

Abstract
Furan is a heterocyclic organic compound produced in the chemical manufacturing industry and also found in a broad range of food products, including infant formulas and baby foods. Previous reports have indicated that the adverse biological effects of furan, including its liver tumorigenicity, may be associated with epigenetic abnormalities. In the present study, we investigated the persistence of epigenetic alterations in rat liver. Male F344 rats were treated by gavage 5 days per week with 8 mg furan/kg body weight (bw)/day for 90 days. After the last treatment, rats were divided randomly into 4 groups; 1 group of rats was sacrificed 24 h after the last treatment, whereas other groups were maintained without further furan treatment for an additional 90, 180, or 360 days. Treatment with furan for 90 days resulted in alterations in histone lysine methylation and acetylation, induction of base-excision DNA repair genes, suggesting oxidative damage to DNA, and changes in the gene expression in the livers. A majority of these furan-induced molecular changes was transient and disappeared after the cessation of furan treatment. In contrast, histone H3 lysine 9 and H3 lysine 56 showed a sustained and time-depended decrease in acetylation, which was associated with formation of heterochromatin and altered gene expression. These results indicate that furan-induced adverse effects may be mechanistically related to sustained changes in histone lysine acetylation that compromise the ability of cells to maintain and control properly the expression of genetic information.
Keywords: microarrays, gene expression profiling, genetic toxicology, liver carcinogenesis
Furan, a colorless, volatile organic compound, has been found in a broad range of food products (US Food and Drug Administration [US-FDA], 2004), including coffee and canned and jarred foods (Maga, 1976), as a result of thermal degradation of natural food components, including sugars, polyunsaturated fatty acids, and ascorbic acid (Perez Locas and Yaylayan, 2004). Importantly, relatively high levels of furan have been detected in infant formulas and baby foods (US-FDA, 2004).
The presence of furan in foods has caused concern for regulatory public health agencies, such as the US Food and Drug Administration and European Food Safety Authority, and has led to research and evaluation of furan by organizations, including the National Toxicology Program (NTP, 1993) and the International Agency for Research on Cancer (IARC, 1995). The results from 2-year rodent bioassays have indicated that a lifetime exposure of rats and mice to furan at a dose 2 mg/kg body weight (bw)/day resulted in the development of liver cholangiocarcinomas in F344 rats, whereas exposure to 4 mg/kg bw/day and greater induced hepatocellular adenomas, hepatocellular carcinomas, and mononuclear cell leukemia in F344 rats, and hepatocellular adenoma and carcinoma in B6C3F1 mice (Moser et al., 2009; NTP, 1993). Importantly, rats treated with higher doses of furan (30 mg/kg bw/day) for 90 days developed cholangiocarcinomas and hepatocellular carcinomas within a year without further treatment (NTP, 1993). Based on this evidence, IARC classified furan as “possibly carcinogenic” to humans (group 2B) (IARC, 1995).
The carcinogenicity of furan has been attributed to genotoxic and non-genotoxic changes (Chen et al., 2010, 2012; Fransson-Steen et al., 1997; Hickling et al., 2010; Jackson et al., 2014) in the liver. The exposure of rats and mice to furan at tumorigenic doses caused hepatocellular necrosis and apoptosis, sustained regenerative cell proliferation, and inflammation, which are key pathophysiological events involved in liver carcinogenesis (Fransson-Steen et al., 1997). Also, furan exposure altered the expression of protein-coding and protein-non-coding genes, for example, short and long non-coding RNAs, and induced various epigenetic alterations (Chen et al., 2010, 2012; Jackson et al., 2014; Recio et al., 2013).
In a previous study, we demonstrated that treating male F344 rats by gavage 5 days/week with furan at 0, 0.92, 2.0, or 4.4 mg/kg bw/day for 90, 180, and 360 days resulted in dose- and time-dependent epigenetic changes consisting of alterations in DNA methylation and histone lysine methylation and acetylation, altered expression of chromatin modifying genes, and gene-specific methylation (Conti et al., 2014). This suggests the possible involvement of epigenetic mechanisms in the hepatotoxicity and carcinogenicity of furan; however, it is not clear (1) which of the epigenetic alterations are directly associated with furan treatment and (2) which may be involved in furan-induced liver tumorigenesis, since it is highly unlikely that all the epigenetic changes are equally important in the carcinogenic process.
Based on these considerations, in the present study we investigated the possible role of epigenetic events in furan-induced liver carcinogenesis by examining the extended evolution of epigenetic alterations in the livers of rats after exposure to furan had ceased. This approach should allow discrimination between treatment-related carcinogenic and non-carcinogenic changes (Bannasch, 1986), including epigenetic alterations (Christman et al., 1993; Pogribny et al., 2006), because only alterations that persist after removal of carcinogen are presumably involved in the tumorigenic process.
MATERIALS AND METHODS
Animals, experimental design, and treatments
Male F344 rats were obtained from the breeding colony at the National Center for Toxicological Research (NCTR). This strain of rats was used in a furan carcinogenicity bioassay conducted by the NTP 1993. Rats were housed in a temperature controlled room (24°C) with a 12-h light/dark cycle, and given ad libitum access to water and NIH-41 irradiated pellet diet. Forty-six-week-old male F344 rats were randomly distributed into control and experimental groups. Rats from the experimental groups were treated by gavage 5 days/week with 8 mg/kg bw/day furan in corn oil for 90 days. This dose was selected based on evidence that a lifetime exposure to furan at doses 4 mg/kg bw/day and greater induced hepatocellular adenomas and hepatocellular carcinomas in male F344 rats (NTP, 1993). Rats from control group received only corn oil. Five rats from control and furan groups were euthanized by exsanguination following deep isoflurane anesthesia after the 90-day treatment period (time point 0). The remaining rats were then maintained without further furan treatment (stop-exposure groups) and 5 rats from each group were euthanized after an additional 90, 180, or 360 days. Age-matched rats were maintained during the recovery periods and were used as controls. The livers were excised, and a slice of the median lobe was snap-frozen immediately in liquid nitrogen and stored at −80°C for subsequent analyses. All experimental procedures were reviewed and approved by the NCTR Animal Care and Use Committee.
