

Abstract
Transforming growth factor–β (TGF-β) functions to suppress tumorigenesis in normal mammary tissues and early-stage breast cancers and, paradoxically, acts to promote the metastasis and chemoresistance in late-stage breast cancers, particularly triple-negative breast cancers (TNBCs). Precisely how TGF-β acquires oncogenic characteristics in late-stage breast cancers remains unknown, as does the role of the endogenous mammalian target of rapamycin (mTOR) inhibitor, Dep domain–containing mTOR-interacting protein (Deptor), in coupling TGF-β to TNBC development and metastatic progression. Here we demonstrate that Deptor expression was downregulated in basal-like/TNBCs relative to their luminal counterparts. Additionally, Deptor expression was 1) inversely correlated with the metastatic ability of human (MCF10A) and mouse (4T1) TNBC progression series and 2) robustly repressed by several inducers of epithelial-mesenchymal transition programs. Functional disruption of Deptor expression in 4T07 cells significantly inhibited their proliferation and organoid growth in vitro, as well as prevented their colonization and tumor formation in the lungs of mice. In stark contrast, elevated Deptor expression was significantly associated with poorer overall survival of patients harboring estrogen receptor α–negative breast cancers. Accordingly, enforced Deptor expression in MDA-MB-231 cells dramatically enhanced their 1) organoid growth in vitro, 2) pulmonary outgrowth in mice, and 3) resistance to chemotherapies, an event dependent on the coupling of Deptor to survivin expression. Collectively, our findings highlight the dichotomous functions of Deptor in modulating the proliferation and survival of TNBCs during metastasis; they also implicate Deptor and its stimulation of survivin as essential components of TNBC resistance to chemotherapies and apoptotic stimuli.
Abbreviations: Deptor, Dep domain–containing mTOR-interacting protein; EMT, epithelial-mesenchymal transition; ER, estrogen receptor; MEC, mammary epithelial cell; mTOR, mammalian target of rapamycin; NMuMG, normal murine mammary gland cell; PyMT, polyoma middle T antigen; S6K, S6 kinase; TβR-I, TGF-β type I receptor; TGF-β, transforming growth factor–β; TNBC, triple-negative breast cancer
Introduction
Transforming growth factor–β (TGF-β) is a multifunctional cytokine that governs essentially all aspects of mammary epithelial cell (MEC) biology, which coalesce in the proper maintenance of mammary gland homeostasis and structural integrity [1], [2]. Importantly, this pleiotropic cytokine exhibits bifurcating activities that manifest in its suppression of mammary tumorigenesis in normal MECs and early-stage breast cancers and, conversely, in the induction of disease progression and metastasis in their late-stage counterparts [3], [4], [5]. This functional metamorphosis is known as the “TGF-β Paradox,” whose molecular underpinnings remain mysterious and a major barrier toward effectively targeting the TGF-β pathway as a means to alleviate breast cancer development and metastatic progression.
TGF-β governs the behaviors of normal and malignant MECs by engaging the receptor Ser/Thr protein kinases, TGF-β type I (TβR-I) and type II receptors, which coalesce in phosphorylating and activating the latent transcription factors, Smad2 and Smad3, leading to their nuclear accumulation with Smad4 and global alterations in gene expression [5], [6], [7]. Collectively, TGF-β activities that are dependent on the functions of Smad2/3 are referred to as canonical TGF-β signaling, which is complemented by its ability to activate a variety of noncanonical signaling systems that are independent of Smad2/3. Included in this list of noncanonical TGF-β effectors are small GTPases (e.g., RhoA, Rac1, and Cdc42), Mitogen-activated protein (MAP) kinases (e.g., Extracellular signal-regulated kinases [ERKs], c-Jun N-terminal kinases [JNKs], and p38 Mitogen-activated protein kinase [MAPK]), Phosphoinositide 3-kinase (PI3K):AKT:mammalian target of rapamycin (mTOR), Nuclear factor-kappa B (NF-κB):Cox-2, and integrins and adhesion complexes [6], [8]. Importantly, imbalances between canonical and noncanonical TGF-β signaling are associated with its acquisition of oncogenic and metastatic activities [3], [5]. Canonical and noncanonical TGF-β signaling inputs also coalesce in stimulating epithelial-mesenchymal transition (EMT), which endows polarized epithelial cells with mesenchymal and invasive phenotypes [5], [9], [10] and with stem cell–like properties [11], [12], [13]. The initiation of EMT programs in breast carcinoma cells contributes to their metastasis, as well as to their resistance to traditional chemotherapies, leading to incurable disease relapse [14], [15]. Thus, deciphering the molecular mechanisms and functions of EMT programs driven by TGF-β may provide new insights and therapies capable of improving overall survival rates in patients with metastatic disease.
Recently, Dep domain–containing mTOR-interacting protein (Deptor) has emerged as an important player during the development and progression of human malignancies [16]. Functionally, Deptor interacts physically with mTOR and is a natural antagonist of mTOR complex (mTORCs) 1 and 2 [17], and as such, aberrant Deptor expression promotes the activation of AKT, the induction of autophagy, and the acquisition of chemoresistance [18], [19]. Additionally, upregulated Deptor expression associates with lymph node and metastasis status [20] and predicts for poor overall survival of cancer patients [20], [21]. Interestingly, although elevated expression of Deptor coincides with the appearance of metastatic phenotypes, this same event is sufficient to inhibit the expression of Snail and its stimulation of EMT programs [22], suggesting that Deptor mediates both metastasis-suppressing and metastasis-promoting activities in developing carcinomas, including those of the breast. Given the parallels between the dichotomous functions of Deptor and TGF-β in developing mammary tumors, our objective was to determine whether TGF-β coupled to Deptor expression during mammary tumorigenesis, and if so, how this event transpired and impacted the acquisition of metastatic and chemoresistant phenotypes of breast cancers. Herein we demonstrate dual functions for Deptor during the metastatic cascade, such that 1) downregulated Deptor expression in primary tumors is essential for the acquisition of EMT and invasive phenotypes coupled to cell cycle arrest and 2) upregulated Deptor expression in metastatic lesions is essential for the initiation of anti-apoptotic and chemoresistant activities dependent on the induction of survivin expression.
Materials and Methods
Cell Lines and Reagents
Normal murine mammary gland cells (NMuMG), murine 4T1 cells, and human MCF7, MCF10A, MDA-MB-231, and MDA-MB-468 cells were obtained from American Type Culture Collection (ATCC) (Manassas, VA), while murine 67NR and 4T07 and human MCF10Ca1h and MCF10Ca1a were all obtained from Dr Fred Miller (Wayne State University, Detroit, MI). We previously described the expression of polyoma middle T antigen (PyMT) or human β3 integrin in NMuMG cells [23], [24], while those engineered to express Twist were subjected to Vesicular stomatitis virus glycoprotein (VSVG) retroviral transduction of pBabe (Addgene, Cambridge, MA) and subsequent selection with puromycin (5 μg/ml) as described previously [25]. Human MDA-MB-468 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% FBS, while all additional cell lines were cultured as described previously [26], [27], [28]. 4T07 and MDA-MB-231 cells were engineered to stably express firefly luciferase by transfection with pNifty-CMV-luciferase and selection with Zeocin (500 μg/ml; Invitrogen, Carlsbad, CA). Deptor-deficient cells were produced by pLKO.1-puro lentiviral transduction using a scrambled shRNA (i.e., nonsilencing shRNA) or two independent and verified Deptor-specific shRNAs, all of which were obtained from Open Biosystems (Lafayette, CO). Alternatively, Deptor overexpression was undertaken by subcloning the human Deptor cDNA (Addgene) cloned into the Gateway G418 destination vector (Life Technologies, Carlsbad, CA) using the BamHI and XhoI restriction sites as follows: 1) forward 5′-CCGGCCGGATCCATGGACTACAAGGATGAC and 2) reverse 5′-CCGGCCCTCGAGTCAGCACTCTAACTCCTC. The selection of stable polyclonal populations of Deptor-deficient NMuMG, 4T07, MCF-7, or MDA-MB-231 cells was accomplished by continuous culture of over a span of 14 days in puromycin (5 μg/ml), while Deptor-expressing MDA-MB-231 cells were isolated following 14 days of continuous culture in G418 (500 μg/ml; EMD Millipore, Billerica, MA). In all cases, the extent of Deptor deficiency or overexpression was determined by immunoblot analysis for Deptor as described below.
