

Abstract
Deletion of the LIMK1 gene is associated with Williams syndrome, a unique neurodevelopmental disorder characterized by severe defects in visuospatial cognition and long-term memory (LTM). However, whether LIMK1 contributes to these deficits remains elusive. Here, we show that LIMK1-knockout (LIMK1−/−) mice are drastically impaired in LTM but not short-term memory (STM). In addition, LIMK1−/− mice are selectively defective in late-phase long-term potentiation (L-LTP), a form of long-lasting synaptic plasticity specifically required for the formation of LTM. Furthermore, we show that LIMK1 interacts and regulates the activity of cyclic AMP response element-binding protein (CREB), an extensively studied transcriptional factor critical for LTM. Importantly, both L-LTP and LTM deficits in LIMK1−/− mice are rescued by increasing the activity of CREB. These results provide strong evidence that LIMK1 deletion is sufficient to lead to an LTM deficit and that this deficit is attributable to CREB hypofunction. Our study has identified a direct gene-phenotype link in mice and provides a potential strategy to restore LTM in patients with Williams syndrome through the enhancement of CREB activity in the adult brain.
INTRODUCTION
Williams syndrome (WS) is a neurodevelopmental disorder caused by the hemizygous deletion of a 1.5-million-bp segment of human chromosome 7q11.23 (1). Although individuals with WS have global cognitive impairments, they demonstrate a consistent pattern of strengths and weaknesses characterized by a relatively preserved concrete vocabulary and verbal short-term memory (STM) paired with dramatic deficits in visuospatial construction and long-term memory (LTM) (2–6). Another notable feature of WS patients is their hypersociability and enhanced empathy toward others, contrasting sharply with autism, which is characterized by impaired communication and social interaction (3, 4, 6). These apparently bidirectional phenotypes of WS and autism suggest that these two brain disorders may share common mechanisms, a notion that is also supported by human genetic studies (7). Thus, WS, with its clearly defined genetic abnormalities, provides an important window to understanding human cognition and behavior.
The hemizygous deletion in patients with WS spans 28 genes, but which of these genes are responsible for the cognitive deficits remains elusive (7–10). Human genetic studies, however, have suggested that the LIMK1 gene may be particularly important in relation to the visuospatial memory deficit (9–13). Recent in vitro and animal studies have also provided evidence that supports such a possibility (14–16). LIMK1 encodes a serine/threonine protein kinase whose main function is to regulate the actin cytoskeleton by phosphorylating and inactivating the actin depolymerization factor (ADF)/cofilin (17–24). Indeed, we have demonstrated that LIMK1-knockout (LIMK1−/−) mice exhibit reduced cofilin phosphorylation and altered actin networks (14, 25), but whether these actin aberrations are related to the synaptic and behavioral deficits seen in these mice remains unknown.
In addition to its role in actin regulation, LIMK1 is also known to interact with protein kinase C, neuregulin, and cyclic AMP response element-binding protein (CREB) (20, 22, 26). However, the functional significance of these protein interactions is unknown. The interaction with CREB is particularly interesting because CREB is a key transcription factor critically involved in the formation of LTM and hippocampal plasticity (27–30), both of which are profoundly impaired in patients with WS (3, 31–33). In this study, we provide evidence that LIMK1 regulates LTM and long-lasting synaptic plasticity through interacting with and activating CREB. Our study has identified a novel signaling pathway and provides a potential therapeutic strategy to improve LTM through enhancing the activity of CREB in patients with WS.
MATERIALS AND METHODS
Animals.
The generation and initial characterization of LIMK1−/− mice were described previously (14). The mutant mice were backcrossed with C57BL/6 mice for more than 8 generations to obtain a cogenic genetic background. The mice were housed under a standard 12-h light/12-h dark cycle condition. All the procedures used for this study were approved by the Animal Use Committee at The Hospital for Sick Children, Toronto, Ontario, Canada.
Electrophysiology.
The preparation and recovery of hippocampal slices were described previously (34). Briefly, a single slice was placed in a recording chamber and continuously perfused with oxygenated artificial cerebrospinal fluid (ACSF) containing 120 mM NaCl, 3.0 mM KCl, 1.2 mM MgSO4, 1.0 mM NaH2PO4, 26 mM NaHCO3, 2.0 mM CaCl2, and 1 mM 1 d-glucose saturated with 95% O2 and 5% CO2 at a rate of 2 to 2.5 ml/min. The Schaffer collateral pathway was stimulated, and the response of the CA1 synapse was recorded. The slices were typically stimulated at 60% of the maximum evoked field excitatory postsynaptic potential (fEPSP). For high-frequency stimulation (HFS)-induced late-phase long-term potentiation (L-LTP), four trains of high-frequency stimulations of 100 Hz lasting 1 s each were given at 5-min intervals. The response was then recorded for at least 3 h after the last HFS. For theta burst stimulation-induced L-LTP, three trains of 10 theta bursts with an intertrain interval of 10 s were used, and the response was then recorded for at least 3 h after the last stimulation. For the tetra HFS stimulation protocol, four trains of HFS were given at intervals of 20 s. L-LTP was verified by its sensitivity to the protein synthesis inhibitor anisomycin. Early-phase long-term potentiation (E-LTP) was induced by one train of HFS at 100 Hz lasting 1 s, and the response was recorded for 40 min following the HFS stimulation. For rescue experiments, slices were treated with either 200 μM cell-permeant cofilin peptides [for the control, NFVKIVGDSVAVGSAMRQIKIWFQNRRMKWKK; for phospho-serine 3 (pS3), MAS(p)GVAVSDGVIKVFNRQIKIWFQNRRMKWKK; for the serine 3 (S3) peptide, MASGVAVSDGVIKVFNRQIKIWFQNRRMKWKK; purity, 99%; Biomatik, Canada] or 50 μM forskolin for 1 h prior to electrophysiological recordings. All recording data were collected using pCLAMP (version 8 or 7) software (Axon Instruments). fEPSPs were measured by taking the slope of the rising phase between 5% and 60% of the peak response. The size of the response was normalized using the mean baseline response value as 100%. All data were statistically evaluated by using Student's t test.
Slice treatment and extraction of protein lysate.
Total protein lysates were extracted from whole brain, hippocampal slices, or dissected CA1 regions as described previously (35). Briefly, hippocampal slices were prepared in a manner identical to that used for electrophysiology. After recovery for 2 h in oxygenated ACSF and/or after appropriate stimulations (i.e., HFS) or drug treatment (30 μM N-methyl-d-aspartic acid [NMDA] and 3 μM glycine for 0 to 10 min or 200 μM S3, 200 μM pS3, 50 μM forskolin, or 50 μM rolipram for 30 min), the slices were lysed for protein extraction. For direct analysis of protein samples, 140 μl of lysis buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 5 mM EDTA, 0.5% NP-40, 1% Triton X-100, 0.5 mM phenylmethylsulfonyl fluoride) was used. For immunoprecipitation experiments, a buffer containing 1% Triton X-100, 137 mM NaCl, 5 mM EDTA, 5 mM EGTA, and 20 mM Tris was used. Both buffers contained a combination of inhibitors for proteases and protein phosphatases, as recommended by the supplier (Calbiochem). The slices were then mechanically broken down and allowed to be digested for at least 1 h on ice. The samples were then centrifuged at 12,500 rpm for 20 min, and the supernatants were then either used immediately or stored at −70°C.
