesis therapies using the antibody against VEGF and the antagonists of VEGFR/integrin are being increasingly investigated for malignant glioma.
VEGF/VEGFR
The VEGF/VEGFR system is the major and critical regulator of angiogenesis on malignant gliomas. The level of VEGF expression has been positively related to the tumor’s malignancy degree. Hence, targeting VEGF/VEGFR is proposed to be an effective means to control glioma growth [26], [27], [38].
Given that bevacizumab has not been demonstrated to increase OS in GBM, investigators have shifted strategies towards combined therapies, including bevacizumab with radiotherapy and other chemotherapeutics. A multicenter study and a meta-analysis of bevacizumab in combination with irinotecan in recurrent malignant glioma only demonstrated higher PFS at 6 months (PFS-6) and response rate and similar OS compared with bevacizumab monotherapy [39], [40]. Similar results were also obtained from a phase II trial, which compared the bevacizumab + TMZ + radiotherapy with TMZ + radiotherapy for newly diagnosed GBM [41].
In addition, the clinical benefits of bevacizumab seem obvious (e.g., decreasing peritumoral edema, reducing amount of steroid, and improving neurologic symptoms), but it still has not been approved by the European Medical Agency for malignant glioma treatment [2]. Moreover, recent clinical studies have implicated that bevacizumab only had a transient antiangiogenic antiglioma effect and made a subset of GBM patients develop multifocal or diffuse recurrence during the course of bevacizumab therapy. This phenomenon appears to be more prevalent in malignant glioma patients with bevacizumab over-usage [28], [29]. Likewise, patients who have failed bevacizumab have particularly dismal outcomes, as the median OS and PFS-6 have been reported to be 2 to 5 months and 0% to 4%, respectively [42]. Thus, there is a clear need for salvage therapies for GBM patients with rebound of intracranial edema and diffuse invasive recurrence patterns.
Notably, Aguilera et al. reported that two cases with diffuse intrinsic pontine glioma had ongoing PFS of 37 and 47 months from diagnosis and decreased tumor size by > 65% [43] with bevacizumab + radiation + TMZ. Clinical trials are expected to re-evaluate response of the therapeutic schedule in malignant gliomas based on the positive impact of bevacizumab on survival.
Aflibercept is a novel agent that targets VEGF with a soluble decoy VEGFR fused to an immunoglobulin constant region and also binds placental growth factor [2]. However, a phase II trial of aflibercept monotherapy in recurrent malignant glioma showed minimal evidence of single-agent activity with PFS-6 of 7.7% and median PFS of 12 weeks for GBM patients and PFS-6 of 25% and median PFS of 24 weeks for anaplastic glioma patients [44]. Additionally, long-term treatment with aflibercept seemed to result in acquisition of an invasive phenotype of glioma, which seemed to coincide with preliminary data in GBM patients treated with bevacizumab [45].
RTK inhibitors (TKIs) targeting VEGFR include cediranib, vatalanib, pazopanib, cabozanitib, and several other multitargeted kinase inhibitors. Recently, a phase II study of cediranib [inhibitor of VEGFR, PDGFR, fibroblast growth factor receptor 1 (FGFR1), and v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog (c-KIT)] monotherapy in patients with recurrent GBM showed that PFS-6 was 25.8% and the radiographic partial responses were 56.7% by magnetic resonance imaging with three-dimensional measurements and had a steroid-sparing effect [46]. Concurrently, results indicated that cediranib could normalize tumor vessels, reduce cellularity in tumor central area, reduce peritumor edema, and inhibit a second wave of angiogenesis [47], [48], which presumes that vascular normalization can potentially be beneficial for chemotherapy delivery and provide a rationale for combined therapy. Another study demonstrated that cediranib enhanced the effectiveness of TMZ in both wild-type EGFR and EGFRvIII expressing glioma cells [49]. Notably, the structural and functional normalization of tumor vessels induced by cediranib also improved tumor blood perfusion for 1 month and was associated with longer survival in GBM patients [50]. These results implicate that anti-VEGFR may improve glioma patient survival by feeding the cancer with normalized vasculature and improving chemotherapy delivery rather than starve the tumor through elimination or reduction of tumor vasculature.
Concomitant and adjuvant TMZ and radiotherapy with vatalanib (inhibitor of VEGFR, PDGFR, and c-KIT) was evaluated in newly diagnosed GBM patients in phase I/II trials. The result showed that the median OS and median PFS were 17.3 and 6.8 months, respectively [51]. Similar to vatalanib, pazopanib (inhibitor of VEGFR, PDGFR, and c-KIT) did not provide any significant benefit to recurrent GBM patients in a phase II trial as the median OS, median PFS, and PFS-6 were 35 weeks, 12 weeks, and 3%, respectively [52].
Cabozanitib also has potent activity against MET, which is important in both angiogenesis and invasion of glioma [53]. In a phase II study, cabozanitib alone achieved modest but promising PFS with a high response rate (21-30%) in recurrent GBM patients without prior antiangiogenic agents [54], [55]. Moreover, recent preclinical data showed significant increase in OS of GBM xenografts [56]. However, GBM would ultimately escape from cabozanitib by diffuse infiltration and blood-brain barrier (BBB) restoration. Thus, cabozanitib delivery was limited [56]. Further trials of cabozanitib are encouraged based on the promising results but not ignoring potential shortcomings of this drug.
Integrin
Integrins are a large family of cell surface adhesion molecules, which mediates the adhesion between cells and cell-extracellular matrix. Recently, they have also been identified as an important factor for glioma-associated angiogenesis. So far, two isoforms of integrins (ανβ3 and ανβ5) have been identified as having high expression levels in both glioma cells and the endothelial lining of blood vessels and mediating glioma invasion and migration [57].
Cilengitide can selectively block activation of ανβ3 and ανβ5 and exert a bimodal phenotypic antitumor effect by inhibiting angiogenesis and glioma cell invasion in vivo[58], [59]. Although monotherapy had only limited effect in GBM, cilengitide with standard radiation and TMZ appeared to improve PFS and OS in newly diagnosed GBM patients with methylated MGMT status [57], [58], [60]. Apparent significant activity of cilengitide plus standard of care was also seen in a randomized phase II study, demonstrating median survival of 30 and 17.4 months in methylated and unmethylated MGMT newly diagnosed GBM patients, respectively [61].
Additionally, high-dose cilengitide seemed to have more benefits [61]. Promising results in these trials resulted in a broader interest in cilengitide for glioma therapy. However, no significant improved median OS was found in a recent study evaluating cilengitide combined with standard treatment for patients with newly diagnosed GBM with a methylated MGMT promoter. The cilengitide-treated group had a median OS of 26.3 months, while that of the control group was 26.3 months [62]. Subsequently, efficacy and safety of cilengitide in conjunction with radiotherapy and chemotherapy are currently being assessed in a randomized phase II study for newly diagnosed GBM patients with an unmethylated MGMT gene promoter (NCT00813943).
Moreover, a case report suggested that cilengitide had significant antitumor activity in treating bevacizumab-refractory high-grade glioma [63]. This finding might be due to the upregulated integrin signaling in bevacizumab-treated glioma. Further clinical trials could evaluate the antitumor effect of combined VEGF and integrins in glioma patients, especially for those where bevacizumab was ineffective.
Thalidomide can also interfere with the expression of ανβ3 and ανβ5. Moreover, it plays an important role in inhibiting basic fibroblast growth factor and VEGF-mediated angiogenesis [64], [65]. However, a few phase II trials of thalidomide observed negative results in either monotherapy or in combination with TMZ, procarbazine, or irinotecan in recurrent high-grade gliomas [66], [67], [68], [69].
Ongoing Experimental Options for Molecular Targeted Therapy
The loss of p53 and RB tumor suppressor pathways accompanies pro-growth signaling RTK/RAS/PI3K pathway to induce glioma formation [2]. The genetically engineered mouse models have definitively implicated this combination [70]. Specific targeting of these signaling pathways is therefore a rational treatment strategy for molecular therapy of glioma. However, the p53 and RB pathways are difficult to target. In contrast, targeting gain-of-function molecules is relatively easier. Current efforts are mainly focused on amplified, mutated, and/or overexpressed effectors of the RTK/RAS/PI3K pathway and other novel targets such as proteasome and so on. The pro-growth signaling pathways and their designed intervention are shown in Figure 2, A and B.
Figure 2.
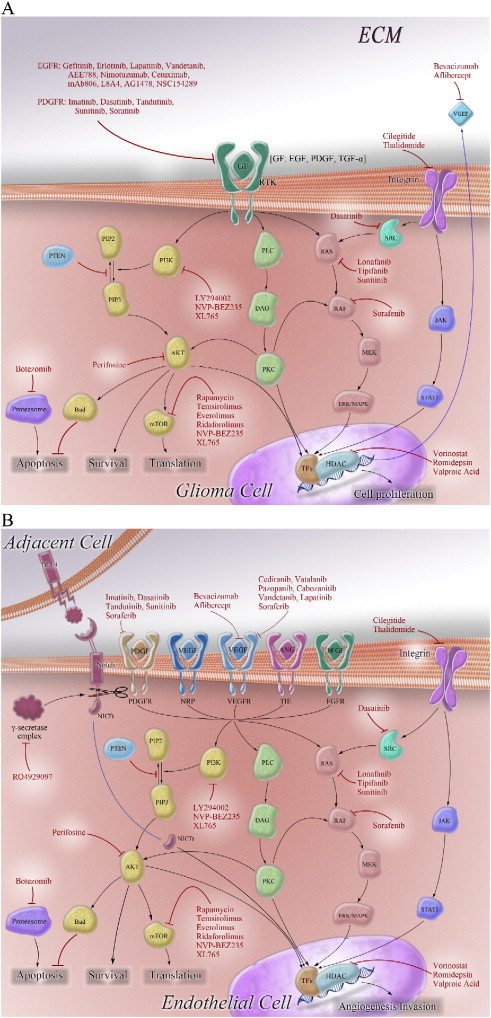
Molecular targets in glioma cells (A) and glioma-associated endothelial cells (B) and designed intervention in molecular targeted therapies for malignant glioma. Growth factor receptors, glioma-associated transmembrane proteins, and their downstream intracellular signaling pathways are commonly altered in glioma and have been implicated in gliomagenesis. Several molecules of them have been explored as the targets to inhibit glioma growth and angiogenesis. GF indicates growth factor; ECM, extracellular matrix; TF, transcription factor; NICD, Notch intracellular domain.
Growth Factor Receptors
Growth factor receptors are critical regulatory proteins in signaling networks of malignant glioma. Novel agents designed to target specific proteins such as EGFR and PDGFR are currently in clinical trials.
EGFR
EGFR activated by EGF or transforming growth factor α and the constitutively activated variant EGFRvIII promote growth signal transduction through activating several downstream signaling pathways and contribute to tumor progression and poor prognosis [71]. EGFR- and/or EGFRvIII-targeted therapies are suggested to produce anticancer effect and have been extensively evaluated preclinically and clinically in malignant gliomas.
Recently, a phase II clinical trial of an EGFR monoclonal antibody cetuximab in patients with recurrent GBM with EGFR amplification showed that patients with positive EGFRvIII had a worse survival compared with negative ones (i.e., median PFS: 1.63 vs 3.03 months; median OS: 3.27 vs 5.57 months) [72]. Another phase II monotherapy trial in patients with recurrent high-grade gliomas demonstrated limited activity of cetuximab (PFS-6 of 9.2%; median OS of 5 months) and failed to find significant correlation between response or survival and EGFR amplification [73]. In short, cetuximab monotherapy has not yet been demonstrated to have clinical benefits, while the outcome of glioma patients treated with cetuximab seems to be associated with EGFRvIII but not amplification alone. Thus, future investigations of EGFR status in EGFR-targeted therapies are indicated.
Gefitinib’s effect in newly diagnosed GBM patients was also neither affected by EGFR amplification nor mutation (i.e., EGFRvIII) in a phase II trial [74]. The post-radiation gefitinib application did not improve survival and neither OS at 1 year nor PFS at 1 year (PFS-1) was statistically different compared with historical control data from the North Central Cancer Treatment Group (OS-1: 54.2% vs 48.9%; PFS-1: 16.7% vs 30.3%) [74]. Furthermore, addition of gefitinib to radiation demonstrated little improvement compared with radiotherapy alone in terms of median survival in patients with newly diagnosed GBM [75].
Another TKI erlotinib whose activity was also not associated with EGFR amplification or EGFRvIII status had an unacceptable toxicity but did not have any clinical benefit as a monotherapy or in combination with radiotherapy and TMZ in several phase II trials of malignant glioma [76], [77], [78]. Lapatinib suffered the same fate as erlotinib [79].
With respect to the failures of reversible EGFR inhibitors, which are susceptible to acquired resistance in EGFR-targeted therapies, other current trials in patients with malignant glioma are focusing on irreversible EGFR inhibitors such as humanized monoclonal antibody nimotuzumab [80], [81] and RTK afatinib (NCT00727506; NCT00977431). Nimotuzumab has the advantage of preferentially binding to areas with a high density of EGFR [82]. A randomly controlled trial revealed that nimotuzumab had better activity than radiotherapy and chemotherapy alone, as improved treatment efficacy and prolonged survival were observed in malignant glioma patients with nimotuzumab [median OS: 16.5 vs 10.5 months (control groups)] [80]. A 5-year institutional experience also demonstrated evident clinical benefit in children with high-grade glioma treated by regimens containing prolonged administration of nimotuzumab (median OS: 32.66 months; OS-2: 54.2%) [81]. In combination, nimotuzumab shows a potential antiglioma activity, which may be worth further investigation. Meanwhile, two ongoing clinical trials are evaluating the safety and efficacy of afatinib either in monotherapy or in combination with radio-chemotherapy in patients with recurrent or newly diagnosed malignant glioma (NCT00727506; NCT00977431).