Western blotting
The levels of mono-, di-, and trimethylation of histone H3 lysine 4 (H3K4), trimethylation of histone H3 lysine 9 (H3K9), H3 lysine 27 (H3K27), mono- and trimethylation of H4 lysine 20 (H4K20), and of acetylation of histone H3 lysine 9 (H3K9ac), H3 lysine 27 (H3K27ac), and H3 lysine 56 (H3K56ac) were determined by Western blot analysis as described in Conti et al. (2014). Equal protein loading was confirmed by immunostaining against the total histone H4.
Quantitative reverse transcription-PCR
Total RNA (2 μg) was reverse transcribed using random primers and High Capacity cDNA Reverse Transcription kits (Life Technologies, Grand Island, New York) according to the manufacturer’s protocol. cDNA was analyzed in a 96-well plate assay format using a 7900HT Fast Real-Time PCR System (Life Technologies). Each plate contained 1 experimental gene and a housekeeping gene. All primers for the gene expression analysis were obtained from Life Technologies and are listed in Supplementary Table 1. The cycle threshold (Ct) for each sample was determined from the linear region of the amplification plot. The ΔCt values for all genes were determined relative to the endogenous control β-actin (Actb). The ΔΔCt values were calculated using treated group means relative to the control group means (Schmittgen and Livak, 2008). The fold change data were calculated from the ΔΔCt values. All quantitative reverse transcription-PCR (qRT-PCR) reactions were conducted in triplicate and the experiments were repeated twice.
Methylation-sensitive analysis of chromatin structure
The structure of hepatic chromatin was determined by a modified methylation-based analysis of nucleosomal DNA accessibility (Miranda et al., 2010). This technique is based on the fact that CpG sites in DNA are protected from methylation when these sequences are wrapped around histones (Kladde and Simpson, 1994; Miranda et al., 2010). Briefly, nuclei from 40 mg of liver tissue of control and furan-exposed rats were isolated and purified. After purification, the nuclei were incubated with 60 U of methylase SssI (M. SssI; New England Biolabs, Ipswich, Massachusetts) for 15 min at 37°C. The reaction was terminated by the addition of 150 µl of a solution containing 10mM Tris HCl, 300mM NaCl, 1% SDS, and 5mM disodium EDTA, and genomic DNA was extracted using a standard digestion with proteinase K, followed by phenol/chloroform/isoamyl alcohol extraction and ethanol precipitation (Strauss, 1989). The extent of CCGG methylation was evaluated with a [3H]dCTP extension assay after digestion of DNA with HpaII (Pogribny et al., 1999).
Gene expression analysis using microarray technology
Total RNA was extracted from liver tissue samples using RNeasy Mini kits (Qiagen, Valencia, California) according to the manufacturer’s instructions. Gene expression profiles of rat livers were determined using Agilent whole genome 8x60K rat microarrays (Agilent Technologies, Santa Clara, California). Sample labeling and microarray processing were performed as detailed in the “One-Color Microarray-Based Gene Expression Analysis” Version 5.5 (Agilent Technologies) protocol. The hybridized slides were scanned with an Agilent DNA Microarray scanner (Agilent Technologies) at 5 µm resolution. The resulting images were analyzed by determining the Cy3 fluorescence intensity of all gene spots (features) on each array using the Agilent Feature Extraction Software (Version 10.7). The raw data were then uploaded into the ArrayTrack database (Fang et al., 2009). The median fluorescence intensity of all the pixels within one feature was taken as the intensity value for that feature. The raw intensity values were then normalized using 75 percentile channel scaling normalization within ArrayTrack. A list of differentially expressed genes was generated using a Student’s t test at a P-value < 0.05 and a fold change >2. The microarray gene expression data were deposited in the NCBI’s Gene Expression Omnibus database (accession number GSE63740).
Functional analysis of significant genes
The Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Kanehisa and Goto, 2000) was used to determine pathways that were enriched for the significant mRNA transcripts identified from the t test analysis using ArrayTrack. Significance values were calculated based on a right-tailed Fisher’s exact test that determined whether a pathway was overrepresented by calculating whether or not the genes in a given pathway were enriched within the dataset compared with all genes on the array in the same pathway; a P < 0.05 was selected as the cutoff for significance based on KEGG threshold recommendations. Only those pathways with a P-value below the threshold and having more than 3 representative genes in the dataset were considered to be significant.
Methylated DNA immunoprecipitation analysis
Methylated DNA immunoprecipitation was performed with MethylMiner Methylated DNA Enrichment kits (Invitrogen, Carlsbad, California) according to the manufacturer’s instructions. The methylation status of CpG sites located within the promoter/first exon region of Tmtc3, Mon1b, Pde5a, Stk39, and Gfap genes was determined by qPCR of DNA from immunoprecipitates and input DNA.
Chromatin immunoprecipitation assay
Formaldehyde cross-linking and chromatin immunoprecipitation (ChIP) assays with primary antibodies against H3K9ac (Abcam, Cambridge, Massachusetts) were performed by using a ChIP Assay kit (Millipore Corporation, Billerica, Massachusetts). Purified DNA from immunoprecipitates and input DNA were analyzed by quantitative PCR (qPCR) with primers for Tmtc3, Mon1b, Pde5a, Stk39, and Gfap genes. The primer sequences are presented in Supplementary Table 2. The results were normalized to the amount of input DNA and presented as fold change for each DNA in liver of rats from experimental groups relative to age-matched control rats.