HEK293 cell culture, immunoprecipitation, and Western blot analysis.
HEK293 cells at 20 to 40% confluence were transfected by use of the calcium phosphate protocol. The culture medium (Dulbecco modified Eagle medium) was changed on the next day, and cells were harvested 4 days later and resuspended in either lysis buffer or immunoprecipitation buffer. Standard protocols for immunoprecipitation and Western blot analysis were used (35). The extracted protein (600 to 800 μg) was mixed with 50 μl of a primary antibody, and the mixture was incubated overnight at 4°C. Fifty microliters of protein A-agarose beads was then added to the protein-antibody mix, and the mixture was incubated for a further 2 h at 4°C. The samples were then centrifuged, the supernatant was discarded, and the beads were washed three times with lysis buffer and resuspended in 70 μl of lysis buffer, 20 μl of 6× loading buffer, and 10 μl of 1 M dithiothreitol. Fifteen to 20 μl of the samples was separated on 10% SDS-polyacrylamide gels, electrotransferred to nitrocellulose membranes, and probed with one of the following primary antibodies: polyclonal anti-CREB (Cell Signaling), anti-phospho-CREB (anti-pCREB; Cell Signaling), anti-LIMK1 (Cell Signaling), anticofilin (Cytoskeleton), and antiphosphocofilin (Santa Cruz). The blots were then probed with appropriate secondary antibodies linked with horseradish peroxidase, and the proteins were visualized by enhanced chemiluminescence (GE Healthcare). The amounts of the proteins were estimated by measuring the density of the luminescence signals using AlphaEaseFC software per the manufacturer's instruction. The levels of the phosphorylated forms of CREB and cofilin were normalized to the density values of total CREB and cofilin, respectively. The data were then statistically evaluated using Student's t test.
Neuronal culture, immunostaining, and image analysis.
Hippocampal neuronal cultures were prepared from postnatal day 1 pups as previously described (30, 34). For immunostaining, the cells were fixed with ice-cold 4% paraformaldehyde–4% sucrose for 20 min and permeabilized with 0.25% Triton X-100 for 20 min. Cells were blocked with 3% donkey serum and 3% bovine serum albumin in phosphate-buffered saline (PBS) for 1 h and incubated with primary antibodies overnight at 4°C, followed by incubation with appropriate secondary antibodies (Invitrogen, Jackson ImmunoResearch) for 1 h at room temperature. After washing with PBS, coverslips were mounted using Dako mounting medium for image collection. The primary antibodies used included anti-LIMK1 (Cell Signaling Technology), anti-CREB (Cell Signaling Technology), and anti-microtubule-associated protein 2 (anti-MAP2) (Millipore). Images were obtained using a Zeiss LSM 510 laser scanning system and confocal microscope under a 63× (numerical aperture, 1.4) objective lens.
Fear conditioning test.
The previously described apparatus and procedures for fear conditioning training and testing (14, 35) were followed, but the following modifications were made in order to minimize the potential effect of enhanced short-term memory on the assessment of long-term memory in LIMK1−/− mice. Individual mice were placed in a conditioning chamber with controlled contextual cues and an electrified shock floor (Coulbourn Instruments). During the training session, the mice were allowed to acclimate to the chamber for 2 min. After the 2 min, a 30-s tone (85 dB, 18,000 Hz) was played and was paired with a mild foot shock (0.5 mA) in the last 2 s of the tone. The mouse was allowed to remain in the chamber for 1 more minute after the shock. Short-term memory was tested 2 h after the training. Since LIMK1−/− mice have enhanced early LTP and short-term memory, as shown previously (14), we tested long-term memory 48 h or 1 week after the training in order to avoid any potential effect of the enhanced short-term memory on long-term memory. This also applied to the water maze test (see below). During the contextual test, the mouse was placed back in the original training chamber for 5 min. During the cued test, the mouse was placed in a new chamber with different contextual cues, and after a 2-min acclimation, the same tone used for training was presented without shocks, and then freezing behavior was monitored for 2 min. Freezing is defined as a lack of any movement with the exception of respiration. For rescue experiments, the mice received an intraperitoneal injection of rolipram or vehicle (dimethyl sulfoxide [DMSO]) at 0.1 μmol/kg of body weight 1 h before the onset of the training, and the same training/testing procedures described above were then followed.
Morris water maze test.
For the Morris water maze test, the previously described procedures and water maze apparatus were used with some modifications (14). Briefly, the mice were handled for at least 3 days prior to the beginning of the experiments. All mice were subjected to training on the visible platform (10 cm in diameter raised 2 cm above the surface of the water), which consisted of 3 days of trials and 4 trials per day, before training on the hidden platform (10 cm in diameter submerged 0.5 to 1.0 cm under the water level) began. The hidden platform training also lasted for 3 days with 4 trials per day. All training started by placing an individual mouse in the water maze (diameter, 1.3 m) with the arranged spatial cues. The starting position of the mice was randomized. The mice were then left to swim until the platform was found or until 60 s elapsed. In cases where the platform was not found, the mice were directed and placed on the platform and allowed to stay on the platform for 5 s. The position of the mice was tracked and recorded continuously using a camera connected to a video tracking system (Noldus Information Technology). During probe trials (2 h, 48 h, or 1 week after the training), the platform was removed and mice were placed in the quadrant opposite that from the platform and allowed to swim for 60 s. The platform zone was defined as a zone of 20 cm in diameter, with the center being located at the position of the platform. The amount of time that the animals spent searching the platform zone was recorded and compared to the amount of time that the animals spent searching similar zones in the other quadrants. For rolipram rescue experiments, the drug or vehicle (DMSO) at 0.1 μmol/kg was administered by intraperitoneal or bilateral intrahippocampal injections 1 h before the training on each training day. Intrahippocampal injections were made using stainless steel guide cannulae inserted in the dorsal hippocampus (distances from bregma: anteroposterior (AP) = −4.3 mm, mediolateral (ML) = ±3.5 mm, dorsoventral (DV) = −2.0 mm). The cannulae were secured using dental cement.
RESULTS
LIMK1 deletion specifically impairs long-term memory.