Given that EGFRvIII overexpression concurrent with EGFR amplification is a hallmark for high invasion and resistance to therapy and that EGFRvIII-positive glioma cells can release microvesicles containing EGFRvIII to surround EGFRvIII-negative glioma cells and lead to transfer of oncogenic activity and enhanced tumorigenicity, EGFRvIII-targeted therapy may have more potential to be effective [83], [84].
These agents mentioned previously mainly target wild-type EGFR. Only a limited amount of EGFRvIII-specific or preferential agents are in development, such as monoclonal antibodies (L8A4 and mAb806) and TKIs (AG1478 and NSC154829) [85]. Several preclinical studies demonstrated antiglioma activities of L8A4, mAb806, and AG1478 in glioma cells and models [86], [87], [88], [89]. AG1478 can also enhance the antitumor efficacy of mAb806 and cytotoxic agents, which indicates the promising future of AG1478 in combination with application prospects [89]. Another novel molecule NSC154829 was shown to selectively inhibit EGFRvIII-expressing GBM cell growth and increase apoptosis [90]. Further testing is under way. Unfortunately, few clinical trials have been carried out in glioma patients with these EGFRvIII-specific agents. Only a phase I trial has been published in which patients with several human cancer types received mAb806 monotherapy and only one of the participants was a glioma patient [91]. This area needs further exploration.
PDGFR
PDGFRs (α and β) interact with different PDGF subunits (-A,-B and -C) to form an autocrine and paracrine stimulation loop between cancer cells and tumor blood vessels and are therefore important for tumor growth and angiogenesis [17], [92]. Blocking PDGFR can inhibit PI3K/mTOR and RAS/MAPK pathways and has an antitumor activity. Thus far, several TKIs have been evaluated in preclinical or clinical studies in malignant glioma.
Imatinib is a TKI of PDGFR, c-KIT, and BCR-ABL. Imatinib can enhance chemosensitivity and its response is associated with increased PDGFRα expression [93], [94]. However, a recent study revealed that imatinib had no significant inhibitory effects against malignant glioma and cautioned on its use [95]. Consistent with this finding, a few phase II studies of imatinib plus hydroxyurea demonstrated negligible antitumor activity in patients with either recurrent/progressive low-grade glioma or recurrent GBM. The radiographic response rates were 0 and 3.4%, respectively [93], [96].
Dasatinib, an ATP-competitive inhibitor of PDGFR, c-KIT, BCR-ABL, and SRC [97], is undergoing evaluation as a monotherapy in a phase II trial for patients with recurrent GBM (NCT00423735). However, dasatinib in combination with lomustine (CCNU) in patients with recurrent GBM showed no significant effect in a phase I/II trial, of which the median PFS was 1.35 months and PFS-6 was only 7.7% [98].
The study of a second-generation PDGFR inhibitor tandutinib is also ongoing (NCT00379080). Notably, the adverse effect on neuromuscular junction may limit anti-PDGFR’s usage [99].
Intracellular Signaling Pathways
Several intracellular pathways mediate signals transduced by upstream RTKs and promote diverse cellular effects such as proliferation, survival, and angiogenesis. Targeting specific signaling mediators will block signal transduction and may lead to inhibition of glioma growth.
PI3K/AKT/mTOR Pathway
Activated by EGFR, EGFRvIII, PDGFR, and RAS pathways, the PI3K/AKT/mTOR pathway has emerged as a central player in glioma pathogenesis by promoting growth and survival [71].
A preclinical study showed that inhibiting PI3K by LY294002 could decrease TMZ resistance by promoting p-AKT and Bcl-2 and enhance cytotoxicity of TMZ by downregulating PI3K/AKT pathway in glioma. LY294002 plus TMZ significantly suppressed proliferation of glioma cells compared with monotherapy of either drug [100]. Furthermore, inhibition of the PI3K/AKT signaling pathway also radiosensitizes cancer cells and delays DNA repair after irradiation [101]. However, radiation-induced up-regulation of telomerase (a ribonucleoprotein complex that elongates telomeric DNA and regulates the cellular immortalization of cancers) activity could escape PI3K inhibition in LY294002-treated glioma cells, which suggests that specific suppression of PI3K when combined with radiation may be optimized by additional treatments inhibiting telomerase in malignant glioma [101], [102].
An AKT-targeted agent perifosine can also cooperate with TMZ to exert strong antitumor activity. However, it failed to enhance radiosensitivity in glioma cells and models [103], [104], [105].
In terms of mTOR inhibitors, previous preclinical studies have revealed that PTEN-deficient gliomas were more sensitive to mTOR inhibition [71]. Due to the high frequency of PTEN gene abnormality in malignant glioma, trials of mTOR inhibitors are very promising. However, recently, a GBM xenograft test panel suggested no relationship between loss of PTEN function and responsiveness to mTOR inhibitors, which is consistent with the data from a phase I trial of patients with recurrent PTEN-deficient GBM treated with rapamycin that observed substantially variable mTOR inhibition [106], [107].
Several rapamycin analogs are undergoing evaluation, such as temsirolimus, everolimus, and deforolimus. Although trials have shown that these agents were well tolerated by patients, monotherapies have generally failed without major clinical benefit seen in malignant glioma patients including children with high-grade glioma [108], [109], [110], [111]. For example, temsirolimus only produced minimal clinical activity with low PFS-6 (7.8%) and median OS (4.4 months) in recurrent GBM patients [112]. Preclinical trials demonstrated that mTOR inhibitors could also sensitize tumor cells to radiotherapy or TMZ in combined therapies. One recent study demonstrated that temsirolimus plus radiotherapy prolonged survival of glioma-bearing mice [113]. However, the combination of temsirolimus with radiotherapy and TMZ might increase infections in patients with newly diagnosed GBM due to the suppression of several immune system components as reported by a North Central Cancer Treatment Group phase I trial, and no definitive conclusions were drawn regarding efficacy of the regime because of the limited number of patients and short follow-up [114]. During a median follow-up of 8.4 months, everolimus and TMZ in combination with radiation resulted in only one partial response in 18 patients with newly diagnosed GBM [115]. A larger sample pool and a longer follow-up are needed to further evaluate the effect of temsirolimus.
Recent studies have indicated that the limited clinical activity of mTOR inhibitors may result from the mTOR-p70S6K-PI3K feedback loop. This resulted in the development of inhibitors dually targeting PI3K and mTOR, such as NVP-BEZ235 and XL765 [116]. In in vitro and in vivo studies, NVP-BE235 significantly prolonged the survival of tumor-bearing animals and displayed multifaceted antiglioma activity such as down-regulation of VEGF, radiosensitization of glioma cells and glioma stem cells (GSCs), and autophagy [117], [118], [119]. In addition, XL765 showed a trend toward improvement in survival in GBM xenografts when combined with TMZ [120]. These results suggest that further investigation of these drugs may be warranted in glioma clinical trials.
RAS/RAF/MAPK Pathway
RAS proteins are recruited to the inner plasma membrane by its upstream growth factor receptors [121]. When bound to the membrane, RAS undergoes posttranslational modification by farnesyl transferase (FT) and becomes ready for signal transmission within the cell [20]. Downstream of RAS lies the RAF/MEK/MAPK pathway. Retrospective studies have reported that activated MAPK predicts radiotherapy resistance and poor outcomes in patients with GBM [122]. Thus, targeting RAS signal pathway is a rational treatment strategy.
FT inhibitors (FTIs) can not only inhibit RAS/RAF/MAPK pathway but also partially block RhoB and the PI3K/AKT pathway [123]. Moreover, the FTI lonafarnib was found to have a significant inhibitory activity and improve effectiveness of TMZ and radiation in malignant glioma cells [124]. Recently, a phase I trial of lonafarnib + TMZ combination in glioma patients reported median OS and median PFS of 14.3 and 4.5 months, respectively. The phase II trials are expected [125].
Simultaneously, another FTI tipifarnib in monotherapy demonstrated discouraging results with a median OS of 7.7 months in a phase II trial for newly diagnosed GBM patients. The study was stopped due to no tumor response and progression of 48% patients [126]. Whereas tipifarnib plus TMZ and radiation was well tolerated in patients with newly diagnosed GBM as shown by a phase I trial, phase II studies are needed to evaluate the therapeutic strategy [127].
Other Molecular Targets in Development
Proteasome
Imbalance of the ubiquitin-proteasome degradation system would lead to cancer cells escaping cell cycle control, inhibition of chemotherapy-induced apoptosis, and development of drug resistance. Thus, targeting this pathway is a viable option [128], [129]. Bortezomib is a proteasome inhibitor that can cause apoptosis and cell cycle arrest in human GBM cells [130]. A phase I trial showed that bortezomib had limited improvement in response rate (3%) and survival time (PFS-6 of 15%; median OS of 6 months) in patients with recurrent high-grade gliomas, and its maximum tolerated dose was affected by enzyme-inducing antiepileptic drugs [131]. Another phase I study using bortezomib, concurrent TMZ, and radiotherapy appeared to be effective in high-grade glioma patients when compared to historical norms: newly diagnosed high-grade glioma patients seemed to have a slightly longer median OS than that of historical controls (16.9 months vs 14.4 months) [132]. Further formal phase II studies are needed to evaluate the potential effect of bortezomib in combination with standard of care.
Histone Deacetylase
Histone deacetylases (HDACs) and histone acetyltransferases play opposite roles in the regulation of acetylation of histone and non-histone substrates including tumor suppressor proteins and oncogenes [133], [134]. Inhibition of HDAC’s function can activate silent tumor suppressor genes and result in cell cycle arrest, induce differentiation, and promote apoptosis in cancer cells [135]. Vorinostat and romidepsin are two HDAC inhibitors. Vorinostat showed modest single-agent activity in treating patients with recurrent GBM, with a median OS of 5.7 months and PFS-6 of 15.2% in a phase I/II trial. Romidepsin was also not effective in another trial where median OS for recurrent GBM was only 34 weeks and median PFS was 8 weeks [136], [137]. Nevertheless, preclinical studies have demonstrated that HDAC inhibitors that help unravel the DNA are effective radiosensitizers in cancers including GBM [134]. Combination regimens are under investigation, such as vorinostat with radiotherapy and TMZ in newly diagnosed GBM and vorinostat combined with radiation therapy in recurrent glioma [138] (NCT00731731; NCT01378481).
Recently, a study showed that the HDAC inhibitory properties of valproic acid, which is a known antiepileptic agent and radiotherapy sensitizer, could mediate the prolonged survival derived from radio-chemotherapy seen in GBM patients [139]. A phase II trial of valproic in combination with TMZ and radiotherapy for GBM patients is ongoing (NCT00302159). Notably, a case of GBM receiving an experimental protocol of concurrent valproic acid, TMZ, and radiation has lived for 3.5 years since the initial GBM diagnosis with no disease progression [134].
Issues with Resistance and Multiple Targets in Therapy and Strategies to Overcome These Barriers
Multiple Targets in Therapy
Reviewing these clinical trials, various molecularly targeted single-agent therapies have failed to demonstrate a significantly better survival compared with current standard treatment regimens. These disappointing results may be due to several factors, including the presence of multiply mutated tyrosine kinases, redundant signaling pathways, and pathway co-activation in most malignant gliomas. Thus, targeting multiple signaling pathways simultaneously by multitargeted kinase inhibitors or combinations of targeted agents may improve outcomes.
Multitargeted Kinase Inhibitor
Multitargeted kinase inhibitors are more convenient to administer and avoid the potential pitfalls of drug-drug interactions and synergistic toxicity. Antiangiogenesis drugs such as cediranib, pazopanib, vatalanib, and cabozanitib have inhibitory activities against multiple kinases. Several other multitargeted kinase inhibitors such as the dual EGFR and VEGFR inhibitors vandetanib and AEE788, VEGFR, PDGFR, c-KIT and FLT-3 inhibitor sunitinib, and RAF, VEGFR, PDGFR, c-KIT, and FLT-3 inhibitor sorafenib have been under development as well [25], [140]. Recently, vandetanib monotherapy demonstrated insignificant activity in unselected patients with recurrent malignant glioma in a phase I/II trial, whereas combined therapy of vandetanib and TMZ could provide a marked 94% tumor volume reduction in a glioma xenograft model. Further studies of combination treatment are warranted based on the results of xenograft and several other phase I trials [141], [142], [143], [144]. For other agents, monotherapies of AEE788 and sunitinib did not demonstrate significant antitumor activity in patients with recurrent malignant gliomas in respective clinical trial. Neither did combined therapies of sorafenib plus TMZ and sunitinib plus irinotecan. Notably, AEE788 had unacceptable toxicity and a clinical trial was discontinued prematurely [145], [146], [147], [148]. Although a phase I trial of sorafenib with traditional treatment for both primary and recurrent high-grade gliomas showed impressive results that cell viability was significantly reduced, and the median OS of the entire population was 18 months, a phase II trial of the regimen was recommended [149]. In all, the studies of multitargeted kinase inhibitors for treatment of malignant glioma patients are at the initial stage. However, there are still some factors, such as modest benefit and potential dangers of synergistic or additive toxicities, which need further investigation.