Immunoblot Analyses
Immunoblot analyses were performed as previously described [29]. Briefly, parental and Deptor-manipulated murine and human breast cancer cells were seeded into six-well plates (500,000 cells per well) and allowed to adhere overnight. Cells were subsequently incubated in the absence or presence of the pharmacological inhibitors described in Supplementary Table 1. Afterward, detergent-solubilized whole-cell extracts were prepared by lysing the cells in Buffer H/1% Triton X-100 (50 mM β-glycerophosphate, 1.5 mM EGTA, 1 mM DTT, 0.2 mM sodium orthovanadate, 1 mM benzamidine, 10 μg/ml leupeptin, and 10 μg/ml aprotinin, pH 7.3), and clarified whole-cell extracts (30 μg/lane) were fractionated through 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels, transferred electrophoretically to nitrocellulose, and immunoblotted with the antibodies described in Supplementary Table 2.
Semiquantitative Real-Time Polymerase Chain Reaction Analyses
Real-time polymerase chain reaction (PCR) studies were performed as described previously [26], [29]. Briefly, cells (500,000 cells per well) were seeded overnight onto six-well plates and subsequently stimulated with TGF-β1 (5 ng/ml) for 24 hours. Where indicated, cells were treated with paclitaxel (Sigma, St. Louis, MO; 50 nM) and doxorubicin (Sigma; 50 nM). Afterward, total RNA was isolated using the RNeasy Plus Kit (Qiagen, Germantown, MD) and reverse transcribed using the iScript cDNA Synthesis System (Bio-Rad, Hercules, CA). Semiquantitative real-time PCR was conducted using iQ-SYBR Green (Bio-Rad) according to the manufacturer’s recommendations. In all cases, differences in RNA concentration for individual genes were normalized to their corresponding Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) RNA signals. The oligonucleotide primer pairs used are provided in Supplementary Table 3.
Apoptosis Assay
NMuMG and MDA-MB-231 derivatives were seeded in serum-free media onto 96-well plates (10,000 cells per well). NMuMG cells were allowed to adhere overnight, while MDA-MB-231 cells were cultured over poly-HEMA–coated plates. Seventy-two hours later, the extent of caspase-3/7 activity was quantified using the Caspase-Glo 3/7 luminescence assay system according to the manufacturer’s recommendations (Promega, Madison, WI).
Invasion Assay
The ability of 4T07 cells (50,000 cells per well) to invade reconstituted basement membranes was measured using modified Boyden chambers as previously described [26] and using 2% serum as a chemoattractant.
Three-Dimensional Organotypic Cultures
Three-dimensional (3D) organotypic cultures using the “on-top” method were performed as described [29]. Briefly, cells (2000 cells per well) were cultured in 96-well plates onto Cultrex cushions (50 μl/well; Trevigen, Gaithersburg, MD) in complete media supplemented with 5% Cultrex. Organoid growth was monitored by bright-field microscopy [30] or by longitudinal bioluminescence growth assays as described previously [31].
Anoikis Assay
Anoikis assays were performed as described [32]. Briefly, control (MT) and Deptor-expressing MDA-MB-231 or 4T07 cells were cultured over poly-HEMA–coated plates in serum-free medium. The cells were collected 72 hours after plating, at which point they were prepared for survivin immunoblot analyses.
Annexin V Staining
Paclitaxel (50 nM) and doxorubicin (50 nM) treated MDA-MB-231 derivative cells were trypsinized and resuspended in binding buffer (1 × 106 cells/ml). Subsequently, 100 μl of each treatment group was stained with Annexin V–fluorescein isothiocyanate (BD Biosciences, San Jose, CA) for 15 minutes at room temperature, at which point 400 μl of binding buffer was added to the stained cells before analyzing Annexin V positivity by flow cytometry.
Tumor Growth and Bioluminescence Imaging
Luciferase-expressing parental (scram) or Deptor-deficient 4T07 cells (50,000 cells per mouse) or parental (MT) or Deptor-overexpressing MDA-MB-231 cells (1.5 × 106 cells per mouse; n = 5) were injected into the lateral tail vein of female BALB/c or nude mice, respectively. Afterward, pulmonary outgrowth was monitored and quantified using intravital bioluminescence imaging as described [33]. All animal studies were performed in accordance with the Institutional Animal Care and Use Committee for Case Western Reserve University.
Results
Deptor Expression Is Decreased in Aggressive Breast Cancers
Although elevated mTOR activity has been reported in breast cancers [34], [35], the role that Deptor plays in mediating this event during mammary tumorigenesis remains to be fully elucidated. As such, we performed Oncomine microarray expression analyses and found Deptor expression to be significantly downregulated in breast cancers as compared to normal breast tissue (Figure 1A). Accordingly, Deptor expression was reduced in metastatic 4T1 cells relative to their indolent 67NR and dormant 4T07 counterparts (Figure 1B). Next, we addressed whether this trend in Deptor expression also occurred in human breast cancer cell lines. As shown in Figure 1C, Deptor expression was noticeably absent in low-grade human MCF10aCa1h and high-grade human MCF10aCa1a cells relative to their indolent human MCF10aT1K counterparts. Likewise, Deptor expression was also dramatically reduced in metastatic MDA-MB-231 cells as compared to nonmetastatic MCF7 cells (Figure 1D). Collectively, these findings suggest that diminished Deptor expression is characteristic of invasive breast cancers.
Figure 1.
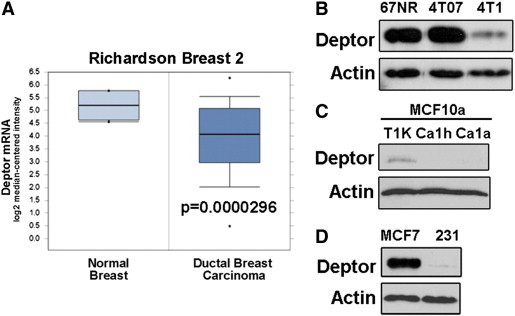
Deptor expression is decreased in aggressive breast cancers. (A) Oncomine data examining Deptor expression in normal and malignant cancer tissues using Richardson Breast 2 data set. (B) Deptor expression is dramatically reduced in metastatic 4T1 cell lines as compared with their nonmetastatic 67NR and 4T07 counterparts. (C) Deptor expression is dramatically reduced in malignant MCF10Ca1h and MCF10Ca1a cells as compared to their weakly tumorigenic MCF10aT1K counterpart. (D) Deptor expression is noticeably absent in basal-like/TNBC MDA-MB-231 cells as compared to luminal MCF7 cells. Immunoblots in B to D are representative of three independent experiments.