Statistical analyses
Results are presented as mean ± SD. The results are presented as an average fold change in the livers of furan-treated rats relative to that in their respective age-matched controls. Data were analyzed by 1-way analysis of variance, with pair-wise comparisons being made by the Student-Newman-Keuls method. Linear regression analysis was used to determine time-related trends. P-values < 0.05 were considered significant.
RESULTS
Effect of Furan Stop-Exposure on Histone Lysine Acetylation and Methylation
The disruption of a normal pattern of covalent histone modifications is an epigenetic event frequently observed upon chemical exposure (Arita et al., 2012; Dik et al., 2012; Sadikovic et al., 2008). In order to determine whether or not furan treatment caused any alterations in histone lysine modifications, the levels of H3K9ac, H3K27ac, H3K56ac, H3K4me1, H3K4me2, H3K4me3, H3K9me3, H3K27me3, H4K20me1, and H4K20me3 were examined using Western immunoblotting in the livers of control rats and rats exposed to furan (Fig. 1 and Supplementary Fig. 1). It has been demonstrated that these lysine modifications are associated with different chromatin states (Ernst et al., 2011). Figure 1 shows that treatment with furan caused transient changes in the levels of histone H3K4me1 and a marked decrease in the levels of histone H3K9ac and H3K56ac. The levels of histone H3K9ac decreased by 62% and 53% in the 180-day and 360-day stop-exposure groups, respectively, and the levels of H3K56ac decreased by 55% and 76% in the 180-day and 360-day stop-exposure groups, respectively, when compared with the age-matched control rats. In contrast, furan treatment resulted in moderate changes in H3K4me2, H4K20me1, and H3K27ac (Fig. 1) and had no effect on hepatic histone H3K4, H3K9, H3K27, or H4K20 trimethylation (Supplementary Fig. 1).
FIG. 1.

The levels of histone H3K4me1, H3K4me2, H4K20me1, H3K9ac, H3K27ac, and H3K56ac in the livers of control rats and rats treated with furan. A, Representative immunoblots images of histone modifications in the livers of control rats and rats treated with furan. B, Densitometric analysis of the Western blot results is shown as percent change in histone modification level in the livers of furan-treated rats relative to that in the control group, which was assigned a value of 100%. Values are mean ± SD, n = 5. Asterisk (*) denotes a significant (P < 0.05) difference from the control rats; dagger (†) denotes a significant (P < 0.05) trend. Equal protein loading was confirmed by immunostaining with anti-histone H4 antibodies.
Effect of Furan Stop-Exposure on DNA Damage
It has been suggested that, in addition to non-genotoxic mechanisms, persistent indirect DNA damage caused by reactive oxygen species may have significance in furan tumorigenicity (Hickling et al., 2010). In light of this, the expression of base-excision DNA repair (BER) genes, a sensitive in vivo biomarker of persistent oxidative DNA damage (Powell et al., 2005; Rusyn et al., 2004), was assessed in the livers of rats treated with furan. Figure 2 shows a significant increase in the expression of apurinic/apirimidinic endonuclease 1 (Apex), ligase 1 (Lig1), x-ray repair complementing defective repair in Chinese hamster cells 1 (Xrcc1), and flap structure-specific endonuclease 1 (Fen1) genes in the livers of rats treated with furan for 90 days. The expression of Lig1, Xrcc1, and Fen1 genes returned to a normal level 90 days after the cessation of furan treatment and Apex by the 180-day time point indicating that furan-induced oxidative DNA lesions had been repaired and DNA integrity restored (Fig. 2).
FIG. 2.

The expression of base-excision DNA repair genes in the livers of control rats and rats treated with furan. The expression of Ogg1, Apex, Lig1, Polb, Xcrcc1, and Fen1 genes was determined by qRT-PCR as detailed in Materials and Methods. The results are presented as an average fold change in the expression of each gene in the livers of furan-treated rats relative to that in the control group, which was assigned a value 1. Values shown are mean ± SD, n = 5. Asterisk (*) denotes a significant (P < 0.05) difference from the control rats.
Effect of Furan Stop-Exposure on Chromatin Structure
The observed changes in hepatic histone lysine acetylation in the furan stop-exposure groups may be indicative of chromatin structure alterations. Hence, the expression of transcriptional corepressors involved in regulation of chromatin and chromatin structure was investigated. Figure 3A shows that the expression of Ncor1, Smrt, and Sin3a was increased in the 360-day stop-exposure group, by 1.9-, 2.0-, and 1.7-fold, respectively, when compared with the age-matched control rats. Analysis of the status of hepatic chromatin, by evaluating the accessibility of nucleosomal DNA to methylation at CpG sites, showed that rats exposed to furan for 90 days and rats from the 90-days post-exposure group did not show changes in the chromatin structure when compared with the respective control groups (Fig. 3B). In contrast, the livers of rats from the 180-day and 360-day stop-exposure groups were characterized by the formation of condensed chromatin. This was evidenced by 30% and 27%, respectively, greater incorporation of [3H]dCTP into HpaII-digested DNA from M. SssI-pretreated nuclei isolated from the livers of furan-exposed rats when compared with age-matched control rats.
FIG. 3.