Combination of Targeted Agents
Combined therapy of kinase inhibitors can target the same kinase or one pathway with synergistic effect and strengthened inhibition. In addition, it can target different kinases and pathways to create multiple antitumor effects and prevent drug resistance. Recently, targeting EGFR and downstream effectors in PI3K/AKT/mTOR pathways has been attractive, for they are complementary targets and among the most commonly aberrant molecules in malignant gliomas [3]. For example, gefitinib in combination with everolimus and erlotinib in combination with sirolimus both target EGFR and mTOR. However, phase II trials of the two combinations had negligible activity among unselected recurrent GBM patients with the median OS being 5.8 and 8.5 months, respectively [150], [151]. Phase I studies of gefitinib plus sirolimus and AEE788 plus everolimus in recurrent GBM patients revealed that the agents could be safely co-administered on a moderate schedule. However, for AEE788 plus everolimus, a dose increase of AEE788 to 200 mg/day and everolimus to 5 mg quaque omni die (qod) caused significant thrombocytopenia, suggesting pharmacokinetic drug interaction [152], [153]. Other studies have found that mTOR inhibition was associated with increasing MAPK signaling [154]; therefore, approaches targeting cross-talk of the two pathways may be potentially beneficial based on significant data in PDGF-driven GBM mice treated with perifosine and temsirolimus [155]. A phase I/II trial of the combined therapy in recurrent or progressive malignant glioma is ongoing (NCT01051557).
Other novel combination therapies are investigated in different phase trials. Some have been shown limited effectiveness, including dasatinib plus erlotinib [156], pazopanib plus lapatinib [157], and vorinostat plus bortezomib [158] in recurrent malignant gliomas, while some others are awaiting safety information in phase I trials in recurrent high-grade glioma, such as bevacizumab in combination with panobinostat [158] and vorinostat combined with bevacizumab and irinotecan [159]. Notably, the combination of cetuximab and bevacizumab in a third-line setting was reported to be successful in a case of relapsed brainstem GBM who achieved a complete radiologic response and a PFS of 20 months [160]. The proposal that the two antibodies exert multifaceted effects mainly in GSCs still needs to be verified, while future research of the combined therapy in this type of difficult clinical situation may be warranted.
Potential Targets in GSCs
Traditional therapies and most of the paradigms discussed above may have limited antiglioma activities, because they may only target proliferating non-tumorigenic cells in malignant glioma. Recently, GSCs, which have the ability of self-renewal and multilineage differentiation, have been considered to be the cellular origin of glioma and account for chemotherapy and radiotherapy resistance, angiogenesis, invasion, and recurrence [3], [25]. Moreover, different types of GSCs can coexist in one GBM, contributing to cellular heterogeneity, which is another main reason resulting in failure of molecularly targeted therapies [161]. Thus, strategies targeting GSCs are urgently needed. Several signaling pathways, such as the Notch, Hedgehog, and Wnt pathways, are essential for function and phenotype maintenance in GSCs [25] and therefore represent novel molecular targets (as shown in Figure 3).
Figure 3.
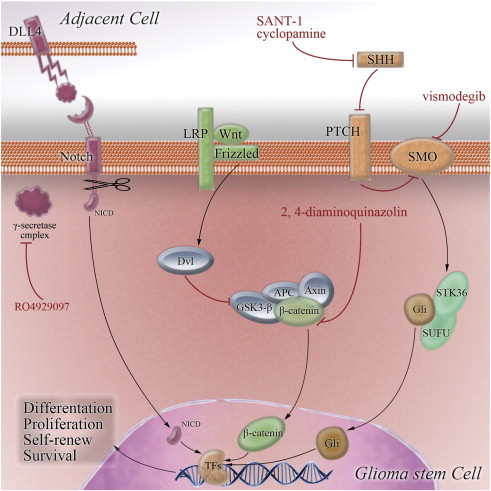
GSC-related signaling pathways represent potential targets for novel treatment strategies. Notch, SHH, and Wnt/β-catenin pathways are altered in malignant glioma and have been shown to regulate GSC function. Constituents of these pathways are potential targets for molecular targeted therapies. NICD indicates Notch intracellular domain; LRP, lipoprotein receptor–related protein; TF, transcription factor.
Notch Pathway
Notch receptors activate notch signaling by binding to transmembrane ligands such as Delta-like 4 by cell-to-cell contact [162]. When activated, Notch is sequentially cleaved and releases its intracellular domain into the nucleus to trigger notch-dependent transcription. This requires the γ-secretase activity of a multiprotein complex [163]. Elevated notch signaling is a contributing factor in glioma angiogenesis and the signaling has been shown to regulate stem cell maintenance, proliferation, and multipotency [164]. In addition, it plays a vital role in regulation of radioresistance and DNA damage response pathway in GSCs [165]. Thus, targeting GSCs by blocking notch signaling using inhibitors of γ-secretase represents a potential therapeutic strategy. In vitro and in vivo experiments have demonstrated that γ-secretase inhibitors negatively regulate GBM clonogenicity and positively prolong survival [166]. RO4929097 is an inhibitor of γ-secretase whose clinical trials are under way as monotherapy in patients with recurrent malignant gliomas and in combined therapy with TMZ and radiation in patients with newly diagnosed GBM in phase I and II trials (NCT01269411, NCT01122901, and NCT01119599).
Sonic Hedgehog Pathway
Sonic hedgehog (SHH) binding to transmembrane receptors protein patched homolog and membrane protein smoothened homolog results in activation of Gli transcription factors that are then transferred into the nucleus to modulate expression of target genes that are essential for GSC self-renewal and survival [25], [163]. Inhibiting SHH pathway may be an effective therapy to prevent malignant gliomas. An SHH inhibitor N-[(1E)-(3,5-Dimethyl-1-phenyl-1H-pyrazol-4yl)methylidene]-4-(phenylmethyl)-1-piperazinamine (SANT-1) has been shown to reduce GSC proliferation, and a RAS/nuclear factor of kappa light polypeptide gene enhancer in B-cells (NF-κB) guggulsterone could sensitize GBM cells to SANT-1–induced apoptosis [167]. Another SHH inhibitor cyclopamine reduced the number of GSCs and suppressed glioma growth in vivo and showed synergistic effects with TMZ [168]. Clinical trials with the smoothened homolog inhibitor vismodegib revealed encouraging antitumor efficiency and safety in medulloblastoma [169], while a phase II trial of vismodegib in treating patients with recurrent GBM is ongoing (NCT00980343).
Wnt/β-Catenin Pathway
Wnt/β-catenin signaling is the canonical Wnt pathway that is responsible for the regulation of stem cell self-renewal in the developing brain [170]. Due to the similarities of pathways regulating in normal stem cells and cancer stem cells, Wnt/β-catenin signaling is regarded to be involved in the regulation of cancer stem cells including GSCs, which has a vital role to play in malignant transformation and tumor progression in gliomas [171], [172]. Although the function of this signaling pathway in GSCs has not been extensively elucidated, blockade of Wnt pathway may effectively target GSCs. Several molecular targeted agents such as 2,4-diaminoquinazolin have been applied in preclinical experiments and in clinical trials in other cancers [171]. The need to evaluate the effects of these agents in malignant gliomas is urgent.
Currently, targeting these pathways in GSCs has not yet delivered convincing results. Destroying glioma vascular niche that is critical for maintenance of the cluster of cells may be therapeutic [173].
Challenges and Future Directions
The power of molecular targeted therapy (partly shown in Table 1) has been limited by diverse factors, ranging from complexity of molecular biology underlying gliomagenesis to challenges of patient selection to specific therapies, drug delivery, and evaluation of treatment response. Exploring these factors in great depth might indicate the appropriate direction for development of molecular targeted therapy in malignant glioma.
Table 1.
Select Molecular Targeted Drugs of Malignant Glioma in Clinical Trials
| Drug | Target(s) | Therapeutic Approach | Type of Glioma | Phase | Number of Subjects | RR | Median OS | Median PFS | PFS-6 (%) | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| VEGF/VEGFR-targeted agents | ||||||||||
| Bevacizumab | VEGF | Bevacizumab + irinotecan | Recurrent MG | II | Grade III 22, IV 93 | 39.1% ORR | Grade III 9 m, IV 8 m | Grade III 6 m, IV 6 m | 46.3 | [40] |
| Bevacizumab + TMZ + RT | ND GBM | II | 70 | 19.6 m | 13.6 m | 88 | [41] | |||
| Aflibercept | VEGF | Monotherapy | Recurrent MG | II | GBM 42, AG 16 | GBM 18%, AG 44% ORR | GBM 39 w, AG 55 w | GBM 12 w, AG24 w | GBM 7.7, AG 25 | [44] |
| Cediranib | VEGFR, PDGFR, FGFR, c-KIT | Monotherapy | Recurrent GBM | II | 31 | 27% PR | 227 d | 117 d | 25.8 | [46] |
| Vatalanib | VEGFR, c-KIT, PDGFR | Vatalanib + TMZ + RT | ND GBM | I/II | 19 | 17.3 m | 6.8 m | 63.2 | [51] | |
| Pazopanib | VEGFR, PDGFR, c-KIT | Monotherapy | Recurrent GBM | II | 35 | 2 PR | 35 w | 12 w | 3 | [52] |
| Integrin-targeted agents | ||||||||||
| Cilengitide | ανβ3/ανβ5 integrin | Cilengitide + TMZ + RT | ND GBM | I/IIa | 52 | 16.1 m | 8 m | 69 | [60] | |
| Cilengitide + TMZ + RT | ND GBM | II | 112 | 19.7 m | 9.97 m | [61] | ||||
| Thalidomide | ανβ3/ανβ5 integrin | Thalidomide + procarbazine | Recurrent MG | II | 18 | 0 | 6.4 m | [68] | ||
| Thalidomide + irinotecan | Recurrent AG | II | 39 | 2 CR, 2 PR | 72 w | 13 w | 36 | [69] | ||
| EGFR-targeted inhibitors | ||||||||||
| Cetuximab | EGFR | Monotherapy | Recurrent GBM | II | 35 | 3.97 m | 1.63 m | [72] | ||
| Monotherapy | Recurrent HGG | II | 55 | 5.5% PR | 5.0 m | 7.3 | [73] | |||
| Gefitinib | EGFR | Monotherapy | ND GBM | II | 96 | 12 m | [74] | |||
| Gefitinib + RT | ND GBM | II | 147 | 11.1 m | 4.9 m | 40 | [75] | |||
| Erlotinib | EGFR | Erlotinib + TMZ + RT | ND GBM | II | 27 | 8.6 m | 2.8 m | 30 | [77] | |
| Monotherapy | Recurrent GBM/AA | I/II | 11 | 0 | 6.9 m | 1.9 m | 20 | [78] | ||
| Lapatinib | EGFR, HER2 | Monotherapy | Recurrent GBM | I/II | 17 | 0 | [79] | |||
| Nimotuzumab | EGFR | Nimotuzumab + TMZ + RT | Grade III to IV glioma | I/II | 20 | 16.5 m | [80] | |||
| PDGFR-targeted inhibitors | ||||||||||
| Imatinib | PDGFR, c-KIT, BCR-ABL | Imatinib + hydroxyurea | Recurrent GBM | II | 231 | 3.4% ORR | 26.0 w | 10.6 | [93] | |
| Imatinib + hydroxyurea | Recurrent LGG | II | 64 | 0 | 11 m | [96] | ||||
| Dasatinib | PDGFR, SRC, c-KIT, BCR-ABL | Dasatinib + CCNU | Recurrent GBM | I/II | 26 | 1.35m | 7.7 | [98] | ||
| PI3K/AKT/mTOR-targeted inhibitors | ||||||||||
| Temsirolimus | mTOR | Monotherapy | Recurrent GBM | II | 65 | 4.4 m | 7.8 | [112] | ||
| Temsirolimus + TMZ + RT | ND GBM | I | 25 | 0 | [114] | |||||
| Everolimus | mTOR | Everolimus + TMZ + RT | ND GBM | I | 18 | 1PR | [115] | |||
| RAS/RAF/MAPK-targeted inhibitors | ||||||||||
| Lonafarnib | RAS (FT) | Lonafarnib + TMZ | MG | I | 36 | 6% PR | 14.3 m | 4.5 m | 41.7 | [125] |
| Tipifarnib | RAS (FT) | Monotherapy | ND GBM | II | 28 | 0 | 7.7 m | [126] | ||
| Other inhibitors | ||||||||||
| Bortezomib | Proteasome | Monotherapy | Recurrent MG | I | 66 | 3% PR | 6.0 m | 2.1 m | 15 | [131] |
| Bortezomib + TMZ + RT | HGG | I | 23 | 0 | 15.0 m | 52 | [132] | |||
| Vorinostat | HDAC | Monotherapy | Recurrent GBM | II | 66 | 2 ORR | 5.7 m | 15.2 | [136] | |
| Romidepsin | HDAC | Monotherapy | Recurrent MG | I/II | GBM 35, AG 5 | GBM 0, AG 0 | GBM 34 w, AG 36 w | GBM 8 w | GBM 3, AG 0 | [137] |
| Vandetanib | EGFR, VEGFR | Monotherapy | Recurrent MG | I/II | GBM 32, AG 32 | GBM 4, AG 2 ORR | GBM 6.3 m, AG 7.6 m | GBM6.5, AG 7 | [141] | |
| AEE788 | EGFR, VEGFR | Monotherapy | Recurrent GBM | I | 64 | 0 | 1.6-2.7 m | [145] | ||
| Sunitinib | PDGFR, VEGFR, c-KIT | Monotherapy | Recurrent MG | II | GBM 16, AG 14 | 0 | GBM 12.6 m, AG 12.1 m | GBM 1.4 m, AG 4.1 m | GBM 16.7, AG 21.5 | [146] |
| Sunitinib + Irinotecan | Recurrent MG | I | 25 | 1 PR | 53.1 w | 6.9 w | 24 | [148] | ||
| Sorafenib | RAF, VEGFR, PDGFR | Sorafenib + TMZ + RT | HGG | I | 18 | 1 CR, 4 PR | 18 m | [149] |
RR, response rate; CR, complete response; PR, partial response; ORR, objective/overall response rate = CR + PR; SD, stable disease; TTP, time to progression; m, months; w, weeks; d, days; NG GBM, newly diagnosed GBM; HGG, high-grade glioma; AA, anaplastic astrocytoma; AG, anaplastic glioma; MG, malignant glioma; HER, human epithelial factor receptor; RT, radiotherapy.