Human genetic studies on WS patients with smaller chromosomal deletions suggest that the LIMK1 gene might be particularly important for the visuospatial cognition (9, 13). To test this hypothesis, we analyzed spatial learning and memory in LIMK1−/− mice. We focused our investigations on hippocampus-dependent memory and synaptic plasticity, as WS patients exhibit profound alterations in hippocampal function (11, 31, 32). We first used the Morris water maze paradigm, a well-established test for hippocampus-dependent spatial learning and memory in mice. As shown in Fig. 1A and B, LIMK1−/− mice performed equally as well as their wild-type (WT) littermates during the learning acquisition phase. Analysis of variance showed no differences in either latency (for WT mice, 29.5 ± 2.5 s, n = 14; for LIMK1−/− mice, 28.9 ± 2.8 s, n = 11; P > 0.05) or travel distance (for WT mice, 715.3 ± 9.7 cm, n = 14; for LIMK1−/− mice, 727.1 ± 11.1 cm, n = 11; P > 0.05) to locate the platform between LIMK1−/− and WT animals. In addition, the probe trial carried out 2 h after the training showed no differences in platform bias between genotypes (Fig. 1C) (for WT mice, 18.4 ± 2.6 s, n = 14; for LIMK1−/− mice, 23.9 ± 2.9 s, n = 11; P > 0.05). However, LIMK1−/− mice were severely impaired in platform searching during the probe trials carried out 48 h (Fig. 1D) (for WT mice, 16.3 ± 2.1 s, n = 14; for LIMK1−/− mice, 8.9 ± 2.9 s, n = 11; P < 0.05) and 1 week (Fig. 1E) (for WT mice, 14.8 ± 1.1 s, n = 14; for LIMK1−/− mice, 7.2 ± 1.2 s, n = 11; P < 0.05) after the training. In fact, the LIMK1−/− mice showed no bias to the platform zone in probe tests carried out both 48 h and 1 week after the training. Importantly, no differences in the swim speed were seen between LIMK1−/− and WT mice (Fig. 1F to K). These results indicate that LIMK1−/− mice are selectively impaired in LTM but exhibit intact learning acquisition and STM. To determine if this deficit also exists in a different learning paradigm, we conducted contextual fear conditioning, another form of hippocampus-dependent task. As shown in Fig. 2, although LIMK1−/− and WT mice performed equally well in both the training phase (Fig. 2A) (before shock, 8.3% ± 4.8% for LIMK1−/− mice [n = 13] and 15.1% ± 6.5% for WT mice [n = 15], P > 0.05; after shock, 49.6% ± 8.1% for LIMK1−/− mice and 55.4% ± 5.3% for WT mice, P > 0.05) and the contextual test carried out 2 h after the training (Fig. 2B) (for LIMK1−/− mice, 75.5% ± 10.0%, n = 13; for WT mice, 85.0% ± 3.5%, n = 15; P > 0.05), the LIMK1−/− mice had a diminished freezing response in the test carried out 48 h (Fig. 2C) (for LIMK1−/− mice, 42.8% ± 6.8%, n = 13; for WT mice, 71.5% ± 4.4%, n = 15; P < 0.05) and 1 week (Fig. 2D) (for LIMK1−/− mice, 27.9% ± 6.2%, n = 13; for WT mice, 46.6% ± 5.7%, n = 15; P < 0.05) after the training. These results together indicate that LIMK1 is specifically required for LTM but not STM.
FIG 1.

Selective deficits in LTM in LIMK1−/− mice in the water maze test. (A) Learning acquisition graph showing that both LIMK1−/− mice and their WT littermates were able to locate the hidden platform equally well during the 3 days of training. (B) Learning acquisition graph showing the distance traveled to locate the platform during the training period. (C) Results of the probe test carried out 2 h after the 3rd day of training showing that both LIMK1−/− mice and their WT littermates exhibited a significant bias toward the target zone, suggesting that STM was intact in LIMK1−/− mice. (D) Results of the probe test done at 48 h posttraining showing that WT mice but not LIMK1−/− mice exhibited a significant bias toward the target zone. (E) Results of a probe test done at 1 week posttraining showing that WT mice but not LIMK1−/− mice exhibited a significant bias toward the target zone, suggesting a deficit in LTM in LIMK1−/− mice. (F) During the probe test carried out 2 h after the training, neither LIMK1−/− mice nor WT mice spent a considerable amount of time searching around the perimeter of the water maze. (G, H) During the probe test carried out 48 h (G) and 1 week (H) after the training, LIMK1−/− mice spent significantly more time than the WT control mice searching around the perimeter of the water maze. (I to K) The swim speed recorded during the probe test at 2 h (I), 48 h (J), and 1 week (K) after the training showed no significant differences between LIMK1−/− mice and their WT littermates. Error bars represent SEMs. *, P < 0.05; n, number of animals; ns, no statistical significance.
FIG 2.

Impaired LTM but not STM in LIMK1−/− mice in the fear conditioning test. (A) During the training phase, LIMK1−/− mice exhibited a similar amount of freezing as their WT littermates before and after the foot shock. (B) In an STM contextual test carried out 2 h after the training, LIMK1−/− mice showed a similar amount of freezing as their WT littermates. (C, D) In LTM tests carried out 48 h (C) or 1 week (D) after the training, LIMK1−/− mice showed significantly reduced freezing compared to their WT littermates. Error bars represent SEMs. *, P < 0.05; n, number of animals; ns, no statistical significance.
LIMK1−/− mice exhibit selective deficits in L-LTP.
To investigate the synaptic basis of this selective LTM deficit in LIMK1−/− mice, we performed electrophysiological recordings in the CA1 region of the hippocampus. At this synapse, two distinct forms of LTP, early-phase LTP (E-LTP) and late-phase LTP (L-LTP), are known to exist and are thought to be important for STM and LTM, respectively (36, 37). Our previous studies indicated that E-LTP is enhanced in LIMK1−/− mice (14), which is consistent with an intact or slightly enhanced STM in these animals. Given the selective deficit in LTM in these mice, we turned our attention to L-LTP specifically. As shown in Fig. 3A, three trains of theta burst stimulation at 10-min intervals, a widely used protocol for eliciting protein synthesis-dependent L-LTP (38), induced a persistent enhancement of synaptic transmission that remained stable during the entire period of recording (>2 h) in WT mice, but this form of L-LTP was significantly reduced in LIMK1−/− mice (for WT mice, 166% ± 14%, n = 7; for LIMK1−/− mice, 132% ± 13%, n = 6; P < 0.05). To confirm this deficit, we employed two additional protocols to induce L-LTP, and in both cases, L-LTP was significantly reduced with an intact E-LTP, and the results are shown in Fig. 3B (for WT mice, 171% ± 11%, n = 6; for LIMK1−/− mice, 130% ± 10%, n = 7; P < 0.05) and Fig. 3C (for WT mice, 178% ± 10%, n = 7; for LIMK1−/− mice, 158% ± 14%, n = 6; P < 0.05). Consistent with previous results (14, 25), E-LTP was enhanced in LIMK1−/− mice (Fig. 3B) (for LIMK1−/− mice, 202% ± 6%, n = 4; for WT mice, 166% ± 5%, n = 4; P < 0.05). Baseline responses without LTP-inducing stimuli were stable for at least 4 h in both WT and LIMK1−/− mice (Fig. 3D). These results indicate that LIMK1 is specifically required for L-LTP, in accordance with its role in LTM.
FIG 3.