Analysis of chromatin structure in the livers of control rats and rats treated with furan. A, The expression of Ncor1, Ncor2, and Sin3a genes was determined by qRT-PCR. The results are presented as an average fold change in the expression of each gene in the livers of furan-treated rats relative to that in the control group, which was assigned a value 1. Values shown are mean ± SD (n = 5). B, Chromatin structure in the livers of control rats and rats treated with furan was determined by analyzing the accessibility of nucleosomal DNA to methylation at Hpa II sites. The extent of [3H]dCTP incorporation into HpaII-digested DNA isolated from SssI methylase-pretreated nuclei is directly proportional to changes in chromatin condensation. Results are presented as an average percent change in the extent of [3H]dCTP incorporation into HpaII-digested in the livers of furan-treated rats relative to that in the control group, which was assigned a value of 100% (n = 5). Asterisk (*) denotes a significant (P < 0.05) difference from the control rats; dagger (†) denotes a significant (P < 0.05) trend. C, A diagram showing an effect of furan effect on chromatin structure. Exposure to furan resulted in the deacetylation of histones and formation of compact chromatin.
Effect of Furan Stop-Exposure on Hepatic Gene Expression Profile
The formation of condensed chromatin may limit accessibility of the transcription factors to DNA and alter the gene expression pattern (Fig. 3C) (Dillon and Festenstein, 2002). To test this hypothesis, hepatic gene expression profiles were examined using high-throughput Agilent whole genome 8x60K rat microarrays in control rats and rats treated with furan for 90 days and in rats 360 days after furan treatment. To identify genes that were differentially expressed between the control and experimental groups, a t test, using a P < 0.05, coupled with a fold-change cutoff >2, was applied. A total of 1134 significantly up-regulated or down-regulated genes was identified in the livers of rats treated with furan for 90 days when compared with control rats (Fig. 4 and Supplementary Table 3). In contrast, the number of differentially expressed genes in the livers of rats 360 days after the exposure has ceased was substantially smaller, totaling only 88 genes (Fig. 4 and Supplementary Fig. 4). Pathway enrichment analysis of the differentially expressed genes in the livers of rats treated with furan for 90 days demonstrated a strong enrichment in genes involved in inflammation, cell cycle, p53 signaling, and drug metabolism. Amino acid metabolism, cancer-associated pathways, and calcium signaling were the top 3 gene-enriched pathways 360 days following furan treatment (Table 1).
FIG. 4.
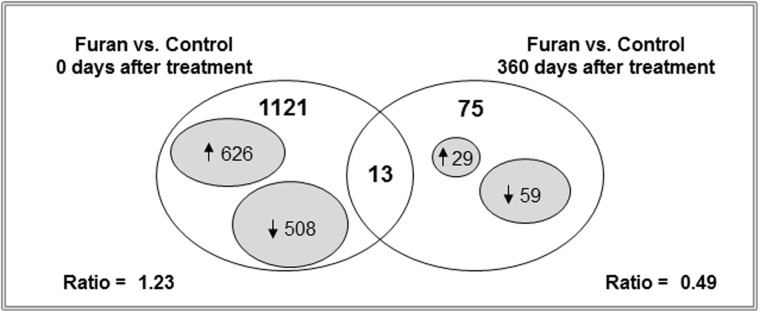
Whole-genome microarray analysis of gene expression in the livers of control rats and rats treated with furan. Venn diagram showing genes that were significantly different in the livers of control rats and rats treated with furan (P < 0.05, fold change > 2; n = 4).
TABLE 1.
Pathway Analysis of Genes in the Liver of Rats Treated with Furan for 90 Days and 360 Days After Furan Treatment
| Category | Pathway | P-Value |
|---|---|---|
| Furan, treatment for 90 daysa | ||
| Immune system | Inflammation | <1.00E + 09 |
| Cell growth and death | Cell cycle and p53 signaling pathway | 9.00E−08 |
| Xenobiotics metabolism and transport | Drug metabolism: cytochrome P450 and ABC transporters | 8.30E−07 |
| Metabolism | Glutathione and amino acid metabolism | 7.33E−06 |
| Fatty acid metabolism and glycolysis | 0.001 | |
| Replication and repair | DNA replication | 0.001 |
| Signal transduction | TGF-beta signaling pathway | 0.05 |
| Furan, 360 after treatmentb | ||
| Metabolism | Amino acid metabolism | 0.006 |
| Cancers | Pathways in cancer | 0.009 |
| Signal transduction | Calcium signaling pathway | 0.012 |
| Digestive system | Carbohydrate digestion and absorption | 0.015 |
a1134 genes, whose expression were significantly different from corn oil-treated controls (fold change >2, P ≤ 0.05; listed in Supplementary Table 3), were analyzed using the KEGG pathway database as described in Materials and Methods.
b88 genes, whose expression were significantly different from corn oil-treated controls (fold change >2, P ≤ 0.05; listed in Supplementary Table 4), were analyzed using the KEGG pathway database as described in Materials and Methods.
Interestingly, there was a marked difference in the pattern of hepatic gene expression in rats treated with furan for 90 days and in rats 360 days after treatment. This was evidenced by a substantially greater number of up-regulated/down-regulated genes (ratio = 1.23) in the livers of rats after 90 days of furan treatment, whereas the number of down-regulated genes was 2 times greater than up-regulated genes (up-regulated/down-regulated gene ratio = 0.49) 360 days after the cessation of furan treatment. Functional analysis of the differentially expressed genes at 360 days after furan treatment indicated that 41% of these genes were cancer-related (Supplementary Table 4).