Intricate Biologic Traits in Glioma
Malignant gliomas are known to harbor complex heterogeneity at the genomic and molecular levels and are driven by intricate signaling cascades. Recently, co-amplification of multiple RTKs such as EGFR with PDGFR or MET has been found within individual gliomas [174], namely, intratumoral heterogeneity. Thus, when treated with EGFR inhibitors, MET and/or PDGFR would maintain activation of downstream pathways, which is a theoretical mechanism of target therapy resistance [174]. TCGA has demonstrated three core pathways, RTK/RAS/PI3K, p53, and RB, which are essential for development of malignant gliomas, and other undiscovered canonical pathways cannot be ruled out in gliomagenesis. However, these pathways are not as simple as vector-to-vector models. Instead, this model features cross-talk and feedback loops that can significantly affect therapy outcome [70]. Several studies have found complicated interplay among PI3K-MAPK-p53-RB pathways, which can compensate for any single pathway perturbation [23]. Moreover, recent data show that EGFR-VEGF(R) cross-talk exists in both tumor and tumor-associated endothelial cells and is involved in tumor survival and angiogenesis in an intracrine fashion [175]. Hence, multiple lines of evidence suggest that successful targeted therapy should require designed combinations of inhibitors to block pathway cross-talk and feedback and suppress upfront and acquired resistance.
Malignant gliomas are also characterized by genomic instability, which favors gene mutations and chromosomal alterations, and cytotoxic agents and radiotherapy would accelerate the mutagenesis [70], [176]. Recently, oncogene addiction, whereby tumors depend on a single oncogenic activity for maintenance of malignant phenotype and cell survival, has been proposed in gliomas with complex genetic aberrations. After exposure to therapeutic agents, glioma cells can escape from one established oncogene addiction to another oncogene, which may explain the reason why previous drugs cease to be effective and the tumor acquires drug resistance [177]. Thus, simultaneously targeting the existing oncogene addiction and oncogene addiction transition may have feasibility. Another highly significant finding is that a small subset of GBMs harbors chromosomal translocations that lead to production of oncogenic fusion proteins demonstrating a novel mechanism of pathogenicity [178]. For instance, FGFR3-TACC3 fusion oncogene greatly enhances tumor progression relative to wild-type FGFR3 and an inhibitor targeting FGFR prolongs survival of mice harboring intracranial FGFR3-TACC3–initiated glioma [179]. These results powerfully suggest new therapeutic approaches for the subset of malignant gliomas.
Biomarkers in Molecular Targeted Therapy
Due to the high heterogeneity of gliomas, each of which may respond differently to one targeted therapy, there has been considerable interest in identifying molecular markers of glioma that predict a response to a particularly molecular targeted therapy, similar to MGMT status predicting better glioma response to the alkylating agent TMZ [37]. The goal would be that patients are parsed into different groups based on the biomarkers that are most likely to predict benefit from the particular treatment. Originally, researchers thought that the presence of EGFR overexpression and EGFR mutations in gliomas could predict activity of EGFR-targeted drugs in patients with gliomas with these aberrations [180]. However, this potential treatment approach still has not been clear with contradictory findings in previous clinical trials (see Growth Factor Receptors section). Some studies also found that tumors with EGFRvIII and intact PTEN and tumors with low phosphorylated Akt levels are more likely to respond to EGFR inhibitors. Unfortunately, subsequent studies did not confirm these initial observations [2]. With EGFR amplification or mutation in gliomas limited in their use as prognostic factors for response to anti-EGFR therapeutics, a study of in situ analysis found that mutant EGFR dimer configurations prevalent in GBMs could evade blockage by anti-EGFR treatments, which suggests further investigation of EGFR mutant dimerization such as EGFRwt-EGFRvIII and EGFRvIII-EGFRc958 heterodimers as a potential parameter for predicting anti-EGFR therapy response [181].
Additionally, previous trials reported that a response to integrin inhibitors is a function of MGMT expression. However, the result of the primary endpoint of the phase III trial (NCT00689221) was reported this year and it did not achieve the expected outcome; newly diagnosed GBM patients with MGMT gene promoter methylation did not live significantly longer when treated with cilengitide plus chemo-radiotherapy (www.merckserono.com). The benefit seen in patients with promoter methylation of MGMT may possibly remain correlated to TMZ; thus, MGMT cannot yet be used as a predictive biomarker for anti-integrin therapy.
As well, there has been no convincing evidence of a correlation between other molecular alterations in glioma and response to molecular targeted therapies. Recent data show that mTOR inhibition may be beneficial in a subpopulation of GBM patients with high baseline tumor levels of phosphorylated ribosomal protein p70S6K, which is a downstream activator of mTOR signaling [112]. Moreover, PIK3R1, a newly identified mutation, increases the likelihood that agents targeting the PI3K pathway would fail [176]. The predictive role of these molecules in treatment response remains to be elucidated in future clinical trials.
Difficulties in Clinical Trials
The success of molecular targeted therapies in clinical trials may be also limited by several factors such as drug delivery, pharmacologic effects, evaluation, and so on.
First, the delivery of agents into the brain is regulated by the BBB that allows lipophilic agents of small molecules to pass through. The BBB becomes the obstacle for entrance of large or water-soluble molecules and limits the ability of drugs to reach sufficient concentration in glioma tissue. Drug development should focus on penetration or bypass of the BBB with techniques like convection-enhanced delivery, liposomal carriers, and nanoscale particles.
Second, when penetrated into the central nervous system, drugs can cause diverse side effects, despite exhibiting therapeutic activities. Furthermore, since normal cells and glioma cells share the same pathways, unacceptable toxicity may derive from the pathway-specific treatments, which may stifle the therapeutic potential of many potential agents. Moreover, targeted agents are evaluated in clinical trials similarly to cytotoxic drugs in phase I trials, whereby the maximum tolerated dose is assessed. These doses may not be the effective concentration to significantly affect tumor growth or induce tumor response in phase II trials. Targeted agents are badly in need of a different type of evaluation. In addition, attention needs to paid to drug interaction when combined with cytotoxic drugs, other molecular targeted agents, or enzyme-inducing antiepileptic drugs.
Third, the difficulty in evaluation of response remains an important component. Radiologic response rate, if low, is a sign of failure in most trials of molecular targeted agents. However, a majority of molecular targeted agents have the ability to inhibit tumor growth but not kill tumor cells. It may be that evaluation of radiologic response is more suitable for cytotoxic drugs than molecular targeted agents. In addition, antiangiogenesis treatment such as bevacizumab rapidly decreases edema and makes assessment of radiographic response difficult. Thus, redefining the criteria for evaluation of response of molecular targeted therapy in malignant glioma is an urgent need.
Opportunities in Other Oncogenic Pathways
Given the limited antitumor activity of current RTK signaling inhibitors in malignant glioma, novel agents targeting the other two major oncogenic pathways, the p53/MDM2-MDM4-p14ARF and RB1/CDK4/p16INK4A/CDKN2B, have increasingly gained attention. However, it is difficult to design small molecules for these loss-of-function targets. Several preclinical studies supported that an inhibitor of CDK4 and CDK6, PD0332991, could efficiently cross the BBB, suppress the growth of intracranial GBM xenograft tumors, and significantly prolong survival. RB status, as well as p16INK4a and CDK4, was a determinant of potential benefit from the therapy [182], [183]. This area remains a great untapped potential strategy.
Recently, metabolic pathways have gained attention as high frequencies of isocitrate dehydrogenase 1 (IDH1)/IDH2 mutation have been found in secondary GBMs and low-grade gliomas [21]. Mutant IDH inhibits wild-type IDH activity, impairs affinity for its substrate, catalyzes the nicotinamide adenine dinucleotide phosphate (NADPH)-dependent reduction of α-ketoglutarate to d-2-hyroxyglutarate, and acquires oncogenic activity possibly through induction of hypoxia induced factor-1α pathway and formation of 2-hyroxyglutarate [184]. This altered tumor metabolic pathway offers the possibility of molecularly targeted therapeutic intervention; however, there still exist difficulties and controversies regarding the largely unknown mechanisms underlying the oncogenic effects of IDH mutations. More studies are warranted to achieve the therapeutic purpose.
Combination of Molecular Targeted Therapy with Other Therapeutic Modalities
Redundancy and complexity of signaling pathways in malignant glioma often lead to failure even with combined molecularly targeted agents. Therefore, adding other therapeutic modalities to molecular targeted therapy may create new avenues for success.
Previous clinical trials of immunotherapy in malignant glioma have reported induction of systemic immune responses and met with success in prolonging survival [185], [186]. Another discovery that mTOR playing a pivotal role in RTK/RAS/PI3K signaling pathway activation may also be a critical regulator of the immune response presents an opportunity to combine molecular targeted therapies and immunotherapeutic approaches with very promising potential [70].
Recent studies have also demonstrated that microRNAs (miRNAs), a class of novel small non-coding RNA molecules, are involved in critical signaling pathways in malignant glioma and present potentially effective therapeutic targets [187]. Regulation of aberrant miRNA could affect sensitivities to molecular targeted therapy [188]. Therefore, combination of miRNA-based therapy with molecular targeted therapy might be able to exert a synergistic effect for treatment of malignant gliomas, particularly for molecular targeted agent–resistant patients.
Finally, global gene expression analysis incorporated into patient glioma analysis and treatment management may identify predicative and therapeutic biomarkers, stratify patients based on molecular characteristics, and provide individualized therapies. Additionally, development in the investigation of novel molecular targeted agents and multiple combined therapies may allow the “molecularly tailored” therapeutic strategies to cure malignant gliomas in the future.
Acknowledgements
The authors thank Andrew S. Chi and Daniel P. Cahill from Massachusetts General Hospital Cancer Center, Harvard Medical School for helpful discussions and professional support.
Footnotes
This work was supported by the National “863” High Technique Project (2007AA02Z483), the National Natural Science Foundation (81272781 and 81201535), the Program for Academic Leaders in Health Science (XBR2011030), and “Shu Guang” Project (11SG37) in Shanghai.
Contributor Information
Da Fu, Email: fu800da900@126.com.
Juxiang Chen, Email: juxiangchen.cz@gmail.com.