Selective L-LTP deficits in LIMK1−/− mice. (A) Plasticity induced by theta burst stimulation showing a significantly reduced L-LTP in LIMK1−/− mice compared to their WT littermates. (B) LTP induced by four trains of 100 Hz lasting 1 s each delivered at 5-min intertrain intervals showing that LIMK1−/− mice were significantly impaired in L-LTP compared to their WT littermates. Note that E-LTP was enhanced in LIMK1−/− mice. (C) Plasticity induced by four trains of 100 Hz lasting 1 s each delivered at 20-s intertrain intervals showing that LIMK1−/− mice were significantly impaired in L-LTP compared to their WT littermates. (D) Baseline recordings without LTP-inducing stimuli showing that synaptic responses were stable for up to 4 h in both WT and LIMK1−/− mice. Representative traces shown above the graphs were taken at the time points indicated by their respective numbers (1 and 2). Dashed lines indicate 100% and are shown for reference. Error bars represent SEMs. Arrows, L-LTP-inducing protocols; *, P < 0.05; n, number of animals.
LIMK1 regulation of L-LTP is independent of cofilin.
To elucidate the molecular mechanisms by which LIMK1 regulates LTM and L-LTP, we analyzed the actin binding protein cofilin. Earlier in vitro studies indicated that cofilin is the predominant substrate and the key mediator of LIMK1 (22, 39). Indeed, our previous in vivo studies showed that LIMK1−/− mice exhibited a decreased level of phosphorylated cofilin (i.e., increased cofilin activity), which, consequently, reduced the amounts of actin filaments (14, 25). Because actin reorganization is required for long-lasting synaptic plasticity (40), we reasoned that the L-LTP and/or LTM deficits in LIMK1−/− mice might be due to the elevated cofilin activity and altered actin. To test this hypothesis, we manipulated cofilin activity by using two TAT-conjugated, cell-permeant short peptides, pS3 and S3, which are known to increase and decrease cofilin phosphorylation in cultured neurons, respectively (41, 42). First, we confirmed that these peptides had the expected effects in hippocampal slices. As shown in Fig. 4A and C, the pS3 peptide increased cofilin phosphorylation to a similar level in both LIMK1−/− and WT mice (for WT mice with pS3 treatment, 1.94 ± 0.15, n = 7; for LIMK1−/− mice with pS3 treatment, 1.62 ± 0.13, n = 7; P > 0.05), whereas the S3 peptide decreased cofilin phosphorylation in WT mice but had no effect in LIMK1−/− mice (for WT mice with S3 treatment, 0.47 ± 0.07, n = 7, P < 0.05 compared to WT control mice; for LIMK1−/− mice with S3 treatment, 0.62 ± 0.06, n = 7, P > 0.05 compared to LIMK1−/− control mice). Neither the pS3 nor the S3 peptide had any effect on CREB activity (Fig. 4B and D). Then, we assessed the effects of these peptides on LTP. As shown in Fig. 4E and consistent with earlier results (Fig. 3B), E-LTP in LIMK1−/− mice was enhanced (for LIMK1−/− mice, 202% ± 6%, n = 4; for WT mice, 166% ± 5%, n = 4; P < 0.05). Treatment with the S3 peptide enhanced E-LTP in WT mice but had little effect on E-LTP in LIMK1−/− mice (Fig. 4F) (for LIMK1−/− mice, 238 ± 12, n = 4; for WT mice, 226 ± 10, n = 4; P < 0.05 compared to the results for WT control mice in Fig. 4E). Importantly, treatment with the pS3 peptide reduced the enhanced E-LTP in LIMK1−/− mice to the level found in WT mice (Fig. 4G) (for LIMK1−/− mice with pS3 treatment, 144% ± 8%; for WT mice with pS3 treatment, 139% ± 6%; P > 0.05). These results suggest that the enhanced E-LTP in LIMK1−/− mice was due to decreased cofilin phosphorylation or enhanced cofilin activity. However, despite its rescuing effect on cofilin phosphorylation and E-LTP, the pS3 peptide treatment did not improve L-LTP in LIMK1−/− mice (Fig. 4H) (for WT mice with pS3 treatment, 196% ± 11%, n = 4; for LIMK1−/− mice with pS3 treatment, 130% ± 8%, n = 4; P < 0.05). In fact, in LIMK1−/− mice L-LTP was indistinguishable with or without treatment with the peptide. These results suggest that altered cofilin/actin is not likely responsible for the L-LTP or LTM deficits in LIMK1−/− mice.
FIG 4.

Rescue of E-LTP but not L-LTP in LIMK1−/− mice by reduced cofilin activity. (A) Western blot analysis of total protein lysates prepared from hippocampal slices treated for 30 min with pS3 or S3 peptide or not treated (control) showing that pS3 increased the amount of phosphorylated cofilin (pCofilin) in WT and LIMK1−/− mice and that S3 decreased the amount of phosphorylated cofilin in WT but not LIMK1−/− mice. The level of total cofilin (A) and CREB or phosphorylated CREB (B) was not affected by either the pS3 or S3 peptide. (C) Summary graph of the Western blot shown in panel A showing a significant increase in the phosphorylated cofilin/cofilin relative density (RD) in slices from both WT and LIMK1−/− mice treated with pS3 peptide and a significant decrease in phosphorylated cofilin/cofilin in slices from WT but not LIMK1−/− mice treated with S3 peptide. Note that the basal phosphorylated cofilin/cofilin was significantly lower in slices from LIMK1−/− mice than those from WT mice. (D) Summary graph of the Western blot in panel B showing that neither the pS3 nor the S3 peptide had an effect on CREB or pCREB. (E) E-LTP induced by one train of HFS (100 Hz lasting 1 s) showing a significant enhancement of E-LTP in LIMK1−/− mice compared to that in WT mice. (F) E-LTP induced by one train of HFS (100 Hz lasting 1 s) in hippocampal slices treated with S3 peptide showing that the E-LTP in WT mice was increased to the level in LIMK1−/− mice. The S3 peptide had no effect on LIMK1−/− mice. (G) E-LTP induced by one train of HFS (100 Hz lasting 1 s) in hippocampal slices treated with pS3 peptides showing that enhanced E-LTP in LIMK1−/− mice was reduced to the level found in WT mice. The pS3 peptide had no effect on slices from WT mice. (H) L-LTP induced by four trains of HFS (100 Hz each lasting 1 s at 20-s intertrain intervals) showing that the pS3 peptide did not rescue the L-LTP deficit in LIMK1−/− mice. Traces above the graphs are representative responses taken from the indicated time points. Representative traces shown above the graphs were taken at the time points indicated by their respective numbers (1 and 2). Dashed lines indicate 100% and are shown for reference. Error bars represent SEMs. Arrows, LTP-inducing protocols; *, P < 0.05; ns, no significant difference; n, number of independent experiments (A to D) or number of animals (E to H).
LIMK1 regulates L-LTP and LTM via interaction with the transcription factor CREB.