Deptor Expression Is Regulated by Estrogen Receptor α
Given the differences in Deptor expression that exist between the estrogen receptor α (ERα)–positive MCF7 cells and ERα-negative MDA-MB-231 cells (Figure 1D), we surveyed Deptor mRNA expression in additional breast cancer cell lines categorized as being positive (e.g., luminal subtypes) or negative (e.g., basal-like/triple-negative subtypes) for expression of ERα. As shown in Figure 2A, Deptor expression was significantly decreased in ERα-negative breast cancers (e.g., basal-like/triple-negative) relative to those classified as ERα-positive (e.g., luminal). Indeed, upon examination of the Deptor promoter sequence, we discovered two ER-binding elements (data not shown), suggesting that the expression of Deptor mRNA may be regulated by ERα. Accordingly, Oncomine analyses showed that Deptor expression is indeed elevated and associated with that of ERα (Figure 2B). Moreover, treating MCF7 cells with the ERα antagonist tamoxifen or fulvestrant (ICI) decreased Deptor expression (Figure 2C), while administration of the ERα agonist estradiol significantly increased Deptor expression in MCF-7 cells (Figure 2D). Finally, in considering the role of TGF-β in driving breast cancer pathogenesis [4], [5], [6], we monitored Deptor expression in MCF7 cells before and after their stimulation with TGF-β. As shown in Figure 2E, TGF-β treatment of MCF7 cells failed to affect their expression of either ERα or Deptor. Collectively, these findings suggest that basal-like/triple-negative breast cancers (TNBCs) express significantly decreased levels of Deptor that may reflect their loss of ERα expression.
Figure 2.
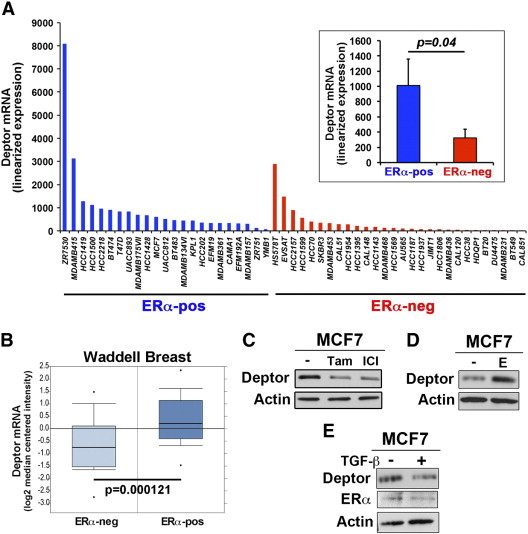
Deptor expression is regulated by ERα. (A) Deptor mRNA expression is significantly lower in ERα-negative (ERα-neg) breast cancer subtypes as compared to ERα-positive (ERα-pos) breast cancer subtypes as determined by the Broad-Novartis Cancer Cell Line Encyclopedia. Inset shows mean (± SEM) of Deptor mRNA expression in ERα-negative and ERα-positive breast cancer cells. (B) Oncomine analyses associated elevated Deptor mRNA expression levels with that of ERα-positive breast cancers using the Waddell Breast data set. (C) MCF7 cells were treated with tamoxifen (0.1 nM) or fulvestrant (ICI, 0.1 nM) for 4 days, at which point the cells were harvested and probed for Deptor expression by immunoblot analysis. MCF7 cells were treated with either estradiol (10 nM) or TGF-β1 (5 ng/ml) for 4 days before monitoring Deptor or ERα expression by immunoblot analysis. Immunoblots in C to E are representative of three independent experiments.
TGF-β Downregulates Deptor Expression through a Smad3-Dependent Pathway
Previous studies observed ERα expression to be downregulated in response to TGF-β [36]. Accordingly, we too observed TGF-β to downregulate ERα expression in normal murine NMuMG MECs (Figure 3A), which occurs concomitantly with a dramatic reduction of Deptor mRNA (Figure 3B) and protein (Figure 3C). Since the pathophysiology of TGF-β reflects its activation of both canonical and noncanonical effector molecules [6], [8], we next sought to elucidate which branch of the TGF-β signaling system elicits diminished Deptor expression in normal and malignant MECs. To do so, we used pharmacological inhibitors against Smad3 (SIS3; [37]) and p38 MAPK (SB203580; [38]) in conjunction with TGF-β administration. In doing so, we observed SIS3 to antagonize the ability of TGF-β to downregulate Deptor expression in nontumorigenic NMuMG cells (Figure 3D). Interestingly, the coupling of TGF-β to downregulated Deptor expression was prevented by antagonism of both p38 MAPK and Smad3 in the 4T1 cells (Figure 3E), suggesting that both branches of the TGF-β signaling system drive the loss of Deptor expression in ERα-negative breast cancer models. We also examined the coupling of TGF-β to Deptor expression in human MDA-MB-468 cells, which lack expression of both ERα and Smad4. Interestingly, Deptor expression was unaffected in TGF-β–treated MDA-MB-468 cells (Figure 3F), confirming the requirement of Smad-dependent signals in downregulating Deptor expression. Finally, we inhibited TGF-β signaling by treating 4T1 cells with two distinct TGF-β antagonists: 1) TβR-I type II inhibitor, which specifically inhibits Alk5 [39], and 2) SB431542, which specifically inhibits Alk4, Alk5, and Alk7 [40]. Importantly, administration of either TβR-I inhibitor induced Deptor expression in 4T1 cells (Figure 3G), suggesting that autocrine TGF-β signaling serves in driving constitutive down-regulation of Deptor expression in basal-like/TNBCs. Collectively, these findings demonstrate that the ability of TGF-β to downregulate Deptor expression in normal and malignant cells is context-specific and dependent on loss of ERα expression.
Figure 3.
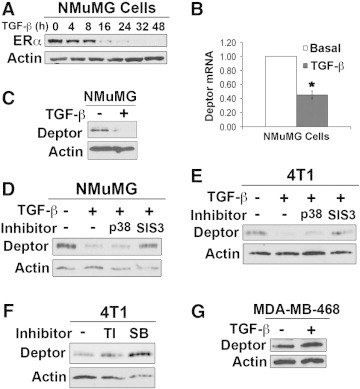
TGF-β downregulates Deptor expression through a Smad3-dependent pathway. (A) NMuMG cells were treated with TGF-β1 (5 ng/ml) as indicated and subsequently subjected to anti-ERα immunoblot analysis. Immunoblots are representative of three independent experiments. TGF-β1 (5 ng/ml for 48 hours) downregulates the expression of Deptor mRNA (B) and protein (C) in NMuMG cells. Data are mean (± SE; *P < .007). NMuMG (D) and 4T1 (E) cells were stimulated with TGF-β1 for 48 hours in the absence or presence of the p38 MAPK inhibitor, SB203580 (p38; 10 μM), or the Smad3 inhibitor, SIS3 (10 μM). Data are representative of three independent experiments. (F) 4T1 cells were cultured in the absence or presence of the TβR-I antagonist TβR-I type II inhibitor (10 μg/ml) or SB431452 (SB; 10 μM). Immunoblots are representative of three independent experiments. (G) MDA-MB-468 cells were stimulated with TGF-β1 (5 ng/ml) for 4 days. Immunoblots are representative of three independent experiments.