References
- 1.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin. 2010;60:166–193. doi: 10.3322/caac.20069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patrick Y, Wen SK. Malignant gliomas in adults. N Engl J Med. 2008:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 4.Auffinger B, Thaci B, Nigam P, Rincon E, Cheng Y, Lesniak MS. New therapeutic approaches for malignant glioma: in search of the Rosetta stone. F1000 Med Rep. 2012;4:18. doi: 10.3410/M4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith J. Erlotinib: small-molecule targeted therapy in the treatment of non-small-cell lung cancer. Clin Ther. 2005;27:1513–1534. doi: 10.1016/j.clinthera.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 6.Ribas A, Hersey P, Middleton MR, Gogas H, Flaherty KT, Sondak VK, Kirkwood JM. New challenges in endpoints for drug development in advanced melanoma. Clin Cancer Res. 2012;18:336–341. doi: 10.1158/1078-0432.CCR-11-2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Habeck M. FDA licences imatinib mesylate for CML. Lancet Oncol. 2002;3:6. doi: 10.1016/s1470-2045(01)00608-8. [DOI] [PubMed] [Google Scholar]
- 8.Cohen MH, Shen YL, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist. 2009;14:1131–1138. doi: 10.1634/theoncologist.2009-0121. [DOI] [PubMed] [Google Scholar]
- 9.Kreisl TN, Kim L, Moore K, Duic P, Royce C, Stroud I, Garren N, Mackey M, Butman JA, Camphausen K. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009;27:740–745. doi: 10.1200/JCO.2008.16.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, Yung WK, Paleologos N, Nicholas MK, Jensen R. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27:4733–4740. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 11.Chamberlain MC. Emerging clinical principles on the use of bevacizumab for the treatment of malignant gliomas. Cancer. 2010;116:3988–3999. doi: 10.1002/cncr.25256. [DOI] [PubMed] [Google Scholar]
- 12.Khasraw M, Simeonovic M, Grommes C. Bevacizumab for the treatment of high-grade glioma. Expert Opin Biol Ther. 2012;12:1101–1111. doi: 10.1517/14712598.2012.694422. [DOI] [PubMed] [Google Scholar]
- 13.Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S, Won M. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370:699–708. doi: 10.1056/NEJMoa1308573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea D. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014;370:709–722. doi: 10.1056/NEJMoa1308345. [DOI] [PubMed] [Google Scholar]
- 15.Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hatanpaa KJ, Burma S, Zhao D, Habib AA. Epidermal growth factor receptor in glioma: signal transduction, neuropathology, imaging, and radioresistance. Neoplasia. 2010;12:675–684. doi: 10.1593/neo.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nazarenko I, Hede SM, He X, Hedren A, Thompson J, Lindstrom MS, Nister M. PDGF and PDGF receptors in glioma. Ups J Med Sci. 2012;117:99–112. doi: 10.3109/03009734.2012.665097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wen PY, Lee EQ, Reardon DA, Ligon KL, Alfred Yung WK. Current clinical development of PI3K pathway inhibitors in glioblastoma. Neuro Oncol. 2012;14:819–829. doi: 10.1093/neuonc/nos117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krakstad C, Chekenya M. Survival signalling and apoptosis resistance in glioblastomas: opportunities for targeted therapeutics. Mol Cancer. 2010;9:135. doi: 10.1186/1476-4598-9-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lo HW. Targeting Ras-RAF-ERK and its interactive pathways as a novel therapy for malignant gliomas. Curr Cancer Drug Targets. 2010;10:840–848. doi: 10.2174/156800910793357970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohgaki H, Kleihues P. Genetic alterations and signaling pathways in the evolution of gliomas. Cancer Sci. 2009;100:2235–2241. doi: 10.1111/j.1349-7006.2009.01308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Masui K, Cloughesy TF, Mischel PS. Review: molecular pathology in adult high-grade gliomas: from molecular diagnostics to target therapies. Neuropathol Appl Neurobiol. 2012;38:271–291. doi: 10.1111/j.1365-2990.2011.01238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 24.Rich JN, Bigner DD. Development of novel targeted therapies in the treatment of malignant glioma. Nat Rev Drug Discov. 2004;3:430–446. doi: 10.1038/nrd1380. [DOI] [PubMed] [Google Scholar]
- 25.Tanaka S, Louis DN, Curry WT, Batchelor TT, Dietrich J. Diagnostic and therapeutic avenues for glioblastoma: no longer a dead end? Nat Rev Clin Oncol. 2013;10:14–26. doi: 10.1038/nrclinonc.2012.204. [DOI] [PubMed] [Google Scholar]
- 26.Sharma PS, Sharma R, Tyagi T. VEGF/VEGFR pathway inhibitors as anti-angiogenic agents: present and future. Curr Cancer Drug Targets. 2011;11:624–653. doi: 10.2174/156800911795655985. [DOI] [PubMed] [Google Scholar]
- 27.Reardon DA, Turner S, Peters KB, Desjardins A, Gururangan S, Sampson JH, McLendon RE, Herndon JE, II, Jones LW, Kirkpatrick JP. A review of VEGF/VEGFR-targeted therapeutics for recurrent glioblastoma. J Natl Compr Canc Netw. 2011;9:414–427. doi: 10.6004/jnccn.2011.0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hardee ME, Zagzag D. Mechanisms of glioma-associated neovascularization. Am J Pathol. 2012;181:1126–1141. doi: 10.1016/j.ajpath.2012.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takano S, Yamashita T, Ohneda O. Molecular therapeutic targets for glioma angiogenesis. J Oncol. 2010;2010:351908. doi: 10.1155/2010/351908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Plate KH, Scholz A, Dumont DJ. Tumor angiogenesis and anti-angiogenic therapy in malignant gliomas revisited. Acta Neuropathol. 2012;124:763–775. doi: 10.1007/s00401-012-1066-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tate MC, Aghi MK. Biology of angiogenesis and invasion in glioma. Neurotherapeutics. 2009;6:447–457. doi: 10.1016/j.nurt.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gatson NN, Chiocca EA, Kaur B. Anti-angiogenic gene therapy in the treatment of malignant gliomas. Neurosci Lett. 2012;527:62–70. doi: 10.1016/j.neulet.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arbab AS. Activation of alternative pathways of angiogenesis and involvement of stem cells following anti-angiogenesis treatment in glioma. Histol Histopathol. 2012;27:549–557. doi: 10.14670/hh-27.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caruso C, Carcaterra M, Donato V. Role of radiotherapy for high grade gliomas management. J Neurosurg Sci. 2013;57:163–169. [PubMed] [Google Scholar]
- 35.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 36.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 37.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 38.Kuczynski EA, Patten SG, Coomber BL. VEGFR2 expression and TGF-β signaling in initial and recurrent high-grade human glioma. Oncology. 2011;81:126–134. doi: 10.1159/000332849. [DOI] [PubMed] [Google Scholar]
- 39.Zhang G, Huang S, Wang Z. A meta-analysis of bevacizumab alone and in combination with irinotecan in the treatment of patients with recurrent glioblastoma multiforme. J Clin Neurosci. 2012;19:1636–1640. doi: 10.1016/j.jocn.2011.12.028. [DOI] [PubMed] [Google Scholar]
- 40.Demirci U, Tufan G, Aktas B, Balakan O, Alacacioglu A, Dane F, Engin H, Kaplan MA, Gunaydin Y, Ozdemir NY. Bevacizumab plus irinotecan in recurrent or progressive malign glioma: a multicenter study of the Anatolian Society of Medical Oncology (ASMO) J Cancer Res Clin Oncol. 2013;139:829–835. doi: 10.1007/s00432-013-1390-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lai A, Tran A, Nghiemphu PL, Pope WB, Solis OE, Selch M, Filka E, Yong WH, Mischel PS, Liau LM. Phase II study of bevacizumab plus temozolomide during and after radiation therapy for patients with newly diagnosed glioblastoma multiforme. J Clin Oncol. 2011;29:142–148. doi: 10.1200/JCO.2010.30.2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reardon DA, Herndon JE, II, Peters K, Desjardins A, Coan A, Lou E, Sumrall A, Turner S, Sathornsumetee S, Rich JN. Outcome after bevacizumab clinical trial therapy among recurrent grade III malignant glioma patients. J Neurooncol. 2012;107:213–221. doi: 10.1007/s11060-011-0740-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aguilera DG, Mazewski C, Hayes L, Jordan C, Esiashivilli N, Janns A, Macdonald TJ. Prolonged survival after treatment of diffuse intrinsic pontine glioma with radiation, temozolamide, and bevacizumab: report of 2 cases. J Pediatr Hematol Oncol. 2013;35:e42–e46. doi: 10.1097/MPH.0b013e318279aed8. [DOI] [PubMed] [Google Scholar]
- 44.de Groot JF, Lamborn KR, Chang SM, Gilbert MR, Cloughesy TF, Aldape K, Yao J, Jackson EF, Lieberman F, Robins HI. Phase II study of aflibercept in recurrent malignant glioma: a North American Brain Tumor Consortium study. J Clin Oncol. 2011;29:2689–2695. doi: 10.1200/JCO.2010.34.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yung WK. Moving toward the next steps in angiogenesis therapy? Neuro Oncol. 2008;10:939. doi: 10.1215/15228517-2008-091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Batchelor TT, Duda DG, di Tomaso E, Ancukiewicz M, Plotkin SR, Gerstner E, Eichler AF, Drappatz J, Hochberg FH, Benner T. Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J Clin Oncol. 2010;28:2817–2823. doi: 10.1200/JCO.2009.26.3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.di Tomaso E, Snuderl M, Kamoun WS, Duda DG, Auluck PK, Fazlollahi L, Andronesi OC, Frosch MP, Wen PY, Plotkin SR. Glioblastoma recurrence after cediranib therapy in patients: lack of "rebound" revascularization as mode of escape. Cancer Res. 2011;71:19–28. doi: 10.1158/0008-5472.CAN-10-2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gerstner ER, Chen PJ, Wen PY, Jain RK, Batchelor TT, Sorensen G. Infiltrative patterns of glioblastoma spread detected via diffusion MRI after treatment with cediranib. Neuro Oncol. 2010;12:466–472. doi: 10.1093/neuonc/nop051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wachsberger PR, Lawrence RY, Liu Y, Xia X, Andersen B, Dicker AP. Cediranib enhances control of wild type EGFR and EGFRvIII-expressing gliomas through potentiating temozolomide, but not through radiosensitization: implications for the clinic. J Neurooncol. 2011;105:181–190. doi: 10.1007/s11060-011-0580-y. [DOI] [PubMed] [Google Scholar]
- 50.Batchelor TT, Sorensen AG, di Tomaso E, Zhang WT, Duda DG, Cohen KS, Kozak KR, Cahill DP, Chen PJ, Zhu M. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11:83–95. doi: 10.1016/j.ccr.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brandes AA, Stupp R, Hau P, Lacombe D, Gorlia T, Tosoni A, Mirimanoff RO, Kros JM, van den Bent MJ. EORTC study 26041-22041: phase I/II study on concomitant and adjuvant temozolomide (TMZ) and radiotherapy (RT) with PTK787/ZK222584 (PTK/ZK) in newly diagnosed glioblastoma. Eur J Cancer. 2010;46:348–354. doi: 10.1016/j.ejca.2009.10.029. [DOI] [PubMed] [Google Scholar]
- 52.Iwamoto FM, Lamborn KR, Robins HI, Mehta MP, Chang SM, Butowski NA, Deangelis LM, Abrey LE, Zhang WT, Prados MD. Phase II trial of pazopanib (GW786034), an oral multi-targeted angiogenesis inhibitor, for adults with recurrent glioblastoma (North American Brain Tumor Consortium Study 06-02) Neuro Oncol. 2010;12:855–861. doi: 10.1093/neuonc/noq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, Qian F, Chu F, Bentzien F, Cancilla B. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther. 2011;10:2298–2308. doi: 10.1158/1535-7163.MCT-11-0264. [DOI] [PubMed] [Google Scholar]
- 54.Wen PY. American Society of Clinical Oncology 2010: report of selected studies from the CNS tumors section. Expert Rev Anticancer Ther. 2010;10:1367–1369. doi: 10.1586/era.10.117. [DOI] [PubMed] [Google Scholar]
- 55.Zhang Y, Guessous F, Kofman A, Schiff D, Abounader R. XL-184, a MET, VEGFR-2 and RET kinase inhibitor for the treatment of thyroid cancer, glioblastoma multiforme and NSCLC. IDrugs. 2010;13:112–121. [PMC free article] [PubMed] [Google Scholar]
- 56.Navis AC, Bourgonje A, Wesseling P, Wright A, Hendriks W, Verrijp K, van der Laak JA, Heerschap A, Leenders WP. Effects of dual targeting of tumor cells and stroma in human glioblastoma xenografts with a tyrosine kinase inhibitor against c-MET and VEGFR2. PLoS One. 2013;8:e58262. doi: 10.1371/journal.pone.0058262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tabatabai G, Tonn JC, Stupp R, Weller M. The role of integrins in glioma biology and anti-glioma therapies. Curr Pharm Des. 2011;17:2402–2410. doi: 10.2174/138161211797249189. [DOI] [PubMed] [Google Scholar]