We then turned our attention to CREB, an extensively studied transcriptional factor critical for the establishment of L-LTP and LTM (27, 28, 43, 44). An early in vitro study showed that LIMK1 can directly interact with and phosphorylate CREB (45), but whether this interaction occurs in the brain remains unknown. We therefore set out to test whether alterations in CREB function are related to the synaptic and memory deficits in LIMK1−/− mice. We first showed that in hippocampal CA1 neurons, LIMK1 is not only expressed in spines and dendrites but also highly expressed in the cell bodies, where it is colocalized with CREB (Fig. 5A). To determine whether LIMK1 interacts with CREB in the brain, we performed reciprocal immunoprecipitation experiments using hippocampal protein lysates and showed that LIMK1 and CREB exist in one immunoprotein complex (Fig. 5B). Consistent with the findings of a previous study (45), we also showed that in HEK293 cells cotransfected with LIMK1 and CREB, these two proteins could coimmunoprecipitate with each other (Fig. 5C). Additionally, we demonstrated that the LIMK1 and CREB interaction occurs in the nuclear fraction of hippocampal brain lysate (Fig. 5D).
FIG 5.

LIMK1 interacts with and regulates CREB. (A) (Top) WT mouse brain sectioned 10 μm thick coimmunostained with anti-CREB (red) and anti-LIMK1 (green) showing the colocalization of LIMK1 and CREB in the cell bodies of hippocampal CA1 neurons; (middle) cultured WT mouse hippocampal neuron (21 days in vitro) coimmunostained with anti-CREB (red) and anti-LIMK1 (green) showing coexpression of CREB and LIMK1 in the nucleus of the neuron; (bottom) LIMK1−/− mouse brain section 10 μm thick costained with anti-CREB (red) and anti-LIMK1 (green) showing no detectable LIMK1 immunoactivities. (B) Western blot analysis with anti-LIMK1 (top) or anti-CREB (bottom) of total brain lysates (input) and immunoprecipitates prepared by using anti-LIMK1 (LIMK1 IP), anti-CREB (CREB IP), or control IgG (IgG), showing that LIMK1 and CREB exist in one protein complex. Note that anti-LIMK1 pulled down both LIMK1 and CREB in WT mouse (+/+) but not in LIMK1−/− mouse (−/−) protein samples and that anti-CREB pulled down LIMK1 in WT mouse but not LIMK1−/− mouse samples. (C) LIMK1 and CREB interact in transfected HEK293 cells. Protein lysates were prepared from HEK293 cells cotransfected with either hemagglutinin (HA)-tagged or green fluorescent protein (GFP)-tagged LIMK1 and CREB, immunoprecipitated with hemagglutinin and green fluorescent protein, and probed with anti-LIMK1 (top) and anti-CREB (bottom), and the analyses show that LIMK1 and CREB exist in one protein complex. Inputs were WT and LIMK1−/− mouse brain lysates. (D) LIMK1 and CREB interact in hippocampal nuclear fraction. Total protein lysate (L), nuclear (N), and cytosolic (C) fractions were prepared from the hippocampus, immunoprecipitated with anti-LIMK1, and probed with anti-LIMK1 (top) and anti-CREB (bottom), and the analyses show that LIMK1 and CREB interact in the hippocampal nuclear fraction. Note that a small amount of LIMK1 was detected in the nuclear fraction, whereas a small amount of CREB was found in the cytosolic fraction. (E, F) Western blot analysis of hippocampal protein lysates (E) and summary graph (F) showing that NMDA treatment (30 μM NMDA plus 10 μM glycine) for 0 to 10 min (the times are indicated above the lanes in panel E) induced a significant increase in the amount of pCREB at serine 133 in slices from WT mice and that this NMDA-induced pCREB upregulation was significantly reduced in slices from LIMK1−/− mice at 10 min following treatment. The level of total CREB in slices from either WT or LIMK1−/− mice was not affected by NMDA treatment. (G, H) Western blot analysis of dissected hippocampal CA1 areas (G) and summary graph (H) showing that an L-LTP-inducing protocol (HFS) elicited a significant increase in pCREB in slices from WT mice and that this HFS-induced pCREB upregulation was significantly reduced in slices from LIMK1−/− mice. The level of total CREB was not affected by HFS in slices from either WT or LIMK1−/− mice. Error bars represent SEMs. *, P < 0.05; n, number of independent experiments.
To assess the functional consequence of LIMK1 deletion on CREB activity, we compared the amount of phosphorylated (or activated) CREB at serine 133. As shown in Fig. 5E and F, although the basal levels of phospho-CREB (pCREB) were the same between LIMK1−/− and WT mice, the level of CREB activation induced by NMDA treatment (Fig. 5E and F) (for LIMK1−/− mice, 0.99 ± 0.01, n = 9; for WT mice, 138.1 ± 0.04, n = 8; P < 0.05) or by a L-LTP-inducing protocol, high-frequency stimulation (HFS) (Fig. 5G and H) (for LIMK1−/− mice, 1.28 ± 0.07, n = 7; for WT mice, 1.58 ± 0.1, n = 7; P < 0.05), was significantly reduced. These results indicate that LIMK1 is a positive in vivo regulator for activity-dependent CREB activation and suggest that reduced CREB activation might be responsible for the L-LTP and LTM deficits in LIMK1−/− mice. To directly test this possibility, we performed rescue experiments using forskolin and rolipram, compounds commonly used to enhance CREB activity (46–48). First, we confirmed that treatment of hippocampal slices with forskolin or in vivo injection of rolipram increased CREB activity in both LIMK1−/− and WT mice without affecting cofilin activity (Fig. 6A to H). Then, we assessed the effects of the same treatments on L-LTP and LTM. As shown in Fig. 6I, forskolin treatment fully restored the L-LTP deficits found in the LIMK1−/− mice to the WT level (for LIMK1−/− mice, 202% ± 10%, n = 7; for WT mice, 204% ± 9%, n = 6; P > 0.05). A similar rescuing effect on LTP was obtained by rolipram (Fig. 6J). Importantly, rolipram treatment either by systematic injections (Fig. 7) or by direct bilateral hippocampal injections (Fig. 8) also rescued the LTM deficits of LIMK1−/− mice in the water maze test (e.g., the results of the searching time at 48 h after training in the probe test are shown in Fig. 7D) (for LIMK1−/− mice treated with DMSO, 8.9 ± 2.1 s, n = 7; for LIMK1−/− mice treated with rolipram, 13.9 ± 2.4 s, n = 7; P < 0.05; for WT mice treated with DMSO, 15.0 ± 2.1 s, n = 8; for WT mice treated with rolipram, 18.8 ± 2.8 s, n = 8; P > 0.05). It is important to note that the rolipram treatment had no effect on learning acquisition (Fig. 7A and B) or swim speed (Fig. 7E to H). We also examined the effect of rolipram on fear memory and found that it rescued the LTM deficit tested at 48 h after the training (Fig. 9C) (for LIMK1−/− mice treated with DMSO, 48.0% ± 2.4%, n = 7; for LIMK1−/− mice treated with rolipram, 70.9% ± 4.7%, n = 7; P < 0.05; for WT mice treated with DMSO, 79.0% ± 3.2%, n = 8; for WT treated with rolipram, 84.9% ± 2.7%, n = 7; P > 0.05). These results suggest that the reduced CREB activation in the hippocampus is likely responsible for the L-LTP and LTM deficits in LIMK1−/− mice.