Loss of Deptor Expression Is Associated with EMT Programs
The loss of ERα expression or function elicits EMT programs in MECs [41], [42], as does activation of the TGF-β signaling system [5], [9], [10]. As such, we monitored the extent to which Deptor expression is downregulated in NMuMG cells stimulated to undergo EMT programs by TGF-β or by overexpression of either PyMT, Twist, or β3 integrin [23], [24], [43]. As expected, TGF-β stimulated NMuMG cells to acquire EMT phenotypes (Figure 4A), as well as downregulated Deptor expression (Figure 4B). Additionally, transgenic expression of PyMT (Figure 4, A and B), Twist (Figure 4, C–E), or β3 integrin (Figure 4, F and G) also elicited EMT programs and downregulated expression of Deptor in NMuMG cells, events that were greatly enhanced by administration of TGF-β. Thus, these findings implicate diminished Deptor expression as a component of EMT programs in MECs. However, these analyses fail to address whether Deptor deficiency is sufficient to elicit EMT reactions in breast cancer cells. To answer this question, we rendered 4T07 and MCF7 cells deficient in Deptor expression using two independent murine and human shRNAs against Deptor mRNA. As expected, Deptor deficiency elicited elevated activation of S6 kinase (S6K) in 4T07 (Supplementary Figure 1A) and MCF7 (Supplementary Figure 1D) cells, as well as produced an inconsistent and less robust stimulation of AKT (data not shown). Moreover, Deptor deficiency was sufficient to promote partial EMT-like phenotypes in both 4T07 and MCF7 cells (Supplementary Figure 1, B and E), particularly with respect to down-regulation of the epithelial markers, E-cadherin and CK19 (Supplementary Figure 1, C and E). Interestingly, Deptor inactivation failed to impact the expression of the mesenchymal markers, β3 integrin and vascular endothelial growth factor (data not shown), suggesting a preferential role of Deptor in regulating epithelial gene expression patterns during EMT. Finally, we determined whether Deptor down-regulation during EMT reactions reflects the inherent stress experienced by cells as they traverse this transdifferentiation process [44]. As shown in Supplementary Figure 2A, Deptor expression was unaffected in NMuMG cells cultured under several stress conditions, including growth factor deprivation or administration of either 5-fluorouracil or hydrogen peroxide. Along these lines, Deptor deficiency failed to impact the invasiveness of 4T07 cells (Supplementary Figure 2B), nor did this same cellular event impact the activation status of S6K and AKT in NMuMG cells (Supplementary Figure 2C), or their survival upon growth factor deprivation (Supplementary Figure 2D). Collectively, these findings demonstrate that EMT programs repress Deptor expression, an event that is sufficient to elicit partial EMT phenotypes and altered epithelial gene expression patterns that are uncoupled from motility and programmed cell death pathways in normal and malignant MECs.
Figure 4.
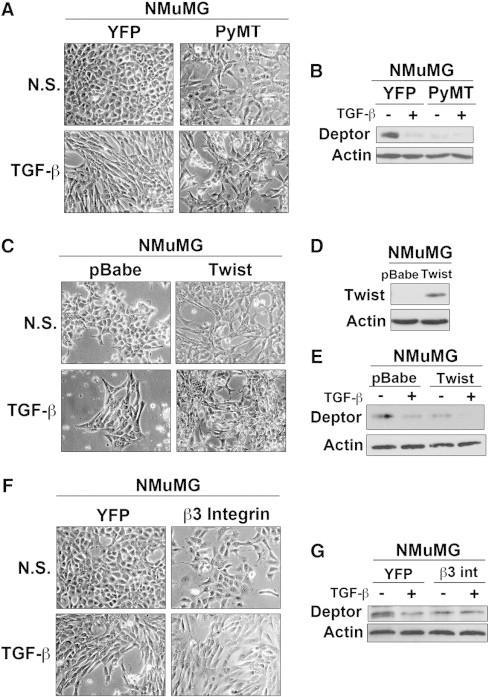
Loss of Deptor expression is associated with EMT programs. Deptor expression was evaluated in different models of EMT induction: (A and B) PyMT (n = 3), (C–E) Twist (n = 3), and (F and G) β3 integrin (n = 2). In all cases, initiation of EMT programs was sufficient to downregulate Deptor expression. Photomicrographs are representative images obtained using a 10 × objective.
Deptor Deficiency Inhibits Breast Cancer Growth Both In Vitro and In Vivo
In addition to exhibiting alterations in cell morphology and motility, cells acquiring EMT phenotypes also display reduced capacities to proliferate [4]. As such, we examined the role of Deptor in regulating cell growth and proliferation. As shown in Figure 5, A and B, Deptor deficiency significantly inhibited DNA synthesis in NMuMG and MCF7 cells, as well as in 4T07 cells propagated in 2D (data not shown) and 3D cultures (Figure 5C). To examine the role of Deptor in vivo, we inoculated parental (scram) and Deptor-deficient 4T07 cells into the lateral tail vein of BALB/c mice. In accordance with our in vitro results, Deptor-deficient 4T07 cells grew less efficiently in the lungs of mice as compared to their parental (scram) counterparts (Figure 5D). Indeed, large tumor nodules were readily apparent by gross anatomy (Figure 5E) and Hematoxylin & Eosin (H&E) staining (Figure 5F) of the lungs of mice inoculated with parental (scram) 4T07 cells, an event that was noticeably absent in the lungs of mice inoculated with Deptor-deficient 4T07 cells. Ex vivo culturing of these 4T07 derivatives demonstrated that Deptor deficiency was retained in the shDeptor group, whereas the parental (scram) group exhibited robust Deptor expression (Figure 5G). Taken together, these findings suggest that Deptor down-regulation elicits diminished proliferative capacity associated with EMT programs in normal and malignant MECs, thereby inhibiting the metastatic outgrowth of breast cancer cells in preclinical settings.
Figure 5.
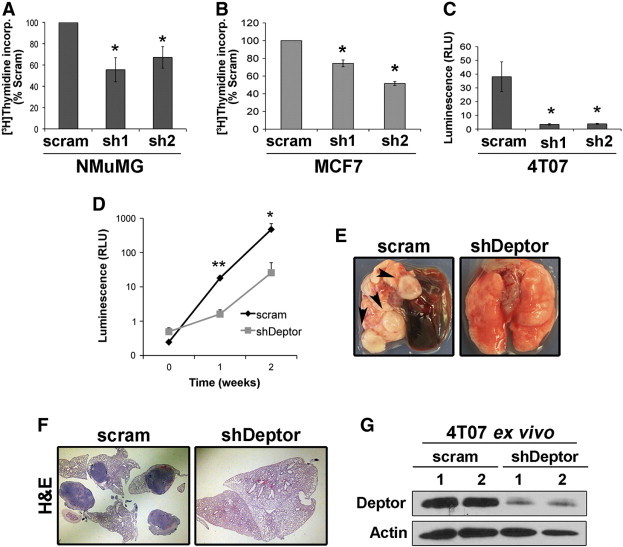
Deptor deficiency inhibits breast cancer growth both in vitro and in vivo. Deptor deficiency (sh1 and sh2) inhibits DNA synthesis in NMuMG (A), MCF7 (B), and 4T07 (C) cells as determined by [3H]thymidine incorporation. Data are mean (± SEM; n = 3; *P < .05), except for C (n = 2). (D) Parental (scram) and Deptor-deficient (shDeptor) 4T07 cells were inoculated into the lateral tail vein of syngeneic BALB/c mice, and pulmonary tumor growth was monitored by longitudinal bioluminescence imaging as indicated. Data are mean (± SEM; n = 5; *P < .0005, **P < .03). Lungs were isolated at the time of sacrifice to visualize surface (E, arrowheads) and embedded metastases by H&E staining (F). (G) Lungs were dissociated into single cells and cultured ex vivo to monitor Deptor expression by immunoblot analysis, which demonstrated maintenance of Deptor deficiency in pulmonary-derived 4T07 cells.
Deptor Expression Is Necessary for Pulmonary Outgrowth of Metastatic Breast Cancers
The aforementioned findings led us to hypothesize that Deptor down-regulation is associated with EMT and the egress of breast cancer cells out of the primary tumor and that the re-expression of Deptor at metastatic lesions facilitates their efficient outgrowth. As an initial test of this intriguing hypothesis, we discovered that high levels of Deptor expression correlated with poor overall survival in patients harboring ERα-negative breast cancers (Figure 6A). Interestingly, engineering MDA-MB-231 cells to overexpress Deptor resulted in a dramatic decrease in AKT activation concomitant with increased activation of S6K (Figure 6B). As compared to their parental counterparts, Deptor-expressing MDA-MB-231 cells exhibited significantly greater outgrowth in 3D organotypic cultures (Figure 6C) and in the lungs of nude mice inoculated intravenously with these TNBCs as determined by bioluminescence imaging (Figure 6D), by gross anatomy (Figure 6E, top), and by H&E staining (Figure 6E, bottom). As shown in Figure 6F, engineered expression of Deptor was retained and readily apparent in pulmonary metastases derived from Deptor-expressing MDA-MB-231 cells, supporting the conclusion that Deptor expression is necessary for and drives the metastatic outgrowth of ERα-negative breast cancer cells both in vitro and in vivo.