- 58.Scaringi C, Minniti G, Caporello P, Enrici RM. Integrin inhibitor cilengitide for the treatment of glioblastoma: a brief overview of current clinical results. Anticancer Res. 2012;32:4213–4223. [PubMed] [Google Scholar]
- 59.Onishi M, Ichikawa T, Kurozumi K, Fujii K, Yoshida K, Inoue S, Michiue H, Chiocca EA, Kaur B, Date I. Bimodal anti-glioma mechanisms of cilengitide demonstrated by novel invasive glioma models. Neuropathology. 2013;33:162–174. doi: 10.1111/j.1440-1789.2012.01344.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stupp R, Hegi ME, Neyns B, Goldbrunner R, Schlegel U, Clement PM, Grabenbauer GG, Ochsenbein AF, Simon M, Dietrich PY. Phase I/IIa study of cilengitide and temozolomide with concomitant radiotherapy followed by cilengitide and temozolomide maintenance therapy in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28:2712–2718. doi: 10.1200/JCO.2009.26.6650. [DOI] [PubMed] [Google Scholar]
- 61.Nabors LB, Mikkelsen T, Hegi ME, Ye X, Batchelor T, Lesser G, Peereboom D, Rosenfeld MR, Olsen J, Brem S. A safety run-in and randomized phase 2 study of cilengitide combined with chemoradiation for newly diagnosed glioblastoma (NABTT 0306) Cancer. 2012;118:5601–5607. doi: 10.1002/cncr.27585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stupp R, Hegi ME, Gorlia T, Erridge SC, Perry J, Hong YK, Aldape KD, Lhermitte B, Pietsch T, Grujicic D. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15:1100–1108. doi: 10.1016/S1470-2045(14)70379-1. [DOI] [PubMed] [Google Scholar]
- 63.Lombardi G, Zustovich F, Farina P, Polo V, Farina M, Puppa AD, Bertorelle R, Gardiman MP, Berti F, Zagonel V. Cilengitide in bevacizumab-refractory high-grade glioma: two case reports and critical review of the literature. Anticancer Drugs. 2012;23:749–753. doi: 10.1097/CAD.0b013e3283520e2c. [DOI] [PubMed] [Google Scholar]
- 64.Onishi M, Ichikawa T, Kurozumi K, Date I. Angiogenesis and invasion in glioma. Brain Tumor Pathol. 2011;28:13–24. doi: 10.1007/s10014-010-0007-z. [DOI] [PubMed] [Google Scholar]
- 65.Maruotti N, Cantatore FP, Ribatti D. Thalidomide in treatment of connective diseases and vasculities. Reumatismo. 2006;58:187–190. doi: 10.4081/reumatismo.2006.187. [DOI] [PubMed] [Google Scholar]
- 66.Marx GM, Pavlakis N, McCowatt S, Boyle FM, Levi JA, Bell DR, Cook R, Biggs M, Little N, Wheeler HR. Phase II study of thalidomide in the treatment of recurrent glioblastoma multiforme. J Neurooncol. 2001;54:31–38. doi: 10.1023/a:1012554328801. [DOI] [PubMed] [Google Scholar]
- 67.Riva M, Imbesi F, Beghi E, Galli C, Citterio A, Trapani P, Sterzi R, Collice M. Temozolomide and thalidomide in the treatment of glioblastoma multiforme. Anticancer Res. 2007;27:1067–1071. [PubMed] [Google Scholar]
- 68.Ruiz J, Case D, Enevold G, Rosdhal R, Tatter SB, Ellis TL, McQuellon RP, McMullen KP, Stieber VW, Shaw EG. A phase II trial of thalidomide and procarbazine in adult patients with recurrent or progressive malignant gliomas. J Neurooncol. 2012;106:611–617. doi: 10.1007/s11060-011-0698-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Giglio P, Dhamne M, Hess KR, Gilbert MR, Groves MD, Levin VA, Kang SL, Ictech SE, Liu V, Colman H. Phase 2 trial of irinotecan and thalidomide in adults with recurrent anaplastic glioma. Cancer. 2012;118:3599–3606. doi: 10.1002/cncr.26663. [DOI] [PubMed] [Google Scholar]
- 70.Huang TT, Sarkaria SM, Cloughesy TF, Mischel PS. Targeted therapy for malignant glioma patients: lessons learned and the road ahead. Neurotherapeutics. 2009;6:500–512. doi: 10.1016/j.nurt.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Omuro AM, Faivre S, Raymond E. Lessons learned in the development of targeted therapy for malignant gliomas. Mol Cancer Ther. 2007;6:1909–1919. doi: 10.1158/1535-7163.MCT-07-0047. [DOI] [PubMed] [Google Scholar]
- 72.Lv S, Teugels E, Sadones J, De Brakeleer S, Duerinck J, Du Four S, Michotte A, De Greve J, Neyns B. Correlation of EGFR, IDH1 and PTEN status with the outcome of patients with recurrent glioblastoma treated in a phase II clinical trial with the EGFR-blocking monoclonal antibody cetuximab. Int J Oncol. 2012;41:1029–1035. doi: 10.3892/ijo.2012.1539. [DOI] [PubMed] [Google Scholar]
- 73.Neyns B, Sadones J, Joosens E, Bouttens F, Verbeke L, Baurain JF, D'Hondt L, Strauven T, Chaskis C, In't Veld P. Stratified phase II trial of cetuximab in patients with recurrent high-grade glioma. Ann Oncol. 2009;20:1596–1603. doi: 10.1093/annonc/mdp032. [DOI] [PubMed] [Google Scholar]
- 74.Uhm JH, Ballman KV, Wu W, Giannini C, Krauss JC, Buckner JC, James CD, Scheithauer BW, Behrens RJ, Flynn PJ. Phase II evaluation of gefitinib in patients with newly diagnosed Grade 4 astrocytoma: Mayo/North Central Cancer Treatment Group Study N0074. Int J Radiat Oncol Biol Phys. 2011;80:347–353. doi: 10.1016/j.ijrobp.2010.01.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chakravarti A, Wang M, Robins HI, Lautenschlaeger T, Curran WJ, Brachman DG, Schultz CJ, Choucair A, Dolled-Filhart M, Christiansen J. RTOG 0211: a phase 1/2 study of radiation therapy with concurrent gefitinib for newly diagnosed glioblastoma patients. Int J Radiat Oncol Biol Phys. 2013;85:1206–1211. doi: 10.1016/j.ijrobp.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yung WK, Vredenburgh JJ, Cloughesy TF, Nghiemphu P, Klencke B, Gilbert MR, Reardon DA, Prados MD. Safety and efficacy of erlotinib in first-relapse glioblastoma: a phase II open-label study. Neuro Oncol. 2010;12:1061–1070. doi: 10.1093/neuonc/noq072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Peereboom DM, Shepard DR, Ahluwalia MS, Brewer CJ, Agarwal N, Stevens GH, Suh JH, Toms SA, Vogelbaum MA, Weil RJ. Phase II trial of erlotinib with temozolomide and radiation in patients with newly diagnosed glioblastoma multiforme. J Neurooncol. 2010;98:93–99. doi: 10.1007/s11060-009-0067-2. [DOI] [PubMed] [Google Scholar]
- 78.Kesavabhotla K, Schlaff CD, Shin B, Mubita L, Kaplan R, Tsiouris AJ, Pannullo SC, Christos P, Lavi E, Scheff R. Phase I/II study of oral erlotinib for treatment of relapsed/refractory glioblastoma multiforme and anaplastic astrocytoma. J Exp Ther Oncol. 2012;10:71–81. [PubMed] [Google Scholar]
- 79.Thiessen B, Stewart C, Tsao M, Kamel-Reid S, Schaiquevich P, Mason W, Easaw J, Belanger K, Forsyth P, McIntosh L. A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother Pharmacol. 2010;65:353–361. doi: 10.1007/s00280-009-1041-6. [DOI] [PubMed] [Google Scholar]
- 80.Hong J, Peng Y, Liao Y, Jiang W, Wei R, Huo L, Han Z, Duan C, Zhong M. Nimotuzumab prolongs survival in patients with malignant gliomas: a phase I/II clinical study of concomitant radiochemotherapy with or without nimotuzumab. Exp Ther Med. 2012;4:151–157. doi: 10.3892/etm.2012.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cabanas R, Saurez G, Rios M, Alert J, Reyes A, Valdes J, Gonzalez MC, Pedrayes JL, Avila M, Herrera R. Treatment of children with high grade glioma with nimotuzumab: a 5-year institutional experience. MAbs. 2013;5:202–207. doi: 10.4161/mabs.22970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Boland WK, Bebb G. Nimotuzumab: a novel anti-EGFR monoclonal antibody that retains anti-EGFR activity while minimizing skin toxicity. Expert Opin Biol Ther. 2009;9:1199–1206. doi: 10.1517/14712590903110709. [DOI] [PubMed] [Google Scholar]
- 83.Lo HW. EGFR-targeted therapy in malignant glioma: novel aspects and mechanisms of drug resistance. Curr Mol Pharmacol. 2010;3:37–52. doi: 10.2174/1874467211003010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, Rak J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol. 2008;10:619–624. doi: 10.1038/ncb1725. [DOI] [PubMed] [Google Scholar]
- 85.Gan HK, Kaye AH, Luwor RB. The EGFRvIII variant in glioblastoma multiforme. J Clin Neurosci. 2009;16:748–754. doi: 10.1016/j.jocn.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 86.Yang W, Barth RF, Wu G, Kawabata S, Sferra TJ, Bandyopadhyaya AK, Tjarks W, Ferketich AK, Moeschberger ML, Binns PJ. Molecular targeting and treatment of EGFRvIII-positive gliomas using boronated monoclonal antibody L8A4. Clin Cancer Res. 2006;12:3792–3802. doi: 10.1158/1078-0432.CCR-06-0141. [DOI] [PubMed] [Google Scholar]
- 87.Yang W, Barth RF, Wu G, Ciesielski MJ, Fenstermaker RA, Moffat BA, Ross BD, Wikstrand CJ. Development of a syngeneic rat brain tumor model expressing EGFRvIII and its use for molecular targeting studies with monoclonal antibody L8A4. Clin Cancer Res. 2005;11:341–350. [PubMed] [Google Scholar]
- 88.Ellis AG, Doherty MM, Walker F, Weinstock J, Nerrie M, Vitali A, Murphy R, Johns TG, Scott AM, Levitzki A. Preclinical analysis of the analinoquinazoline AG1478, a specific small molecule inhibitor of EGF receptor tyrosine kinase. Biochem Pharmacol. 2006;71:1422–1434. doi: 10.1016/j.bcp.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 89.Johns TG, Luwor RB, Murone C, Walker F, Weinstock J, Vitali AA, Perera RM, Jungbluth AA, Stockert E, Old LJ. Antitumor efficacy of cytotoxic drugs and the monoclonal antibody 806 is enhanced by the EGF receptor inhibitor AG1478. Proc Natl Acad Sci U S A. 2003;100:15871–15876. doi: 10.1073/pnas.2036503100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Trembath DG, Lal A, Kroll DJ, Oberlies NH, Riggins GJ. A novel small molecule that selectively inhibits glioblastoma cells expressing EGFRvIII. Mol Cancer. 2007;6:30. doi: 10.1186/1476-4598-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Scott AM, Lee FT, Tebbutt N, Herbertson R, Gill SS, Liu Z, Skrinos E, Murone C, Saunder TH, Chappell B. A phase I clinical trial with monoclonal antibody ch806 targeting transitional state and mutant epidermal growth factor receptors. Proc Natl Acad Sci U S A. 2007;104:4071–4076. doi: 10.1073/pnas.0611693104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Board R, Jayson GC. Platelet-derived growth factor receptor (PDGFR): a target for anticancer therapeutics. Drug Resist Updat. 2005;8:75–83. doi: 10.1016/j.drup.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 93.Reardon DA, Dresemann G, Taillibert S, Campone M, van den Bent M, Clement P, Blomquist E, Gordower L, Schultz H, Raizer J. Multicentre phase II studies evaluating imatinib plus hydroxyurea in patients with progressive glioblastoma. Br J Cancer. 2009;101:1995–2004. doi: 10.1038/sj.bjc.6605411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kinsella P, Howley R, Doolan P, Clarke C, Madden SF, Clynes M, Farrell M, Amberger-Murphy V. Characterization and response of newly developed high-grade glioma cultures to the tyrosine kinase inhibitors, erlotinib, gefitinib and imatinib. Exp Cell Res. 2012;318:641–652. doi: 10.1016/j.yexcr.2012.01.014. [DOI] [PubMed] [Google Scholar]
- 95.Dong Y, Jia L, Wang X, Tan X, Xu J, Deng Z, Jiang T, Rainov NG, Li B, Ren H. Selective inhibition of PDGFR by imatinib elicits the sustained activation of ERK and downstream receptor signaling in malignant glioma cells. Int J Oncol. 2011;38:555–569. doi: 10.3892/ijo.2010.861. [DOI] [PubMed] [Google Scholar]
- 96.Reardon DA, Desjardins A, Vredenburgh JJ, Herndon JE, II, Coan A, Gururangan S, Peters KB, McLendon R, Sathornsumetee S, Rich JN. Phase II study of Gleevec plus hydroxyurea in adults with progressive or recurrent low-grade glioma. Cancer. 2012;118:4759–4767. doi: 10.1002/cncr.26541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Morris PG, Abrey LE. Novel targeted agents for platelet-derived growth factor receptor and c-KIT in malignant gliomas. Target Oncol. 2010;5:193–200. doi: 10.1007/s11523-010-0160-7. [DOI] [PubMed] [Google Scholar]
- 98.Franceschi E, Stupp R, van den Bent MJ, van Herpen C, Laigle Donadey F, Gorlia T, Hegi M, Lhermitte B, Strauss LC, Allgeier A. EORTC 26083 phase I/II trial of dasatinib in combination with CCNU in patients with recurrent glioblastoma. Neuro Oncol. 2012;14:1503–1510. doi: 10.1093/neuonc/nos256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lehky TJ, Iwamoto FM, Kreisl TN, Floeter MK, Fine HA. Neuromuscular junction toxicity with tandutinib induces a myasthenic-like syndrome. Neurology. 2011;76:236–241. doi: 10.1212/WNL.0b013e3182074a69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen L, Han L, Shi Z, Zhang K, Liu Y, Zheng Y, Jiang T, Pu P, Jiang C, Kang C. LY294002 enhances cytotoxicity of temozolomide in glioma by down-regulation of the PI3K/Akt pathway. Mol Med Rep. 2012;5:575–579. doi: 10.3892/mmr.2011.674. [DOI] [PubMed] [Google Scholar]