Despite a substantial difference in the number of differentially expressed genes in the livers of rats treated with furan for 90 days and in rats 360 days after furan treatment, 13 genes were expressed in common (Fig. 4). Five of these genes (Tmtc3, Camsap2, Mon1b, Slc17a6, and Cntnap) exhibited lower expression at both time intervals, whereas the expression of 6 genes (Amn, Aldh1a7, Pde5a, Stk39, Fzd3, and Adcyap1) was decreased 360 days after the cessation of furan treatment compared with value observed at 90 days (Table 2). The expression of only 2 genes (Cml2 and Gfap) was increased 360 days after furan treatment compared with the 90-day value.
TABLE 2.
Expression of Common Genes in the Livers of Rats Treated with Furan for 90 Days and 360 Days after Furan Treatment (P < 0.05; Fold Change >2)
| Number | Gene Name | Gene Bank ACC Number | Description | 90 Days of Furan Treatment, Fold Change | 360 Days after Furan Treatment, Fold Change |
|---|---|---|---|---|---|
| 1 | Amna,b | NM_001108061 | Amnion associated transmembrane protein | 8.60 | 2.44 |
| 2 | Aldh1a7b | NM_017272 | Aldehyde dehydrogenase family 1, subfamily A7 | 62.77 | 2.39 |
| 3 | Tmtc3a,b | NM_001135858 | Transmembrane and tetratricopeptide repeat containing 3 | −2.27 | −2.02 |
| 4 | Camsap2 | NM_001134503 | Calmodulin regulated spectrin-associated protein family, member 2 | −2.00 | −2.10 |
| 5 | Mon1ba,b | NM_001107433 | MON1 homolog b (yeast) | −2.38 | −2.43 |
| 6 | Slc17a6a,b | NM_053427 | Solute carrier family 17 (sodium-dependent inorganic phosphate co-transporter), member 6 | −2.12 | −2.60 |
| 7 | Cntnap1 | NM_032061 | Contactin associated protein 1 | −2.02 | −2.16 |
| 8 | Pde5aa,b | NM_133584 | Phosphodiesterase 5A, cGMP-specific | 4.32 | −2.11 |
| 9 | Stk39a,b | NM_019362 | Serine threonine kinase 39 | 3.71 | −2.65 |
| 10 | Fzd3 | NM_153474 | Frizzled family receptor 3 | 3.93 | −3.19 |
| 11 | Adcyap1 | NM_016989 | Adenylate cyclase activating polypeptide 1 | 2.59 | −2.19 |
| 12 | Cml2 | NM_001173449 | Camello-like 2 | −2.05 | 2.03 |
| 13 | Gfapa,b | NM_017009 | Glial fibrillary acidic protein | −2.54 | 2.08 |
aGenes that contain CpG island in the promoter region.
bGenes related to carcinogenesis.
Effect of Furan Stop-Exposure on the Gene-Specific Epigenetic Status
In silico analysis of the common differentially expressed genes, using the CpG Island Searcher program (http://www.cpgislands.com) (Takai and Jones, 2003), revealed the presence of CpG islands in the 7 of the genes, suggesting that they may be regulated by epigenetic mechanisms (Table 2). To determine whether or not these sustained changes in gene expression were associated with furan-induced epigenetic alterations, the status of histone H3K9 acetylation and CpG island methylation was examined. Figure 5 shows that among genes down-regulated in the livers of rats both after 90 days of furan treatment and 360 days after furan treatment ceased (Tmtc3, Mon1b, and Slc17a6), the level of gene-specific histone H3K9 acetylation was lower at both time intervals. Likewise, in genes whose expression was decreased 360 days after treatment compared with the value at 90 days (Pde5a and Stk39), the level of gene-specific histone H3K9 acetylation was lower at either both time intervals (Pde5a) or at 360 days after furan treatment (Stk39). Gene-specific histone H3K9 acetylation was increased in Gfap, the only gene containing GpG island and up-regulated at 360 days after furan treatment. CpG island methylation of these genes in the livers of furan-treated rats did not differ from age-matched control rats at any time interval (Supplementary Fig. 4).
FIG. 5.

Chromatin immunoprecipitation analysis of gene-specific histone H3K9 in the livers of control rats and rats treated with furan. The results are presented as an average fold change in the extent of histone H3K9 acetylation in the livers of furan-treated rats relative to that in the control rats after normalization to input DNA. Asterisk (*) denotes a significant (P < 0.05) difference from the control rats (n = 4).
DISCUSSION
The results of several comprehensive studies have indicated that the adverse biological effects of furan, including its liver tumorigenicity, may be associated with furan-induced epigenetic abnormalities (Chen et al., 2010, 2012; Jackson et al., 2014; Recio et al., 2013). In a previous study (Conti et al., 2014), we demonstrated that long-term exposure of F344 rats to furan at doses 2 and 4 mg/kg bw/day resulted in prominent epigenetic changes in the liver consisting of alterations in DNA methylation and histone lysine modifications, including global DNA demethylation, a marked reduction of the levels of histone H3K9 and H4K20 trimethylation and histone H3K9 and H3K56 acetylation, gene-specific hypermethylation, and an altered expression of chromatin modifying genes. However, there is an insufficient knowledge to clarify whether or not these epigenetic changes are directly related to the furan-treatment or just indirect secondary sequelae of furan-induced cellular alterations. It is important to clarify the role of these changes in the pathogenesis of furan-induced liver toxicity and carcinogenicity.
In this report, we demonstrate that the treatment of F344 rats with furan at 8 mg/kg bw/day for 90 days resulted in alterations of histone lysine methylation and acetylation, induction of BER genes, suggesting oxidative damage to DNA, and changes in the gene expression pattern in the livers. It is well known that for any carcinogen-induced alteration to be associated with the tumorigenic process, it needs to be stable and persist after the factor that caused the changes has been removed (Bannasch, 1986; Christman et al., 1993; Pogribny et al., 2006). In the present study, by using a stop-treatment approach, we show that a majority of furan-induced molecular changes, including DNA damage, histone lysine methylation, and aberrant gene expression were transient and became restored after cessation of the furan treatment. In contrast to these alterations, histone lysine acetylation, especially, histone H3K9ac and H3K56ac, decreased in a time-dependent manner following furan treatment.