Figure 6.
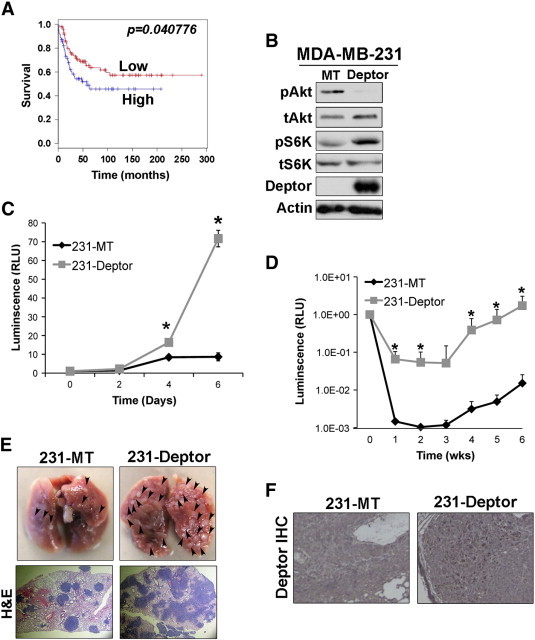
Deptor expression is necessary for pulmonary outgrowth of metastatic breast cancers. (A) ERα-negative breast cancer patient survival is significantly longer if Deptor expression is low as compared to tumors housing high Deptor expression. (B) Monitoring changes in AKT and S6K expression and activation in MDA-MB-231 cells engineered to overexpress Deptor. (C) Parental (MT) or Deptor-expressing MDA-MB-231 cells were propagated in 3D organotypic cultures for 6 days, during which time organoid growth was monitored longitudinally by bioluminescence on the indicated days. Data are the mean (± SE; n = 3; *P < .03). (D) Parental (MT) or Deptor-expressing MDA-MB-231 cells were injected into the lateral tail vein of nude mice. Pulmonary tumor growth was monitored by longitudinal bioluminescence imaging at the indicated times (n = 5; *P < .05). (E) Gross anatomy and H&E staining of lungs bearing parental (MT) or Deptor-expressing MDA-MB-231 tumors. (F) Immunohistochemical staining of Deptor in parental (MT) and Deptor-expressing MDA-MB-231 pulmonary lesions.
Deptor Promotes Breast Cancer Survival through Coupling to Survivin Expression
It should be noted that parental MDA-MB-231 cells exhibit a rapid and sharp decline in their bioluminescent signals when inoculated into the lateral tail veins of mice, an event that was significantly reduced in their Deptor-expressing counterparts (Figure 6D), suggesting that one mechanism whereby elevated Deptor expression may enhance pulmonary outgrowth is by preventing apoptosis. In support of this hypothesis, the magnitude of anoikis and caspase-3/7 activation exhibited by nonadherent MDA-MB-231 cells was significantly reduced by Deptor overexpression (Figure 7A), which also resulted in the robust expression of survivin both in vitro (Figure 7B) and in vivo (Figure 7C). Importantly, the linkages between Deptor and elevated survivin expression were also observed in murine 4T07 cells engineered to stably overexpress Deptor (Figure 7D). We also determined the extent to which elevated Deptor expression contributes to chemotherapy resistance. In doing so, Oncomine analysis demonstrated that elevated Deptor expression is associated with paclitaxel resistance (Figure 7E). Unlike their parental counterparts, Deptor-expressing MDA-MB-231 cells failed to activate caspase3/7 (Figure 7F) and dramatically upregulated their expression of survivin (Figure 7G) in response to paclitaxel administration. Finally, to determine whether elevated survivin expression mediated Deptor-induced chemoresistance, we rendered Deptor-expressing MDA-MB-231 cells deficient in survivin expression (Figure 7H) and subsequently monitored their sensitivity to paclitaxel and doxorubicin. As shown in Figure 7I, inactivation of survivin expression inactivated the pro-survival function of Deptor and, consequently, sensitized MDA-MB-231 cells to the anticancer activities of paclitaxel and doxorubicin as measured by significantly elevated Annexin V staining. Collectively, these findings demonstrate that the ability of Deptor to promote TNBC survival is mediated through upregulated expression of survivin, which contributes to the inherent chemoresistance of TNBCs.
Figure 7.
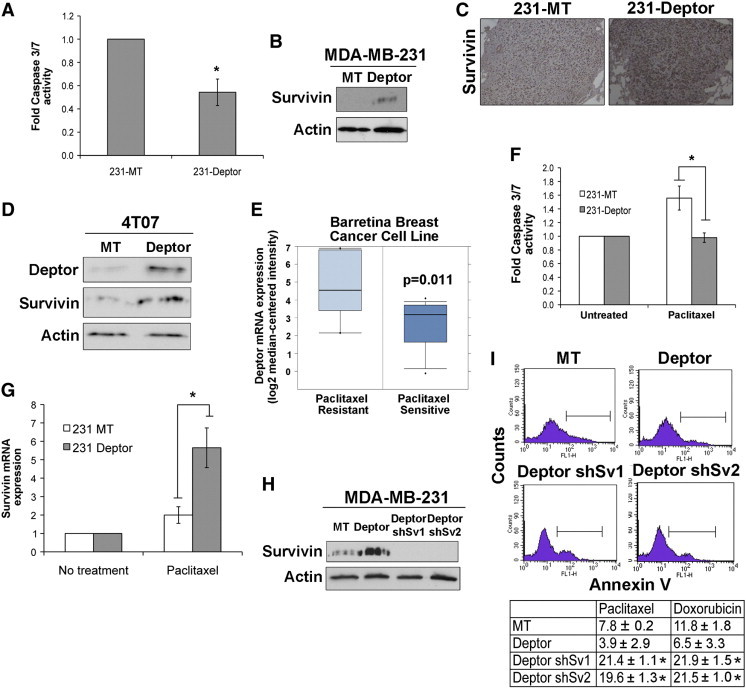
Deptor promotes breast cancer survival through coupling to survivin expression. (A) Caspase-Glo 3/7 analyses were performed to monitor the extent of anoikis and caspase-3/7 activation in control (MT) and Deptor-expressing MDA-MB-231 cells. Data are the mean (± SE; n = 3; *P < .02). (B) Deptor expression induces that of survivin in MDA-MB-231 cells cultured over poly-HEMA–coated plates. Immunoblots are representative of three independent experiments. (C) Immunohistochemical staining of survivin in pulmonary lesions produced by parental (MT) and Deptor-expressing MDA-MD-231 cells inoculated into mice used in Figure 6. (D) Deptor expression induces that of survivin in 4T07 cells cultured over poly-HEMA–coated plates. Immunoblots are representative of two independent experiments. (E) Oncomine data correlating Deptor expression to paclitaxel responsiveness using Barretina Breast Cancer Cell Line. (F) Caspase-Glo 3/7 analyses of paclitaxel-treated (50 nM) control (MT) and Deptor-expressing MDA-MB-231 cells. Data are the mean (± SEM; n = 2; *P < .002). (G) Paclitaxel treatment significantly induces survivin mRNA expression in Deptor-expressing MDA-MB-231 cells as compared to their parental counterparts. Data are the mean (± SEM; n = 4; *P < .04). (H) MDA-MB-231 cells were depleted of survivin expression using two independent lentiviral shRNAs against survivin (shSv1 and shSv2). (I) Annexin V–fluorescein isothiocyanate staining of parental (MT), Deptor-expressing (Deptor), or Deptor-expressing/survivin-deficient (Deptor shSv1/2) MDA-MB-231 cells treated with either paclitaxel (50 nM) or doxorubicin (50 nM) as indicated. The top panels show representative histograms obtained in two independent experiments, while the bottom panel shows the mean (± SEM; n = 2; *P < .05).