- 101.Millet P, Granotier C, Etienne O, Boussin FD. Radiation-induced upregulation of telomerase activity escapes PI3-kinase inhibition in two malignant glioma cell lines. Int J Oncol. 2013;43:375–382. doi: 10.3892/ijo.2013.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mizushina Y, Takeuchi T, Sugawara F, Yoshida H. Anti-cancer targeting telomerase inhibitors: β-rubromycin and oleic acid. Mini Rev Med Chem. 2012;12:1135–1143. doi: 10.2174/138955712802762220. [DOI] [PubMed] [Google Scholar]
- 103.de la Pena L, Burgan WE, Carter DJ, Hollingshead MG, Satyamitra M, Camphausen K, Tofilon PJ. Inhibition of Akt by the alkylphospholipid perifosine does not enhance the radiosensitivity of human glioma cells. Mol Cancer Ther. 2006;5:1504–1510. doi: 10.1158/1535-7163.MCT-06-0091. [DOI] [PubMed] [Google Scholar]
- 104.Becher OJ, Hambardzumyan D, Walker TR, Helmy K, Nazarian J, Albrecht S, Hiner RL, Gall S, Huse JT, Jabado N. Preclinical evaluation of radiation and perifosine in a genetically and histologically accurate model of brainstem glioma. Cancer Res. 2010;70:2548–2557. doi: 10.1158/0008-5472.CAN-09-2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Momota H, Nerio E, Holland EC. Perifosine inhibits multiple signaling pathways in glial progenitors and cooperates with temozolomide to arrest cell proliferation in gliomas in vivo. Cancer Res. 2005;65:7429–7435. doi: 10.1158/0008-5472.CAN-05-1042. [DOI] [PubMed] [Google Scholar]
- 106.Yang L, Clarke MJ, Carlson BL, Mladek AC, Schroeder MA, Decker P, Wu W, Kitange GJ, Grogan PT, Goble JM. PTEN loss does not predict for response to RAD001 (Everolimus) in a glioblastoma orthotopic xenograft test panel. Clin Cancer Res. 2008;14:3993–4001. doi: 10.1158/1078-0432.CCR-07-4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cloughesy TF, Yoshimoto K, Nghiemphu P, Brown K, Dang J, Zhu S, Hsueh T, Chen Y, Wang W, Youngkin D. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008;5:e8. doi: 10.1371/journal.pmed.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.El Habr EA, Adamopoulos C, Levidou G, Saetta AA, Korkolopoulou P, Piperi C. The clinical and prognostic significance of activated AKT-mTOR pathway in human astrocytomas. Neurol Res Int. 2012;2012:454957. doi: 10.1155/2012/454957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Geoerger B, Kieran MW, Grupp S, Perek D, Clancy J, Krygowski M, Ananthakrishnan R, Boni JP, Berkenblit A, Spunt SL. Phase II trial of temsirolimus in children with high-grade glioma, neuroblastoma and rhabdomyosarcoma. Eur J Cancer. 2012;48:253–262. doi: 10.1016/j.ejca.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chang SM, Wen P, Cloughesy T, Greenberg H, Schiff D, Conrad C, Fink K, Robins HI, De Angelis L, Raizer J. Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Invest New Drugs. 2005;23:357–361. doi: 10.1007/s10637-005-1444-0. [DOI] [PubMed] [Google Scholar]
- 111.Reardon DA, Wen PY, Alfred Yung WK, Berk L, Narasimhan N, Turner CD, Clackson T, Rivera VM, Vogelbaum MA. Ridaforolimus for patients with progressive or recurrent malignant glioma: a perisurgical, sequential, ascending-dose trial. Cancer Chemother Pharmacol. 2012;69:849–860. doi: 10.1007/s00280-011-1773-y. [DOI] [PubMed] [Google Scholar]
- 112.Galanis E, Buckner JC, Maurer MJ, Kreisberg JI, Ballman K, Boni J, Peralba JM, Jenkins RB, Dakhil SR, Morton RF. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group Study. J Clin Oncol. 2005;23:5294–5304. doi: 10.1200/JCO.2005.23.622. [DOI] [PubMed] [Google Scholar]
- 113.Weiler M, Pfenning PN, Thiepold AL, Blaes J, Jestaedt L, Gronych J, Dittmann LM, Berger B, Jugold M, Kosch M. Suppression of proinvasive RGS4 by mTOR inhibition optimizes glioma treatment. Oncogene. 2013;32:1099–1109. doi: 10.1038/onc.2012.137. [DOI] [PubMed] [Google Scholar]
- 114.Sarkaria JN, Galanis E, Wu W, Dietz AB, Kaufmann TJ, Gustafson MP, Brown PD, Uhm JH, Rao RD, Doyle L. Combination of temsirolimus (CCI-779) with chemoradiation in newly diagnosed glioblastoma multiforme (GBM) (NCCTG trial N027D) is associated with increased infectious risks. Clin Cancer Res. 2010;16:5573–5580. doi: 10.1158/1078-0432.CCR-10-1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sarkaria JN, Galanis E, Wu W, Peller PJ, Giannini C, Brown PD, Uhm JH, McGraw S, Jaeckle KA, Buckner JC. North Central Cancer Treatment Group Phase I trial N057K of everolimus (RAD001) and temozolomide in combination with radiation therapy in patients with newly diagnosed glioblastoma multiforme. Int J Radiat Oncol Biol Phys. 2011;81:468–475. doi: 10.1016/j.ijrobp.2010.05.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nawroth R, Stellwagen F, Schulz WA, Stoehr R, Hartmann A, Krause BJ, Gschwend JE, Retz M. S6K1 and 4E-BP1 are independent regulated and control cellular growth in bladder cancer. PLoS One. 2011;6:e27509. doi: 10.1371/journal.pone.0027509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Liu TJ, Koul D, LaFortune T, Tiao N, Shen RJ, Maira SM, Garcia-Echevrria C, Yung WK. NVP-BEZ235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomas. Mol Cancer Ther. 2009;8:2204–2210. doi: 10.1158/1535-7163.MCT-09-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cerniglia GJ, Karar J, Tyagi S, Christofidou-Solomidou M, Rengan R, Koumenis C, Maity A. Inhibition of autophagy as a strategy to augment radiosensitization by the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235. Mol Pharmacol. 2012;82:1230–1240. doi: 10.1124/mol.112.080408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang WJ, Long LM, Yang N, Zhang QQ, Ji WJ, Zhao JH, Qin ZH, Wang Z, Chen G, Liang ZQ. NVP-BEZ235, a novel dual PI3K/mTOR inhibitor, enhances the radiosensitivity of human glioma stem cells in vitro. Acta Pharmacol Sin. 2013;34:681–690. doi: 10.1038/aps.2013.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Prasad G, Sottero T, Yang X, Mueller S, James CD, Weiss WA, Polley MY, Ozawa T, Berger MS, Aftab DT. Inhibition of PI3K/mTOR pathways in glioblastoma and implications for combination therapy with temozolomide. Neuro Oncol. 2011;13:384–392. doi: 10.1093/neuonc/noq193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mercer RW, Tyler MA, Ulasov IV, Lesniak MS. Targeted therapies for malignant glioma: progress and potential. BioDrugs. 2009;23:25–35. doi: 10.2165/00063030-200923010-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pelloski CE, Lin E, Zhang L, Yung WK, Colman H, Liu JL, Woo SY, Heimberger AB, Suki D, Prados M. Prognostic associations of activated mitogen-activated protein kinase and Akt pathways in glioblastoma. Clin Cancer Res. 2006;12:3935–3941. doi: 10.1158/1078-0432.CCR-05-2202. [DOI] [PubMed] [Google Scholar]
- 123.Paul S, Mischel TFC. Targeted molecular therapy of GBM. Brain Pathol. 2003;13:52–61. doi: 10.1111/j.1750-3639.2003.tb00006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chaponis D, Barnes JW, Dellagatta JL, Kesari S, Fast E, Sauvageot C, Panagrahy D, Greene ER, Ramakrishna N, Wen PY. Lonafarnib (SCH66336) improves the activity of temozolomide and radiation for orthotopic malignant gliomas. J Neurooncol. 2011;104:179–189. doi: 10.1007/s11060-010-0502-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Desjardins A, Reardon DA, Peters KB, Threatt S, Coan AD, Herndon JE, II, Friedman AH, Friedman HS, Vredenburgh JJ. A phase I trial of the farnesyl transferase inhibitor, SCH 66336, with temozolomide for patients with malignant glioma. J Neurooncol. 2011;105:601–606. doi: 10.1007/s11060-011-0627-0. [DOI] [PubMed] [Google Scholar]
- 126.Lustig R, Mikkelsen T, Lesser G, Grossman S, Ye X, Desideri S, Fisher J, Wright J. Phase II preradiation R115777 (tipifarnib) in newly diagnosed GBM with residual enhancing disease. Neuro Oncol. 2008;10:1004–1009. doi: 10.1215/15228517-2008-070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nghiemphu PL, Wen PY, Lamborn KR, Drappatz J, Robins HI, Fink K, Malkin MG, Lieberman FS, DeAngelis LM, Torres-Trejo A. A phase I trial of tipifarnib with radiation therapy, with and without temozolomide, for patients with newly diagnosed glioblastoma. Int J Radiat Oncol Biol Phys. 2011;81:1422–1427. doi: 10.1016/j.ijrobp.2010.07.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Vlachostergios PJ, Voutsadakis IA, Papandreou CN. The ubiquitin-proteasome system in glioma cell cycle control. Cell Div. 2012;7:18. doi: 10.1186/1747-1028-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Landis-Piwowar KR, Milacic V, Chen D, Yang H, Zhao Y, Chan TH, Yan B, Dou QP. The proteasome as a potential target for novel anticancer drugs and chemosensitizers. Drug Resist Updat. 2006;9:263–273. doi: 10.1016/j.drup.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 130.Yin D, Zhou H, Kumagai T, Liu G, Ong JM, Black KL, Koeffler HP. Proteasome inhibitor PS-341 causes cell growth arrest and apoptosis in human glioblastoma multiforme (GBM) Oncogene. 2005;24:344–354. doi: 10.1038/sj.onc.1208225. [DOI] [PubMed] [Google Scholar]
- 131.Phuphanich S, Supko JG, Carson KA, Grossman SA, Burt Nabors L, Mikkelsen T, Lesser G, Rosenfeld S, Desideri S, Olson JJ. Phase 1 clinical trial of bortezomib in adults with recurrent malignant glioma. J Neurooncol. 2010;100:95–103. doi: 10.1007/s11060-010-0143-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kubicek GJ, Werner-Wasik M, Machtay M, Mallon G, Myers T, Ramirez M, Andrews D, Curran WJ, Jr., Dicker AP. Phase I trial using proteasome inhibitor bortezomib and concurrent temozolomide and radiotherapy for central nervous system malignancies. Int J Radiat Oncol Biol Phys. 2009;74:433–439. doi: 10.1016/j.ijrobp.2008.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.New M, Olzscha H, La Thangue NB. HDAC inhibitor-based therapies: can we interpret the code? Mol Oncol. 2012;6:637–656. doi: 10.1016/j.molonc.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Shabason JE, Tofilon PJ, Camphausen K. Grand rounds at the National Institutes of Health: HDAC inhibitors as radiation modifiers, from bench to clinic. J Cell Mol Med. 2011;15:2735–2744. doi: 10.1111/j.1582-4934.2011.01296.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Huang WJ, Liang YC, Chuang SE, Chi LL, Lee CY, Lin CW, Chen AL, Huang JS, Chiu CJ, Lee CF. NBM-HD-1: a novel histone deacetylase inhibitor with anticancer activity. Evid Based Complement Alternat Med. 2012;2012:781417. doi: 10.1155/2012/781417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Galanis E, Jaeckle KA, Maurer MJ, Reid JM, Ames MM, Hardwick JS, Reilly JF, Loboda A, Nebozhyn M, Fantin VR. Phase II trial of vorinostat in recurrent glioblastoma multiforme: a north central cancer treatment group study. J Clin Oncol. 2009;27:2052–2058. doi: 10.1200/JCO.2008.19.0694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Iwamoto FM, Lamborn KR, Kuhn JG, Wen PY, Yung WK, Gilbert MR, Chang SM, Lieberman FS, Prados MD, Fine HA. A phase I/II trial of the histone deacetylase inhibitor romidepsin for adults with recurrent malignant glioma: North American Brain Tumor Consortium Study 03-03. Neuro Oncol. 2011;13:509–516. doi: 10.1093/neuonc/nor017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lee EQ, Puduvalli VK, Reid JM, Kuhn JG, Lamborn KR, Cloughesy TF, Chang SM, Drappatz J, Yung WK, Gilbert MR. Phase I study of vorinostat in combination with temozolomide in patients with high-grade gliomas: North American Brain Tumor Consortium Study 04-03. Clin Cancer Res. 2012;18:6032–6039. doi: 10.1158/1078-0432.CCR-12-1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Weller M, Gorlia T, Cairncross JG, van den Bent MJ, Mason W, Belanger K, Brandes AA, Bogdahn U, Macdonald DR, Forsyth P. Prolonged survival with valproic acid use in the EORTC/NCIC temozolomide trial for glioblastoma. Neurology. 2011;77:1156–1164. doi: 10.1212/WNL.0b013e31822f02e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bai RY, Staedtke V, Riggins GJ. Molecular targeting of glioblastoma: drug discovery and therapies. Trends Mol Med. 2011;17:301–312. doi: 10.1016/j.molmed.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kreisl TN, McNeill KA, Sul J, Iwamoto FM, Shih J, Fine HA. A phase I/II trial of vandetanib for patients with recurrent malignant glioma. Neuro Oncol. 2012;14:1519–1526. doi: 10.1093/neuonc/nos265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Jo MY, Kim YG, Kim Y, Lee SJ, Kim MH, Joo KM, Kim HH, Nam DH. Combined therapy of temozolomide and ZD6474 (vandetanib) effectively reduces glioblastoma tumor volume through anti-angiogenic and anti-proliferative mechanisms. Mol Med Rep. 2012;6:88–92. doi: 10.3892/mmr.2012.868. [DOI] [PubMed] [Google Scholar]