Acetylation of histone lysine residues, in general, and histone H3K9, in particular, has been shown to play a central role in the formation of active chromatin (Luo and Dean, 1999; Si et al., 2010) and is considered as a main mark of promoter activation (Ernst et al., 2011). Additionally, H3K56 acetylation coordinates a nucleosome assembly (Clemente-Ruiz et al., 2011) and is a key player in DNA replication and repair (Tanaka et al., 2012). Therefore, it is possible that a persistent furan-induced loss of any of these histone marks may compromise chromatin architecture resulting in the formation of a compact heterochromatin structure. The analysis of the structure of hepatic chromatin in control rats and rats treated with furan showed a formation of a more condensed chromatin in the livers of furan-treated rats, as evidenced by a decreased accessibility of nucleosomal DNA to SssI methylase. More importantly, the formation of heterochromatin in the livers of furan-treated rats was negatively correlated with decrease of histone H3K9 acetylation (r = −0.61, P < 0.05).
One of the main consequences of the heterochromatin formation is a subsequent decrease in gene expression (Grewal and Moazed, 2003; Turner, 1998). Indeed, the results of this study showed a greater numbers of down-regulated genes that correlated with heterochromatin formation in the livers of rats after 360 days of furan treatment when compared with those in rats treated with furan for 90 days. Importantly, more than 40% of differentially regulated genes after 360 days of furan treatment and more than 60% common differentially regulated genes, including Stk39, Aldh1, Adcyap1, Pde5a, and Gfap, have been reported to have significance in tumorigenesis (Balatoni et al., 2009; Kleinstreuer et al., 2013; Muzio et al., 2012; Okabe et al., 2009; Wentzensen et al., 2014).
Another important finding of this study is a demonstration that, in addition to the global sustained reduction in acetylation of histone H3K9 and H3K56, furan treatment caused also a gene-specific reduction in the level of histone H3K9 acetylation at genes containing CpG island promoters. Interestingly, a decrease in gene-specific histone H3K9 acetylation correlated with reduced expression of these genes and occurred without changes in DNA methylation. This finding is in good agreement with growing evidence of the importance histone deacetylation as primary epigenetic mark in gene silencing (Liu et al., 2008; Stirzaker et al., 2004; Vaissière et al., 2008). A decrease in gene-specific histone H3K9 acetylation has been associated with gene silencing in several major human cancers (Fetahu et al., 2014; Rubinek et al., 2012). More importantly, Min et al. (2012) have reported that loss of histone H3K9 acetylation at the survivin gene was attributed to liver cancer initiation.
In summary, the results of the present study indicate that furan-induced adverse effects may be mechanistically related to a sustained decrease in histone H3 lysine acetylation and consequent formation of heterochromatin that compromise the ability of cells to maintain and control properly the expression of genetic information, which is well-known indispensable event in the tumorigenic process. Our study sheds light on the involvement of epigenetic changes in the mechanism of furan hepatotoxicity and carcinogenicity. However, future studies focusing on the investigation of these epigenetic changes in furan-induced pathological lesions in the liver are needed to provide a conclusive answer on the role of epigenetic alterations in furan hepatotoxicity and carcinogenicity.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
This work was carried out under the auspices of the National Toxicology Program (NTP) and supported by an Interagency Agreement between FDA and NIEHS (FDA IAG No. 224-12-0003/NIEHS IAG No. AES12013).
Supplementary Material
ACKNOWLEDGMENT
The views expressed in this article do not necessarily represent those of the U.S. Food and Drug Administration.
REFERENCES
- Arita A., Shamy M. Y., Chervona Y., Clancy H. A., Sun H. A., Hall M. N., Qu Q., Gamble M. V., Costa M. (2012). The effect of exposure to carcinogenic metals on histone tail modifications and gene expression in human subjects. J. Trace Elem. Med. Biol. 26, 174–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balatoni C. E., Dawson D. W., Suh J., Sherman M. H., Sanders G., Hong J. S., Frank M. J., Malone C. S., Said J. W., Teitell M. A. (2009). Epigenetic silencing of Stk39 in B-cell lymphoma inhibits apoptosis from genotoxic stress. Am. J. Pathol. 175, 1653–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannasch P. (1986). Preneoplastic lesions as end point in carcinogenicity testing. I. Hepatic preneoplasia. Carcinogenesis 7, 689–695. [DOI] [PubMed] [Google Scholar]
- Chen T., Mally A., Ozden S., Chipman J. K. (2010). Low doses of the carcinogen furan alter cell cycle and apoptosis gene expression in rat liver independent of DNA methylation. Environ. Health Perspect. 118, 1597–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T., Williams T. D., Mally A., Hamberger C., Mirbahai L., Hickling K., Chipman J. K. (2012). Gene expression and epigenetic changes by furan in rat liver. Toxicology 292, 63–70. [DOI] [PubMed] [Google Scholar]