FIG 6.

Rescue of L-LTP by enhanced CREB activity. (A) Western blot analysis of total protein lysates prepared from hippocampal slices treated with forskolin for 10 min (lanes 10) showing increased pCREB but not CREB compared to that in untreated slices (lanes 0) for both LIMK1−/− mice and their WT littermates. (B) Summary graph of the Western blots shown in panel A showing a significant increase in the levels of pCREB and CREB in hippocampal slices treated with forskolin (10 min) compared to the levels in untreated slices. (C) Western blot analysis of total brain protein lysates prepared from mice treated with intraperitoneal injections of rolipram (R) or the DMSO vehicle (V) showing that rolipram enhanced pCREB but not CREB levels in both LIMK1−/− mice and their WT littermates. (D) Summary graph of the Western blots shown in panel C showing a significant increase in the levels of pCREB and CREB in mice treated with rolipram compared to that in mice treated with DMSO. (E to H) Western blot experiments showing that neither forskolin (E, F) nor rolipram (G, H) had an effect on cofilin or phosphorylated cofilin. (I, J) Plasticity induced by HFS (four trains of 100 Hz lasting 1 s each delivered at 20-s intertrain intervals) showing that L-LTP in LIMK1−/− mice was completely rescued to the level in WT mice by coapplication of 50 μM forskolin (solid line) during HFS (arrows) (I) or rolipram injections (J). Representative traces shown above the graphs were taken at the time points indicated by their respective numbers (1 and 2). Dashed lines indicate 100% and are shown for reference. Error bars represent SEMs. *, P < 0.05; n, number of independent experiments (A to H) and number of animals (I and J).
FIG 7.

Rescue of spatial LTM in LIMK1−/− mice by enhanced CREB activity (A, B) Learning acquisition graphs of the results of water maze training showing no differences in latency (A) and travel distance (B) to locate the hidden platform between LIMK1−/− mice and their WT littermates treated with rolipram or vehicle (DMSO). (C) Results of a probe test carried out 2 h after the training showing that all groups showed a significant bias toward the target zone. (D) Results of a probe test carried out 48 h after the training showing that LIMK1−/− mice treated with rolipram but not those treated with DMSO displayed a significant bias toward the target zone. (E to H) Locomotor behavior of LIMK1−/− and WT mice treated with rolipram. (E) Results of a probe test carried out 2 h after the training showing that all mice treated with either vehicle (DMSO) or rolipram spent a similar amount of time searching around the perimeter of the water maze. (F) Results of a probe test carried out 48 h after the training showing that LIMK1−/− mice treated with DMSO spent significantly more time searching around the perimeter of the water maze than LIMK1−/− mice treated with rolipram or WT mice treated or not treated with rolipram. (G, H) Swim speeds in the probe test recorded 2 h (G) and 48 h (H) after training showing no differences between LIMK1−/− mice and their WT littermates treated with DMSO or rolipram. Error bars represent SEMs. *, P < 0.05; ns, no statistical significance; n, number of animals.
FIG 8.

Rescue of spatial LTM in LIMK1−/− mice by enhanced CREB activity with bilateral local hippocampal injections of rolipram (0.1 μmol/kg). (A) Learning acquisition graph of the results of water maze training showing no differences in the latency to locate the hidden platform between LIMK1−/− mice and their WT littermates injected with rolipram or vehicle (DMSO). (B) Results of a probe test carried out 2 h after training showing that all groups showed a significant bias toward the target zone. (C) Results of a probe test carried out 48 h after the training showing that LIMK1−/− mice treated with hippocampal local injections of rolipram displayed a significant bias toward the target zone.
FIG 9.
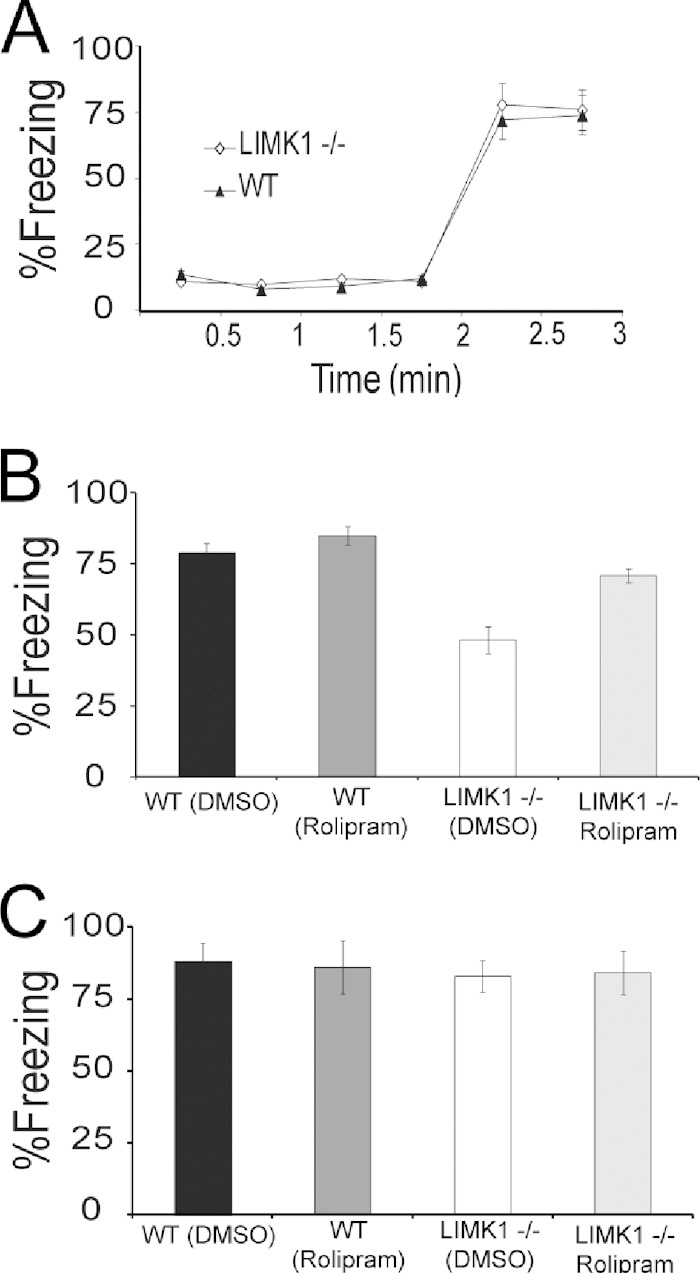
Rescue of LTM of fear in LIMK1−/− mice by rolipram treatment. (A) During the training phase, both LIMK1−/− mice and their WT littermates showed similar amounts of freezing before and after the foot shock. After the training, mice were randomly selected for rolipram or DMSO injections, and then STM and LTM were tested at 2 and 48 h posttraining, respectively. (B) All groups performed equally well in the STM test. (C) In the LTM test, LIMK1−/− mice treated with rolipram performed significantly better than LIMK1−/− mice treated with DMSO and similarly to their WT littermates. Error bars represent SEMs. *, P < 0.05; n, number of animals.