Discussion
The role of mTOR in human breast cancers has been well characterized, leading to the development and implementation of mTOR inhibitors as potential targeted therapies against TNBCs [45]. Despite the intense interest in deploying mTOR inhibitors in the treatment of human mammary tumors, comparatively little information exists related to the precise role played by the endogenous mTOR inhibitor, Deptor, during breast cancer development and metastatic progression. Here, we aimed to determine how Deptor expression was regulated by TGF-β and how these events coalesced to affect breast cancer metastasis, particularly in TNBCs. In doing so, we observed Deptor expression to be governed by two major signaling systems in MECs: TGF-β, which repressed Deptor expression, and ERα, which induced Deptor expression. With respect to the former pathway, we demonstrated the ability of Smad3 to suppress Deptor expression in normal and malignant MECs (Figure 3), presumably through TGF-β inhibitor elements [46]. Although future studies need to determine the extent to which the loss of Deptor expression is mediated either directly by Smad3 or indirectly by a distinct Smad3-inducible gene, we nevertheless established the necessity for noncanonical TGF-β signaling and its coupling to p38 MAPK in maximally suppressing Deptor expression specifically in late-stage breast cancers, not their normal counterparts. Interestingly, the fundamental rewiring of Deptor expression in normal and malignant MECs treated with TGF-β resembles similar disturbances between the canonical and noncanonical TGF-β pathways during mammary tumorigenesis [5], [6], thereby implicating Deptor as a dichotomous regulatory molecule during the development of metastatic disease.
With respect to ERα and its regulation of Deptor expression, we observed the presence of two estrogen response elements (EREs) within the Deptor promoter (data not shown), suggesting that ERα induces Deptor expression at the level of transcription. Accordingly, treating MCF7 cells with estradiol significantly elevated Deptor expression, while pharmacological inactivation of ERα expression or activity significantly depressed Deptor expression (Figure 2). Interestingly, ERα has been documented to antagonize TGF-β signaling, doing so by interacting physically with Smad3 and inhibiting its activation in response to TGF-β [47]. These findings point toward a novel and dynamic post-transcriptional mechanism that enables ERα to modulate the downregulated expression of Deptor induced by TGF-β:Smad3 in ERα-positive tumors, an event that is lacking in their ERα-negative counterparts and likely contributes to their disease progression stimulated by TGF-β. Future studies need to better define post-transcriptional mechanisms coupled to Deptor expression across distinct breast cancer subtypes, particularly distinct subtypes of TNBCs and their varied sensitivities to chemotherapies [48], [49].
Deptor was originally identified as an endogenous inhibitor of mTORCs 1 and 2, whose constitutive activation in Deptor-deficient cells elicits the robust activation of S6K, AKT, and Serum and glucocorticoid regulated-kinase 1 (SGK1) necessary in driving carcinoma growth and survival. Interestingly, this same study also found Deptor overexpression to promote constitutive activation of PI3K and AKT, leading to enhanced cell survival [17]. In stark contrast, we observed Deptor deficiency to elicit a robust stimulation of S6K solely in malignant MECs; however, this same cellular condition produced little to no effect on the activation status of ATK, as did the dramatic overexpression of Deptor in human MDA-MB-231 cells. Although the reasons underlying these experimental discrepancies remain to be fully elucidated, it is tempting to speculate that mammary carcinomas lack expression of additional scaffolding molecules operant in coupling Deptor to AKT activation, an event that appears to transpire in a cell type–specific manner. More intriguingly, we found Deptor deficiency to strongly inhibit breast cancer cell proliferation (Figure 5), which contrasts sharply with the strong survival signals produced by Deptor overexpression (Figure 6). Indeed, these disparate activities of Deptor may reflect its coupling to mTOR-dependent and mTOR-independent pathways in developing and progressing mammary tumors. For instance, Deptor contains a PDZ domain [17] that can 1) homodimerize or heterodimerize with other PDZ domain–containing proteins [50], particularly those localized with junctional complexes, such as zona occludens-1 (ZO-1) [51], and 2) form lipid-protein complexes by binding lipid molecules, particularly phosphoinositides [52]. Therefore, we postulate that in addition to its roles in governing mTOR function and signaling, Deptor also serves in maintaining the junctional integrity and polarity of MECs (Figure 3 and Supplementary Figure 1) and in regulating lipid metabolism, both of which become dysregulated during mammary tumorigenesis [53], [54].
In accordance with the aforementioned reasoning, we (Figure 3 and Supplementary Figure 1) and others [55] have associated diminished Deptor expression with the acquisition of EMT phenotypes in malignant MECs (Figure 1). Likewise, Chen and colleagues [22] found Deptor deficiency to promote the activation of AKT and Snail expression in A549 lung carcinoma cells, leading to their stimulation of EMT programs. Remarkably, Deptor deficiency also functioned as a potential inhibitor of cell proliferation, particularly in post-EMT breast cancer cells. It should be noted that EMT programs promote the selection and expansion of breast cancer cells that exhibit characteristics of cancer stem cells [11], which are poorly proliferative as compared to non–cancer stem cells [56]. Collectively, these findings suggest that Deptor expression suppresses the development of EMT and cancer stem cells, a supposition consistent with the down-regulation of Deptor in luminal breast cancer stem cells [57].
Although the mechanisms that enable Deptor to suppress the development of cancer stem cells remains to be fully elucidated, similar uncertainty relates to precisely how late-stage breast cancers circumvent the loss of Deptor, particularly at sites of metastasis. For example, metastatic MDA-MB-231 and MCF10CA1a breast cancer cells harbor activating mutations in Ras [55] and PI3K [58], respectively, which may aid in offsetting Deptor deficiency. Likewise, restoration of Deptor expression enhances the growth of MDA-MB-231 cells (Figure 6), indicating that Deptor fulfills two distinct roles during mammary tumorigenesis. First, initiation of EMT programs alleviates Deptor expression, resulting in cell cycle arrest and in transitioning breast carcinoma cells as they disseminate to distant locales. Upon colonization of a metastatic site, we propose that low levels of Deptor expression contribute to metastatic dormancy, a phenotype that is overcome upon re-expression of Deptor and the initiation of mesenchymal-epithelial transitions necessary to complete the metastatic cascade.
Finally, the mechanism whereby Deptor induces survivin expression remains to be elucidated. A previous study observed TGF-β to downregulate survivin expression in response to Smad2/3-mediated hypophosphorylation of the tumor suppressor, Rb [59]; however, the extent to which Deptor expression modulates and/or inactivates these is currently unknown. Nevertheless, rendering Deptor-expressing MDA-MB-231 cells deficient in survivin expression clearly enhanced the sensitivity of these TNBCs to paclitaxel and doxorubicin (Figure 7). Indeed, the coupling of Deptor to survivin expression likely engenders chemoresistance in TNBCs in part through the ability of survivin to regulate apoptosis [60], [61], mitosis [62], and autophagy [60]. Future studies need to delineate how Deptor drives survivin expression in late-stage TNBCs, as well as to determine which survivin pathways elicit chemoresistance in recurrent disease settings. Collectively, our findings implicate Deptor as an essential player during TNBC development and metastatic progression and as a potential biomarker to predict for resistance to standard-of-care chemotherapies.
Acknowledgements
We thank members of the Schiemann laboratory for their helpful comments and suggestions. We also acknowledge the expertise provided by members of the Case Comprehensive Cancer Center’s Athymic Animal and Xenograft Core, the Small Animal Imaging Core, and the Tissue Resources Core.