- Christman J. K., Sheikhnejad G., Dizik M., Abileah S., Wainfan E. (1993). Reversibility of changes in nucleic acid methylation and gene expression induced in rat liver by severe dietary methyl deficiency. Carcinogenesis 14, 551–557. [DOI] [PubMed] [Google Scholar]
- Clemente-Ruiz M., González-Prieto R., Prado F. (2011). Histone H3K56 acetylation, CAF1, and Rtt106 coordinate nucleosome assembly and stability of advancing replication forks. PLoS Genet. 7, e1002376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti A. D., Kobets T., Escudero-Lourdes C., Montgomery B., Tryndyak V., Beland F. A., Doerge D. R., Pogribny I. P. (2014). Dose- and time-dependent epigenetic changes in the livers of Fisher344 rats exposed to furan. Toxicol. Sci. 139, 371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dik S., Scheepers P. T., Godderis L. (2012). Effects of environmental stressors on histone modifications and their relevance to carcinogenesis: a systematic review. Crit. Rev. Toxicol. 42, 491–500. [DOI] [PubMed] [Google Scholar]
- Dillon N., Festenstein R. (2002). Unravelling heterochromatin: competition between positive and negative factors regulates accessibility. Trends Genet. 18, 252–258. [DOI] [PubMed] [Google Scholar]
- Ernst J., Kheradpour P., Mikkelsen T. S., Shoresh N., Ward L. D., Epstein C. B., Zhang X., Wang L., Issner R., Coyne M., et al. (2011). Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang H., Harris S. C., Su Z., Chen M., Qian F., Shi L., Perkins R., Tong W. (2009). ArrayTrack: an FDA and public genomic tool. Methods Mol. Biol. 563, 379–398. [DOI] [PubMed] [Google Scholar]
- Fetahu I. S., Höbaus J., Aggarwal A., Hummel D. M., Tennakoon S., Mesteri I., Baumgartner-Parzer S., Kállay E. (2014) Calcium-sensing receptor silencing in colorectal cancer is associated with promoter hypermethylation and loss of acetylation on histone 3. Int. J. Cancer 135, 2014–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransson-Steen R., Goldsworthy T. L., Kedderis G. L., Maronpot R. R. (1997). Furan-induced liver cell proliferation and apoptosis in female B6C3F1 mice. Toxicology 118, 195–204. [DOI] [PubMed] [Google Scholar]
- Grewal S. I. S., Moazed D. (2003). Heterochromatin and epigenetic control of gene expression. Science 301, 798–802. [DOI] [PubMed] [Google Scholar]
- Hickling K. C., Hitchcock J. M., Oreffo V., Mally A., Hammond T. G., Evans J. G., Chipman J. K. (2010). Evidence of oxidative stress and associated DNA damage, increased proliferative drive, and altered gene expression in rat liver produced by the cholangiocarcinogenic agent furan. Toxicol. Pathol. 38, 230–243. [DOI] [PubMed] [Google Scholar]
- International Agency for Research on Cancer (IARC). (1995). Furan. IARC Monogr. Eval. Carcinog. Risks Hum. 63, 393–407 (IARC, Lyon, France). [PMC free article] [PubMed] [Google Scholar]
- Jackson A. F., Williams A., Recio L., Waters M. D., Lambert I. B., Yauk C. L. (2014). Case study on the utility of hepatic global gene expression profiling in the risk assessment of the carcinogen furan. Toxicol. Appl. Pharmacol. 274, 63–77. [DOI] [PubMed] [Google Scholar]
- Kanehisa M., Goto S. (2000). KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kladde M. P., Simpson R. T. (1994). Positioned nucleosomes inhibit Dam methylation in vivo. Proc. Natl. Acad. Sci. U.S.A. 91, 1361–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstreuer N. C., Dix D. J., Houck K. A., Kavlock R. J., Knudsen T. B., Martin M. T., Paul K. B., Reif D. M., Crofton K. M., Hamilton K., et al. (2013). In vitro perturbations of targets in cancer hallmark processes predict rodent chemical carcinogenesis. Toxicol. Sci. 131, 40–55. [DOI] [PubMed] [Google Scholar]
- Liu Y., Hong Y., Zhao Y., Ismail T. M., Wong Y., Eu K. W. (2008). Histone H3 (lys-9) deacetylation is associated with transcriptional silencing of E-cadherin in colorectal cancer cell lines. Cancer Invest. 26, 575–582. [DOI] [PubMed] [Google Scholar]
- Luo R. X., Dean D. C. (1999). Chromatin remodeling and transcriptional regulation. J. Natl. Cancer Inst. 91, 1288–1294. [DOI] [PubMed] [Google Scholar]
- Maga J. A. (1979). Furans in foods. CRC Crit. Rev. Food Sci. Nutr. 11, 355–400. [DOI] [PubMed] [Google Scholar]
- Min L., Ji Y., Bakiri L., Qiu Z., Cen J., Chen X., Chen L., Scheuch H., Zheng H., Qin L., Zatloukal K., Hui L., Wagner E. F. (2012). Liver cancer initiation is controlled by AP-1 through SIRT6-dependent inhibition of survivin. Nat. Cell Biol. 14, 1203–1211. [DOI] [PubMed] [Google Scholar]
- Miranda T. B., Kelly T. K., Bouazoune K., Jones P. A. (2010). Methylation-sensitive single-molecule analysis of chromatin structure. Curr. Protoc. Mol. Biol. Chapter 21, Unit 21.17.1–21.17.16. [DOI] [PubMed] [Google Scholar]
- Moser G. J., Foley J., Burnett M., Goldsworthy T. L., Maronpot R. (2009). Furan-induced dose-response relationships for liver cytotoxicity, cell proliferation, and tumorigenicity (furan-induced liver tumorigenicity). Exp. Toxicol. Pathol. 61, 101–111. [DOI] [PubMed] [Google Scholar]