LIMK1+/− mice exhibit deficits in L-LTP and LTM.
Since WS is caused by a hemizygous deletion, we went on to examine whether LIMK1+/− heterozygous mice also display abnormalities in L-LTP and spatial memory. First, we assessed whether LIMK1+/− mice exhibited a deficit in L-LTP. As shown in Fig. 10A, L-LTP was significantly smaller in LIMK1+/− mice (for LIMK1+/− mice, 116% ± 6%, n = 5, P < 0.05 compared to an L-LTP of 171% ± 11% for WT mice in Fig. 3B). Importantly, similar to the findings for LIMK1−/− mice, the L-LTP deficit in LIMK1+/− mice was rescued by coapplication of 50 μM forskolin during HFS (for LIMK1+/− mice treated with forskolin, 148% ± 7%, n = 5; for LIMK1+/− mice not treated with forskolin, 116% ± 6%, n = 5; P < 0.05). Next, we examined LTM in LIMK1+/− mice. During the training phase of the Morris water maze test, the LIMK1+/− mice learned equally as well as the WT controls, as indicated by latency (Fig. 10B) (for WT mice, 29.5 ± 2.5 s, n = 10; for LIMK+/− mice, 28.9 ± 2.8 s, n = 10; P > 0.05) and travel distance (Fig. 10C) (for WT mice, 811.2 ± 13.2 cm, n = 10; for LIMK+/− mice, 791.1 ± 11.9 cm, n = 10; P > 0.05). The LIMK1+/− mice also performed equally as well as their WT littermates in the probe test carried out 2 h after the training (Fig. 10D) (for WT mice, 27.3 ± 1.0 s, n = 10; for LIMK+/− mice, 25.8 ± 1.3 s, n = 9; P > 0.05). However, just like the LIMK1−/− mice, the LIMK1+/− mice exhibited a specific deficit in LTM, as they showed a significantly reduced bias toward the platform zone in the probe test carried out 48 h after the training (Fig. 10E) (for WT mice, 19.7 ± 1.6 s, n = 10; for LIMK+/− mice, 14.8 ± 1.1 s, n = 9; P < 0.05). These results together indicate that LIMK1+/− mice behave similarly to the LIMK1−/− mutants and suggest that reduced CREB function is responsible for the L-LTP and LTM deficits in both LIMK1−/− and LIMK1+/− mice.
FIG 10.

Impaired L-LTP and LTM in LIMK1+/− mice. (A) LTP induced by HFS (four trains of 100 Hz lasting 1 s each delivered at 20-s intertrain intervals) showing that L-LTP was diminished in LIMK1+/− mice and that this impaired L-LTP was rescued by coapplication of 50 μM forskolin during HFS (arrows). (B, C) Learning acquisition graphs of the results of the water maze test showing no differences in either latency (B) or travel distance (C) to locate the hidden platform between LIMK1+/− mice and their WT littermates. (D) Results of a probe test carried out 2 h after the 3rd day of training showing that both LIMK1+/− mice and their WT littermates exhibited a significant bias toward the target zone, suggesting that STM was intact in LIMK1+/− mice. (E) Results of a probe test done at 48 h posttraining showing that LIMK1+/− mice exhibited a significantly reduced bias toward the target zone compared to their WT littermates. Representative traces shown above the graphs were taken at the time points indicated by their respective numbers (1 and 2). Dashed lines indicate 100% and are shown for reference. Error bars represent SEMs. *, P < 0.05; n, number of animals.
DISCUSSION
In this study, we have utilized a combination of behavioral tests, electrophysiological recordings, and biochemical analysis to demonstrate that LIMK1 mutant mice are selectively impaired in LTM and long-lasting synaptic plasticity. In addition, we have identified CREB to be the key molecular target by which LIMK1 regulates these processes. Our results suggest that LIMK1 deletion is sufficient to lead to an LTM deficit in patients with WS and that this deficit may be rescued by enhancing CREB function.
Although WS is a well-defined genetic disorder, the determination of its molecular and neurobiological mechanisms has been challenging. This is because the disorder is linked to a hemizygous deletion of 28 genes, and many of these genes remain poorly understood. In addition, WS is a multifaceted disease involving multiple organs and systems, of which the cognitive deficits are particularly complex. For example, patients with WS are severely impaired in visuospatial cognition and LTM, but their language skills and STM are relatively preserved. Additionally, certain domains of behavior, such as social interaction and empathy for others, are even enhanced in these patients (2–6, 31, 49). One strategy to unravel the complexity of WS is to use genetically altered mice that have defined alterations in one or more of these genes (16, 50). To this end, we have previously generated LIMK1−/− mice and shown that these mice are altered in learning and memory (14, 25). However, the mechanisms underlying these memory disorders remain a major unresolved question. In addition, the relevance of this mouse model to WS and whether its deficits could be rescued had yet to be determined. In this study, we have strived to address these key questions.
First, we show that both LIMK1−/− and LIMK+/− mice are severely impaired in LTM. In both the water maze and fear conditioning tests, this form of memory was significantly diminished or completely absent at 48 h and 1 week after training (Fig. 1 and 2). It is important to note that in this study we chose to use 48 h or longer posttraining for the LTM test because LIMK1−/− mice exhibit enhanced STM, which may affect accurate assessment of LTM in these mice if they had been tested 24 h after the training. In addition, we employed relatively weaker training procedures (i.e., a reduced foot shock in the fear test and a shorter training duration in the water maze test) to avoid potential differences in STM between the LIMK1−/− and WT mice. Indeed, using these protocols, we demonstrate that LIMK1−/− mice are impaired in LTM but show no significant changes in learning acquisition, shock sensitivity, STM, or swim speed, indicating that LIMK1 is particularly important for LTM. Because the LTM but not the STM of patients with WS is also defective (3, 4, 33), our results support the suggestion that LIMK1 mutant mice may be particularly useful to model the memory aspects of cognitive deficits in patients with WS. Our results also confirm that a LIMK1 hemizygous deletion is sufficient to cause LTM deficits (Fig. 10), suggesting a direct causal link between this gene and a specific phenotype associated with WS.
In accordance with the LTM deficit, LIMK1 mutant mice were also impaired in L-LTP. This L-LTP deficit was consistently found across different induction protocols (Fig. 3), indicating that LIMK1 is an essential component for this form of plasticity. It is important to note that E-LTP was not reduced or was even enhanced in the mutant mice (Fig. 3 and 4). These results suggest that LIMK1 is likely involved in the expression but not the induction of L-LTP. Indeed, both the NMDA and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, which play a larger role in E-LTP, appear to be unaffected in LIMK1−/− mice (14). It is important to emphasize that L-LTP is a well-studied form of long-lasting synaptic enhancement widely regarded to be a synaptic mechanism for LTM storage (37, 51). Thus, the finding that only L-LTP and not E-LTP is impaired in LIMK1 mouse models corroborates the selective role of LIMK1 in LTM.