Footnotes
Research support was provided in part by the National Institutes of Health to W.P.S. (CA129359 and CA177069).
This article refers to supplementary materials, which are designated by Supplementary Tables 1 to 3 and Supplementary Figures 1 and 2 and are available online at www.neoplasia.com.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.neo.2015.02.003.
Appendix A. Supplementary data
Supplementary material
References
- 1.Moses H, Barcellos-Hoff MH. TGF-β biology in mammary development and breast cancer. Cold Spring Harb Perspect Biol. 2011;3:a003277. doi: 10.1101/cshperspect.a003277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Band AM, Laiho M. Crosstalk of TGF-β and estrogen receptor signaling in breast cancer. J Mammary Gland Biol Neoplasia. 2011;16:109–115. doi: 10.1007/s10911-011-9203-7. [DOI] [PubMed] [Google Scholar]
- 3.Tian M, Schiemann WP. The TGF-β paradox in human cancer: An update. Future Oncol. 2009;5:259–271. doi: 10.2217/14796694.5.2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morrison CD, Parvani JG, Schiemann WP. The relevance of the TGF-β paradox to EMT-MET programs. Cancer Lett. 2013;341:30–40. doi: 10.1016/j.canlet.2013.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor MA, Parvani JG, Schiemann WP. The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-β in normal and malignant mammary epithelial cells. J Mammary Gland Biol Neoplasia. 2010;15:169–190. doi: 10.1007/s10911-010-9181-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parvani JG, Taylor MA, Schiemann WP. Noncanonical TGF-β signaling during mammary tumorigenesis. J Mammary Gland Biol Neoplasia. 2011;16:127–146. doi: 10.1007/s10911-011-9207-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saitoh M, Miyazawa K. Transcriptional and post-transcriptional regulation in TGF-β-mediated epithelial-mesenchymal transition. J Biochem. 2012;151:563–571. doi: 10.1093/jb/mvs040. [DOI] [PubMed] [Google Scholar]
- 8.Massague J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Katsuno Y, Lamouille S, Derynck R. TGF-β signaling and epithelial-mesenchymal transition in cancer progression. Curr Opin Oncol. 2013;25:76–84. doi: 10.1097/CCO.0b013e32835b6371. [DOI] [PubMed] [Google Scholar]
- 10.Heldin CH, Vanlandewijck M, Moustakas A. Regulation of EMT by TGFβ in cancer. FEBS Lett. 2012;586:1959–1970. doi: 10.1016/j.febslet.2012.02.037. [DOI] [PubMed] [Google Scholar]
- 11.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, Nikolskaya T, Serebryiskaya T, Beroukhim R, Hu M. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11:259–273. doi: 10.1016/j.ccr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 13.Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, Weinberg RA. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Creighton CJ, Chang JC, Rosen JM. Epithelial-mesenchymal transition (EMT) in tumor-initiating cells and its clinical implications in breast cancer. J Mammary Gland Biol Neoplasia. 2010;15:253–260. doi: 10.1007/s10911-010-9173-1. [DOI] [PubMed] [Google Scholar]
- 15.Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, Rimm DL, Wong H, Rodriguez A, Herschkowitz JI. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A. 2009;106:13820–13825. doi: 10.1073/pnas.0905718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Z, Zhong J, Inuzuka H, Gao D, Shaik S, Sarkar FH, Wei W. An evolving role for DEPTOR in tumor development and progression. Neoplasia. 2012;14:368–375. doi: 10.1593/neo.12542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, Sabatini DM. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137:873–886. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao Y, Xiong X, Sun Y. DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(βTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Mol Cell. 2011;44:304–316. doi: 10.1016/j.molcel.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luo Z, Yu G, Lee HW, Li L, Wang L, Yang D, Pan Y, Ding C, Qian J, Wu L. The Nedd8-activating enzyme inhibitor MLN4924 induces autophagy and apoptosis to suppress liver cancer cell growth. Cancer Res. 2012;72:3360–3371. doi: 10.1158/0008-5472.CAN-12-0388. [DOI] [PubMed] [Google Scholar]
- 20.Pei L, Xie P, Zhou E, Yang Q, Luo Y, Tang Z. Overexpression of DEP domain containing mTOR-interacting protein correlates with poor prognosis in differentiated thyroid carcinoma. Mol Med Rep. 2011;4:817–823. doi: 10.3892/mmr.2011.503. [DOI] [PubMed] [Google Scholar]
- 21.Carrasco DR, Tonon G, Huang Y, Zhang Y, Sinha R, Feng B, Stewart JP, Zhan F, Khatry D, Protopopova M. High-resolution genomic profiles define distinct clinico-pathogenetic subgroups of multiple myeloma patients. Cancer Cell. 2006;9:313–325. doi: 10.1016/j.ccr.2006.03.019. [DOI] [PubMed] [Google Scholar]
- 22.Chen R, Yang Q, Lee JD. BMK1 kinase suppresses epithelial-mesenchymal transition through the Akt/GSK3β signaling pathway. Cancer Res. 2012;72:1579–1587. doi: 10.1158/0008-5472.CAN-11-2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wendt MK, Smith JA, Schiemann WP. Transforming growth factor-β-induced epithelial-mesenchymal transition facilitates epidermal growth factor-dependent breast cancer progression. Oncogene. 2010;29:6485–6498. doi: 10.1038/onc.2010.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galliher AJ, Schiemann WP. β3 integrin and Src facilitate transforming growth factor-β mediated induction of epithelial-mesenchymal transition in mammary epithelial cells. Breast Cancer Res. 2006;8:R42. doi: 10.1186/bcr1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wendt MK, Taylor MA, Schiemann BJ, Schiemann WP. Down-regulation of epithelial cadherin is required to initiate metastatic outgrowth of breast cancer. Mol Biol Cell. 2011;22:2423–2435. doi: 10.1091/mbc.E11-04-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wendt MK, Schiemann WP. Therapeutic targeting of the focal adhesion complex prevents oncogenic TGF-β signaling and metastasis. Breast Cancer Res. 2009;11:R68. doi: 10.1186/bcr2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wendt MK, Smith JA, Schiemann WP. p130Cas is required for mammary tumor growth and transforming growth factor-β-mediated metastasis through regulation of Smad2/3 activity. J Biol Chem. 2009;284:34145–34156. doi: 10.1074/jbc.M109.023614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balanis N, Wendt MK, Schiemann BJ, Wang Z, Schiemann WP, Carlin CR. Epithelial to mesenchymal transition promotes breast cancer progression via a fibronectin-dependent STAT3 signaling pathway. J Biol Chem. 2013;288:17954–17967. doi: 10.1074/jbc.M113.475277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor MA, Amin JD, Kirschmann DA, Schiemann WP. Lysyl oxidase contributes to mechanotransduction-mediated regulation of transforming growth factor-β signaling in breast cancer cells. Neoplasia. 2011;13:406–418. doi: 10.1593/neo.101086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 31.Balanis N, Yoshigi M, Wendt MK, Schiemann WP, Carlin CR. β3 integrin–EGF receptor cross-talk activates p190RhoGAP in mouse mammary gland epithelial cells. Mol Biol Cell. 2011;22:4288–4301. doi: 10.1091/mbc.E10-08-0700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taylor MA, Sossey-Alaoui K, Thompson CL, Danielpour D, Schiemann WP. TGF-β upregulates miR-181a expression to promote breast cancer metastasis. J Clin Invest. 2013;123:150–163. doi: 10.1172/JCI64946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wendt MK, Schiemann BJ, Parvani JG, Lee YH, Kang Y, Schiemann WP. TGF-β stimulates Pyk2 expression as part of an epithelial-mesenchymal transition program required for metastatic outgrowth of breast cancer. Oncogene. 2013;32:2005–2015. doi: 10.1038/onc.2012.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wander SA, Hennessy BT, Slingerland JM. Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. J Clin Invest. 2011;121:1231–1241. doi: 10.1172/JCI44145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cidado J, Park BH. Targeting the PI3K/Akt/mTOR pathway for breast cancer therapy. J Mammary Gland Biol Neoplasia. 2012;17:205–216. doi: 10.1007/s10911-012-9264-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petrel TA, Brueggemeier RW. Increased proteasome-dependent degradation of estrogen receptor-α by TGF-β1 in breast cancer cell lines. J Cell Biochem. 2003;88:181–190. doi: 10.1002/jcb.10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jinnin M, Ihn H, Tamaki K. Characterization of SIS3, a novel specific inhibitor of Smad3, and its effect on transforming growth factor-β1-induced extracellular matrix expression. Mol Pharmacol. 2006;69:597–607. doi: 10.1124/mol.105.017483. [DOI] [PubMed] [Google Scholar]