- Muzio G., Maggiora M., Paiuzzi E., Oraldi M., Canuto R. A. (2012). Aldehyde dehydrogenases and cell proliferation. Free Radic. Biol. Med. 52, 735–746. [DOI] [PubMed] [Google Scholar]
- National Toxicology Program (NTP). (1993). Toxicology and carcinogenesis studies of furan (CAS No. 110-00-9) in F344/N rats and B6C3F1 mice (gavage studies). Natl. Toxicol. Program Tech. Rep. Ser. 402, 1–77. [PubMed] [Google Scholar]
- Okabe H., Beppu T., Hayashi H., Horino K., Masuda T., Komori H., Ishikawa S., Watanabe M., Takamori H., Iyama K., Baba H. (2009). Hepatic stellate cells may relate to progression of intrahepatic cholangiocarcinoma. Ann. Surg. Oncol. 16, 2555–2564. [DOI] [PubMed] [Google Scholar]
- Perez Locas C., Yaylayan V. A. (2004). Origin and mechanistic pathways of formation of the parent furan: a food toxicant. J. Agric. Food Chem. 52, 6830–6836. [DOI] [PubMed] [Google Scholar]
- Pogribny I., Yi P., James S. J. (1999). A sensitive new method for rapid detection of abnormal methylation patterns in global DNA and within CpG islands. Biochem. Biophys. Res. Commun. 262, 624–628. [DOI] [PubMed] [Google Scholar]
- Pogribny I. P., Ross S. A., Wise C., Pogribna M., Jones E. A., Tryndyak V. P., James S. J., Dragan Y. P., Poirier L. A. (2006). Irreversible global DNA hypomethylation as a key step in hepatocarcinogenesis induced by dietary methyl deficiency. Mutat. Res. 593, 80–87. [DOI] [PubMed] [Google Scholar]
- Powell C. L., Swenberg J. A., Rusyn I. (2005). Expression of base excision DNA repair genes as a biomarker of oxidative DNA damage. Cancer Lett. 229, 1–11. [DOI] [PubMed] [Google Scholar]
- Recio L., Phillips S. L., Maynor T., Waters M., Jackson A. F., Yauk C. L. (2013). Differential expression of long noncoding RNAs in the livers of female B6C3F1 mice exposed to the carcinogen furan. Toxicol. Sci. 135, 369–379. [DOI] [PubMed] [Google Scholar]
- Rubinek T., Shulman M., Israeli S., Bose S., Avraham A., Zundelevich A., Evron E., Gal-Yam E. N., Kaufman B., Wolf I. (2012). Epigenetic silencing of the tumor suppressor klotho in human breast cancer. Breast Cancer Res. Treat. 133, 649–657. [DOI] [PubMed] [Google Scholar]
- Rusyn I., Asakura S., Pachkowski B., Bradford B. U., Denissenko M. F., Peters J. M., Holland S. M., Reddy J. K., Cunningham M. L., Swenberg J. A. (2004). Expression of base excision DNA repair genes is a sensitive biomarker for in vivo detection of chemical-induced chronic oxidative stress: identification of the molecular source of radicals responsible for DNA damage by peroxisome proliferators. Cancer Res. 64, 1050–1057. [DOI] [PubMed] [Google Scholar]
- Sadikovic B., Andrews J., Carter D., Robinson J., Rodenhiser D. I. (2008). Genome-wide H3K9 histone acetylation profiles are altered in benzopyrene-treated MCF-7 breast cancer cells. J. Biol. Chem. 283, 4051–4060. [DOI] [PubMed] [Google Scholar]
- Schmittgen T. D., Livak K. J. (2008). Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108. [DOI] [PubMed] [Google Scholar]
- Si J., Boumber Y. A., Shu J., Qin T., Ahmed S., He R., Jelinek J., Issa J. P. (2010). Chromatin remodeling is required for gene reactivation after decitabine-mediated DNA hypomethylation. Cancer Res. 70, 6968–6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirzaker C., Song J. Z., Davidson B., Clark S. J. (2004). Transcriptional gene silencing promotes DNA hypermethylation through a sequential change in chromatin modifications in cancer cells. Cancer Res. 64, 3871–3877. [DOI] [PubMed] [Google Scholar]
- Strauss W. M. (1989). Preparation of genomic DNA from mammalian tissue. In Current Protocols in Molecular Biology (Ausubel F. M., Brent R., Kingston R. E., Moore D. D., Seidman J. A., Smith J. A., Struhl K., Eds.), pp. 2.2.1–2.2.3 Wiley-Interscience, New York, NY. [DOI] [PubMed] [Google Scholar]
- Takai D., Jones P. A. (2003). The CpG island searcher: a new WWW resource. In Silico Biol. 3, 235–240. [PubMed] [Google Scholar]
- Tanaka A., Tanizawa H., Sriswasdi S., Iwasaki O., Chatterjee A. G., Speicher D. W., Lewin H. L., Noguchi E., Noma K. (2012). Epigenetic regulation of condensing-mediated genome organization during the cell cycle and upon DNA damage through histone H3 lysine 56. Mol. Cell. 48, 532–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner B. M. (1998). Histone acetylation as an epigenetic determinant of long-term transcriptional competence. Cell. Mol. Life Sci. 54, 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- US Food and Drug Administration (US-FDA). (2004). Furan in Food, Thermal Treatment. Available at: www.cfsan.fda.gov/lrd/fr040510.html (last accessed November, 2014). [Google Scholar]
- Vaissière T., Sawan C., Herceg Z. (2008). Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat. Res. 659, 40–48. [DOI] [PubMed] [Google Scholar]
- Wentzensen N., Bakkum-Gamez J. N., Killian J. K., Sampson J., Guido R., Glass A., Adams L., Luhn P., Brinton L. A., Rush B., et al. (2014). Discovery and validation of methylation markers for endometrial cancer. Int. J. Cancer 135, 1860–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.