- 143.Drappatz J, Norden AD, Wong ET, Doherty LM, Lafrankie DC, Ciampa A, Kesari S, Sceppa C, Gerard M, Phan P. Phase I study of vandetanib with radiotherapy and temozolomide for newly diagnosed glioblastoma. Int J Radiat Oncol Biol Phys. 2010;78:85–90. doi: 10.1016/j.ijrobp.2009.07.1741. [DOI] [PubMed] [Google Scholar]
- 144.Fields EC, Damek D, Gaspar LE, Liu AK, Kavanagh BD, Waziri A, Lillehei K, Chen C. Phase I dose escalation trial of vandetanib with fractionated radiosurgery in patients with recurrent malignant gliomas. Int J Radiat Oncol Biol Phys. 2012;82:51–57. doi: 10.1016/j.ijrobp.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 145.Reardon DA, Conrad CA, Cloughesy T, Prados MD, Friedman HS, Aldape KD, Mischel P, Xia J, DiLea C, Huang J. Phase I study of AEE788, a novel multitarget inhibitor of ErbB- and VEGF-receptor-family tyrosine kinases, in recurrent glioblastoma patients. Cancer Chemother Pharmacol. 2012;69:1507–1518. doi: 10.1007/s00280-012-1854-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Pan E, Yu D, Yue B, Potthast L, Chowdhary S, Smith P, Chamberlain M. A prospective phase II single-institution trial of sunitinib for recurrent malignant glioma. J Neurooncol. 2012;110:111–118. doi: 10.1007/s11060-012-0943-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Reardon DA, Vredenburgh JJ, Desjardins A, Peters K, Gururangan S, Sampson JH, Marcello J, Herndon JE, II, McLendon RE, Janney D. Effect of CYP3A-inducing anti-epileptics on sorafenib exposure: results of a phase II study of sorafenib plus daily temozolomide in adults with recurrent glioblastoma. J Neurooncol. 2011;101:57–66. doi: 10.1007/s11060-010-0217-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Reardon DA, Vredenburgh JJ, Coan A, Desjardins A, Peters KB, Gururangan S, Sathornsumetee S, Rich JN, Herndon JE, Friedman HS. Phase I study of sunitinib and irinotecan for patients with recurrent malignant glioma. J Neurooncol. 2011;105:621–627. doi: 10.1007/s11060-011-0631-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Den RB, Kamrava M, Sheng Z, Werner-Wasik M, Dougherty E, Marinucchi M, Lawrence YR, Hegarty S, Hyslop T, Andrews DW. A phase I study of the combination of sorafenib with temozolomide and radiation therapy for the treatment of primary and recurrent high-grade gliomas. Int J Radiat Oncol Biol Phys. 2013;85:321–328. doi: 10.1016/j.ijrobp.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Reardon DA, Desjardins A, Vredenburgh JJ, Gururangan S, Friedman AH, Herndon JE, II, Marcello J, Norfleet JA, McLendon RE, Sampson JH. Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. J Neurooncol. 2010;96:219–230. doi: 10.1007/s11060-009-9950-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kreisl TN, Lassman AB, Mischel PS, Rosen N, Scher HI, Teruya-Feldstein J, Shaffer D, Lis E, Abrey LE. A pilot study of everolimus and gefitinib in the treatment of recurrent glioblastoma (GBM) J Neurooncol. 2009;92:99–105. doi: 10.1007/s11060-008-9741-z. [DOI] [PubMed] [Google Scholar]
- 152.Reardon DA, Quinn JA, Vredenburgh JJ, Gururangan S, Friedman AH, Desjardins A, Sathornsumetee S, Herndon JE, II, Dowell JM, McLendon RE. Phase 1 trial of gefitinib plus sirolimus in adults with recurrent malignant glioma. Clin Cancer Res. 2006;12:860–868. doi: 10.1158/1078-0432.CCR-05-2215. [DOI] [PubMed] [Google Scholar]
- 153.Reardon DA, Cloughesy T, Rich J, Alfred Yung WK, Yung L, DiLea C, Huang J, Dugan M, Mietlowski W, Maes A. Pharmacokinetic drug interaction between AEE788 and RAD001 causing thrombocytopenia in patients with glioblastoma. Cancer Chemother Pharmacol. 2012;69:281–287. doi: 10.1007/s00280-011-1754-1. [DOI] [PubMed] [Google Scholar]
- 154.Carracedo A, Baselga J, Pandolfi PP. Deconstructing feedback-signaling networks to improve anticancer therapy with mTORC1 inhibitors. Cell Cycle. 2008;7:3805–3809. doi: 10.4161/cc.7.24.7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Pitter KL, Galban CJ, Galban S, Tehrani OS, Li F, Charles N, Bradbury MS, Becher OJ, Chenevert TL, Rehemtulla A. Perifosine and CCI 779 co-operate to induce cell death and decrease proliferation in PTEN-intact and PTEN-deficient PDGF-driven murine glioblastoma. PLoS One. 2011;6:e14545. doi: 10.1371/journal.pone.0014545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Reardon DA, Vredenburgh JJ, Desjardins A, Peters KB, Sathornsumetee S, Threatt S, Sampson JH, Herndon JE, II, Coan A, McSherry F. Phase 1 trial of dasatinib plus erlotinib in adults with recurrent malignant glioma. J Neurooncol. 2012;108:499–506. doi: 10.1007/s11060-012-0848-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Reardon DA, Groves MD, Wen PY, Nabors L, Mikkelsen T, Rosenfeld S, Raizer J, Barriuso J, McLendon RE, Suttle AB. A phase I/II trial of pazopanib in combination with lapatinib in adult patients with relapsed malignant glioma. Clin Cancer Res. 2013;19:900–908. doi: 10.1158/1078-0432.CCR-12-1707. [DOI] [PubMed] [Google Scholar]
- 158.Friday BB, Anderson SK, Buckner J, Yu C, Giannini C, Geoffroy F, Schwerkoske J, Mazurczak M, Gross H, Pajon E. Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: a north central cancer treatment group study. Neuro Oncol. 2012;14:215–221. doi: 10.1093/neuonc/nor198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chinnaiyan P, Chowdhary S, Potthast L, Prabhu A, Tsai YY, Sarcar B, Kahali S, Brem S, Yu HM, Rojiani A. Phase I trial of vorinostat combined with bevacizumab and CPT-11 in recurrent glioblastoma. Neuro Oncol. 2012;14:93–100. doi: 10.1093/neuonc/nor187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Blesa JM, Molla SB, Esparcia MF, Ortells JM, Godoy MP, Das AM, Magan BM, Pulla MP, Sanchez JL, Canales JB. Durable complete remission of a brainstem glioma treated with a combination of bevacizumab and cetuximab. Case Rep Oncol. 2012;5:676–681. doi: 10.1159/000341852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Stopschinski BE, Beier CP, Beier D. Glioblastoma cancer stem cells—from concept to clinical application. Cancer Lett. 2013;338:32–40. doi: 10.1016/j.canlet.2012.05.033. [DOI] [PubMed] [Google Scholar]
- 162.Karamboulas C, Ailles L. Developmental signaling pathways in cancer stem cells of solid tumors. Biochim Biophys Acta. 2013;1830:2481–2495. doi: 10.1016/j.bbagen.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 163.Ohka F, Natsume A, Wakabayashi T. Current trends in targeted therapies for glioblastoma multiforme. Neurol Res Int. 2012;2012:878425. doi: 10.1155/2012/878425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wang Z, Li Y, Banerjee S, Sarkar FH. Emerging role of Notch in stem cells and cancer. Cancer Lett. 2009;279:8–12. doi: 10.1016/j.canlet.2008.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wang J, Wakeman TP, Lathia JD, Hjelmeland AB, Wang XF, White RR, Rich JN, Sullenger BA. Notch promotes radioresistance of glioma stem cells. Stem Cells. 2010;28:17–28. doi: 10.1002/stem.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Fan X, Khaki L, Zhu TS, Soules ME, Talsma CE, Gul N, Koh C, Zhang J, Li YM, Maciaczyk J. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells. 2010;28:5–16. doi: 10.1002/stem.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Dixit D, Ghildiyal R, Anto NP, Ghosh S, Sharma V, Sen E. Guggulsterone sensitizes glioblastoma cells to Sonic hedgehog inhibitor SANT-1 induced apoptosis in a Ras/NFκB dependent manner. Cancer Lett. 2013;336:347–358. doi: 10.1016/j.canlet.2013.03.025. [DOI] [PubMed] [Google Scholar]
- 168.Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol. 2007;17:165–172. doi: 10.1016/j.cub.2006.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.LoRusso PM, Rudin CM, Reddy JC, Tibes R, Weiss GJ, Borad MJ, Hann CL, Brahmer JR, Chang I, Darbonne WC. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin Cancer Res. 2011;17:2502–2511. doi: 10.1158/1078-0432.CCR-10-2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kalani MY, Cheshier SH, Cord BJ, Bababeygy SR, Vogel H, Weissman IL, Palmer TD, Nusse R. Wnt-mediated self-renewal of neural stem/progenitor cells. Proc Natl Acad Sci U S A. 2008;105:16970–16975. doi: 10.1073/pnas.0808616105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Takahashi-Yanaga F, Kahn M. Targeting Wnt signaling: can we safely eradicate cancer stem cells? Clin Cancer Res. 2010;16:3153–3162. doi: 10.1158/1078-0432.CCR-09-2943. [DOI] [PubMed] [Google Scholar]
- 172.Gong A, Huang S. FoxM1 and Wnt/β-catenin signaling in glioma stem cells. Cancer Res. 2012;72:5658–5662. doi: 10.1158/0008-5472.CAN-12-0953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, Shi Q, Cao Y, Lathia J, McLendon RE. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15:501–513. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Snuderl M, Fazlollahi L, Le LP, Nitta M, Zhelyazkova BH, Davidson CJ, Akhavanfard S, Cahill DP, Aldape KD, Betensky RA. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell. 2011;20:810–817. doi: 10.1016/j.ccr.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 175.Larsen AK, Ouaret D, El Ouadrani K, Petitprez A. Targeting EGFR and VEGF(R) pathway cross-talk in tumor survival and angiogenesis. Pharmacol Ther. 2011;131:80–90. doi: 10.1016/j.pharmthera.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 176.Nicholas MK, Lukas RV, Chmura S, Yamini B, Lesniak M, Pytel P. Molecular heterogeneity in glioblastoma: therapeutic opportunities and challenges. Semin Oncol. 2011;38:243–253. doi: 10.1053/j.seminoncol.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 177.Yan W, Zhang W, Jiang T. Oncogene addiction in gliomas: implications for molecular targeted therapy. J Exp Clin Cancer Res. 2011;30:58. doi: 10.1186/1756-9966-30-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Babic I, Mischel PS. Multiple functions of a glioblastoma fusion oncogene. J Clin Invest. 2013;123:548–551. doi: 10.1172/JCI67658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Singh D, Chan JM, Zoppoli P, Niola F, Sullivan R, Castano A, Liu EM, Reichel J, Porrati P, Pellegatta S. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science. 2012;337:1231–1235. doi: 10.1126/science.1220834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Jansen M, Yip S, Louis DN. Molecular pathology in adult gliomas: diagnostic, prognostic, and predictive markers. Lancet Neurol. 2010;9:717–726. doi: 10.1016/S1474-4422(10)70105-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gajadhar AS, Bogdanovic E, Munoz DM, Guha A. In situ analysis of mutant EGFRs prevalent in glioblastoma multiforme reveals aberrant dimerization, activation, and differential response to anti-EGFR targeted therapy. Mol Cancer Res. 2012;10:428–440. doi: 10.1158/1541-7786.MCR-11-0531. [DOI] [PubMed] [Google Scholar]
- 182.Cen L, Carlson BL, Schroeder MA, Ostrem JL, Kitange GJ, Mladek AC, Fink SR, Decker PA, Wu W, Kim JS. p16-Cdk4-Rb axis controls sensitivity to a cyclin-dependent kinase inhibitor PD0332991 in glioblastoma xenograft cells. Neuro Oncol. 2012;14:870–881. doi: 10.1093/neuonc/nos114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Michaud K, Solomon DA, Oermann E, Kim JS, Zhong WZ, Prados MD, Ozawa T, James CD, Waldman T. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 2010;70:3228–3238. doi: 10.1158/0008-5472.CAN-09-4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kim W, Liau LM. IDH mutations in human glioma. Neurosurg Clin N Am. 2012;23:471–480. doi: 10.1016/j.nec.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Wheeler CJ, Black KL. Vaccines for glioblastoma and high-grade glioma. Expert Rev Vaccines. 2011;10:875–886. doi: 10.1586/erv.11.71. [DOI] [PubMed] [Google Scholar]
- 186.Chang CN, Huang YC, Yang DM, Kikuta K, Wei KJ, Kubota T, Yang WK. A phase I/II clinical trial investigating the adverse and therapeutic effects of a postoperative autologous dendritic cell tumor vaccine in patients with malignant glioma. J Clin Neurosci. 2011;18:1048–1054. doi: 10.1016/j.jocn.2010.11.034. [DOI] [PubMed] [Google Scholar]
- 187.Li M, Li J, Liu L, Li W, Yang Y, Yuan J. MicroRNA in human glioma. Cancers. 2013;5:1306–1331. doi: 10.3390/cancers5041306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Mizoguchi M, Guan Y, Yoshimoto K, Hata N, Amano T, Nakamizo A, Sasaki T. Clinical implications of microRNAs in human glioblastoma. Front Oncol. 2013;3:19. doi: 10.3389/fonc.2013.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]