- 38.Yang SH, Galanis A, Sharrocks AD. Targeting of p38 mitogen-activated protein kinases to MEF2 transcription factors. Mol Cell Biol. 1999;19:4028–4038. doi: 10.1128/mcb.19.6.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gellibert F, Woolven J, Fouchet MH, Mathews N, Goodland H, Lovegrove V, Laroze A, Nguyen VL, Sautet S, Wang R. Identification of 1,5-naphthyridine derivatives as a novel series of potent and selective TGF-β type I receptor inhibitors. J Med Chem. 2004;47:4494–4506. doi: 10.1021/jm0400247. [DOI] [PubMed] [Google Scholar]
- 40.Inman GJ, Nicolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, Laping NJ, Hill CS. SB-431542 is a potent and specific inhibitor of transforming growth factor-β superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2002;62:65–74. doi: 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- 41.Fu J, Zhang L, He T, Xiao X, Liu X, Wang L, Yang L, Yang M, Zhang T, Chen R. TWIST represses estrogen receptor-α expression by recruiting the NuRD protein complex in breast cancer cells. Int J Biol Sci. 2012;8:522–532. doi: 10.7150/ijbs.4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wik E, Raeder MB, Krakstad C, Trovik J, Birkeland E, Hoivik EA, Mjos S, Werner HM, Mannelqvist M, Stefansson IM. Lack of estrogen receptor-α is associated with epithelial-mesenchymal transition and PI3K alterations in endometrial carcinoma. Clin Cancer Res. 2013;19:1094–1105. doi: 10.1158/1078-0432.CCR-12-3039. [DOI] [PubMed] [Google Scholar]
- 43.Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, Hartwell K, Onder TT, Gupta PB, Evans KW. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci U S A. 2010;107:15449–15454. doi: 10.1073/pnas.1004900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taddei ML, Giannoni E, Comito G, Chiarugi P. Microenvironment and tumor cell plasticity: an easy way out. Cancer Lett. 2013;341:80–96. doi: 10.1016/j.canlet.2013.01.042. [DOI] [PubMed] [Google Scholar]
- 45.Turner N, Moretti E, Siclari O, Migliaccio I, Santarpia L, D'Incalci M, Piccolo S, Veronesi A, Zambelli A, Del Sal G. Targeting triple negative breast cancer: is p53 the answer? Cancer Treat Rev. 2013;39:541–550. doi: 10.1016/j.ctrv.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 46.Frederick JP, Liberati NT, Waddell DS, Shi Y, Wang XF. Transforming growth factor β-mediated transcriptional repression of c-Myc is dependent on direct binding of Smad3 to a novel repressive Smad binding element. Mol Cell Biol. 2004;24:2546–2559. doi: 10.1128/MCB.24.6.2546-2559.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matsuda T, Yamamoto T, Muraguchi A, Saatcioglu F. Cross-talk between transforming growth factor-β and estrogen receptor signaling through Smad3. J Biol Chem. 2001;276:42908–42914. doi: 10.1074/jbc.M105316200. [DOI] [PubMed] [Google Scholar]
- 48.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burstein MD, Tsimelzon A, Poage GM, Covington KR, Contreras A, Fuqua S, Savage M, Osborne CK, Hilsenbeck SG, Chang JC. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin Cancer Res. 2014 doi: 10.1158/1078-0432.CCR-14-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chang BH, Gujral TS, Karp ES, BuKhalid R, Grantcharova VP, MacBeath G. A systematic family-wide investigation reveals that ~ 30% of mammalian PDZ domains engage in PDZ-PDZ interactions. Chem Biol. 2011;18:1143–1152. doi: 10.1016/j.chembiol.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ivarsson Y. Plasticity of PDZ domains in ligand recognition and signaling. FEBS Lett. 2012;586:2638–2647. doi: 10.1016/j.febslet.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wawrzyniak AM, Kashyap R, Zimmermann P. Phosphoinositides and PDZ domain scaffolds. Adv Exp Med Biol. 2013;991:41–57. doi: 10.1007/978-94-007-6331-9_4. [DOI] [PubMed] [Google Scholar]
- 53.Baenke F, Peck B, Miess H, Schulze A. Hooked on fat: The role of lipid synthesis in cancer metabolism and tumour development. Dis Model Mech. 2013;6:1353–1363. doi: 10.1242/dmm.011338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Halaoui R, McCaffrey L. Rewiring cell polarity signaling in cancer. Oncogene. 2014;34:939–950. doi: 10.1038/onc.2014.59. [DOI] [PubMed] [Google Scholar]
- 55.Evdokimova V, Tognon C, Ng T, Sorensen PH. Reduced proliferation and enhanced migration: Two sides of the same coin? Molecular mechanisms of metastatic progression by YB-1. Cell Cycle. 2009;8:2901–2906. doi: 10.4161/cc.8.18.9537. [DOI] [PubMed] [Google Scholar]
- 56.Vega S, Morales AV, Ocana OH, Valdes F, Fabregat I, Nieto MA. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004;18:1131–1143. doi: 10.1101/gad.294104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Corominas-Faja B, Cufi S, Oliveras-Ferraros C, Cuyas E, Lopez-Bonet E, Lupu R, Alarcon T, Vellon L, Iglesias JM, Leis O. Nuclear reprogramming of luminal-like breast cancer cells generates Sox2-overexpressing cancer stem-like cellular states harboring transcriptional activation of the mTOR pathway. Cell Cycle. 2013;12:3109–3124. doi: 10.4161/cc.26173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kadota M, Yang HH, Gomez B, Sato M, Clifford RJ, Meerzaman D, Dunn BK, Wakefield LM, Lee MP. Delineating genetic alterations for tumor progression in the MCF10A series of breast cancer cell lines. PLoS One. 2010;5:e9201. doi: 10.1371/journal.pone.0009201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang J, Song K, Krebs TL, Jackson MW, Danielpour D. Rb/E2F4 and Smad2/3 link survivin to TGF-β-induced apoptosis and tumor progression. Oncogene. 2008;27:5326–5338. doi: 10.1038/onc.2008.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coumar MS, Tsai FY, Kanwar JR, Sarvagalla S, Cheung CH. Treat cancers by targeting survivin: just a dream or future reality? Cancer Treat Rev. 2013;39:802–811. doi: 10.1016/j.ctrv.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 61.Cheung CH, Huang CC, Tsai FY, Lee JY, Cheng SM, Chang YC, Huang YC, Chen SH, Chang JY. Survivin—biology and potential as a therapeutic target in oncology. Onco Targets Ther. 2013;6:1453–1462. doi: 10.2147/OTT.S33374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Carmena M, Pinson X, Platani M, Salloum Z, Xu Z, Clark A, Macisaac F, Ogawa H, Eggert U, Glover DM. The chromosomal passenger complex activates Polo kinase at centromeres. PLoS Biol. 2012;10:e1001250. doi: 10.1371/journal.pbio.1001250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material