What, then, is the molecular mechanism by which LIMK1 regulates LTM and L-LTP? Extensive previous studies have shown that LIMK1 is a potent regulator of the actin cytoskeleton through phosphorylating and inactivating the actin binding protein ADF/cofilin (23, 24). Specifically, we have shown before that LIMK1−/− mouse neurons have enhanced cofilin activity and more dynamic actin (14, 25). Given the critical role of actin in synaptic plasticity (40, 52), we had reasoned that LIMK1 might regulate L-LTP and LTM through stabilizing the actin cytoskeleton. Surprisingly, despite the normalization of E-LTP by manipulations of cofilin activity, L-LTP is not restored in LIMK1−/− mice (Fig. 4), indicating that while cofilin regulation by LIMK1 is important for E-LTP, it is not likely the mechanism used to regulate L-LTP.
To identify the molecular target of LIMK1 that mediates L-LTP and LTM, we turned our attention to CREB. CREB is known to be specifically required for the formation of LTM and L-LTP but not E-LTP or STM (27–30). In addition, CREB has been shown to interact with LIMK1 in a cultured cell line (45), but whether these two proteins interact in neurons was still unknown. We therefore hypothesized that LIMK1 might regulate LTM and L-LTP via regulation of CREB. First, we showed that LIMK1 is expressed and colocalizes with CREB in the nucleus of hippocampal neurons in both cultured neurons and brain sections (Fig. 5A). Second, we demonstrated that CREB and LIMK1 exist in one immunoprotein complex (Fig. 5B to D). Third, we show that the absence of LIMK1 results in reduced plasticity-dependent CREB activation, as indicated by reduced CREB phosphorylation in response to HFS or NMDA treatment (Fig. 5E to H). It is important to note that the basal level of CREB phosphorylation is not altered in LIMK1 mutant mice, suggesting that LIMK1 does not play a major role in basal regulation of CREB activity. Finally, we show that manipulating CREB but not cofilin is sufficient to restore L-LTP and LTM deficits in LIMK1 mutant mice (Fig. 6 to 9). Therefore, we have identified a novel mechanism activated by LIMK1 that is independent of the conventional actin regulation process but instead requires the transcriptional factor CREB. Exactly how synaptic activity leads to LIMK1-mediated CREB activation remains unknown, but it is possible that activation of synaptic LIMK1 in the spine induces its translocation into the nucleus, where it binds to and activates CREB. Alternatively, LIMK1 may regulate CREB via indirect pathways, including activation of protein kinase C and mitogen-activated protein kinase. Therefore, it would be important to distinguish these possibilities in future studies.
Conclusion.
In summary, the present results provide strong evidence that LIMK1 regulates hippocampal synaptic plasticity and LTM through two distinct mechanisms mediated by cofilin/actin and CREB, respectively. While cofilin/actin is critical for short-term synaptic plasticity, LIMK1-dependent CREB activation is essential for L-LTP and LTM. These results suggest that the LTM deficit in patients with WS is attributable to LIMK1 deletion and, consequently, reduced CREB function and that this deficit may be treatable by enhancing CREB signaling in the adult brain.
ACKNOWLEDGMENTS
This work is supported by grants from the Canadian Institutes of Health Research (MOP119421 and CCI117959 to Z.J.), the Canadian National Science and Engineering Research Council (RGPIN341498 to Z.J.), The Hospital for Sick Children Foundation (to Z.J.), and the China National Basic Research Program (973 Program 2012CB517903 to W.X. and Z.J.).
We are grateful to Shouping Zhang, Celeste Leung, and Yanghong Meng for technical support and Zikai Zhou for valuable discussion and suggestions during the study.
We declare no competing financial interests.
REFERENCES
- 1.Pober BR. 2010. Williams-Beuren syndrome. N Engl J Med 362:239–252. doi: 10.1056/NEJMra0903074. [DOI] [PubMed] [Google Scholar]
- 2.Mervis CB, Robinson BF, Bertrand J, Morris CA, Klein-Tasman BP, Armstrong SC. 2000. The Williams syndrome cognitive profile. Brain Cogn 44:604–628. doi: 10.1006/brcg.2000.1232. [DOI] [PubMed] [Google Scholar]
- 3.Meyer-Lindenberg A, Mervis CB, Berman KF. 2006. Neural mechanisms in Williams syndrome: a unique window to genetic influences on cognition and behaviour. Nat Rev Neurosci 7:380–393. doi: 10.1038/nrn1906. [DOI] [PubMed] [Google Scholar]
- 4.Bellugi U, Lichtenberger L, Jones W, Lai Z, St George M. 2000. I. The neurocognitive profile of Williams syndrome: a complex pattern of strengths and weaknesses. J Cogn Neurosci 12(Suppl 1):S7–S29. doi: 10.1162/089892900561959. [DOI] [PubMed] [Google Scholar]
- 5.Mervis CB, John AE. 2010. Cognitive and behavioral characteristics of children with Williams syndrome: implications for intervention approaches. Am J Med Genet C Semin Med Genet 154C:229–248. doi: 10.1002/ajmg.c.30263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mervis CB, Velleman SL. 2011. Children with Williams syndrome: language, cognitive, and behavioral characteristics and their implications for intervention. Perspect Lang Learn Educ 18:98–107. doi: 10.1044/lle18.3.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, Mason CE, Bilguvar K, Celestino-Soper PBS, Choi M, Crawford EL, Davis L, Wright NR, Dhodapkar RM, DiCola M, DiLullo NM, Fernandez TV, Fielding-Singh V, Fishman DO, Frahm S, Garagaloyan R, Goh GS, Kammela S, Klei L, Lowe JK, Lund SC, McGrew AD, Meyer KA, Moffat WJ, Murdoch JD, O'Roak BJ, Ober GT, Pottenger RS, Raubeson MJ, Song Y, Wang Q, Yaspan BL, Yu TW, Yurkiewicz IR, Beaudet AL, Cantor RM, Curland M, Grice DE, Günel M, Lifton RP, Mane SM, et al. . 2011. Multiple recurrent de novo CNVs, including duplications of the 7q1123 Williams syndrome region, are strongly associated with autism. Neuron 70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robinson WP, Waslynka J, Bernasconi F, Wang M, Clark S, Kotzot D, Schinzel A. 1996. Delineation of 7q11.2 deletions associated with Williams-Beuren syndrome and mapping of a repetitive sequence to within and to either side of the common deletion. Genomics 34:17–23. doi: 10.1006/geno.1996.0237. [DOI] [PubMed] [Google Scholar]
- 9.Martindale DW, Wilson MD, Wang D, Burke RD, Chen X, Duronio V, Koop BF. 2000. Comparative genomic sequence analysis of the Williams syndrome region (LIMK1-RFC2) of human chromosome 7q11.23. Mamm Genome 11:890–898. doi: 10.1007/s003350010166. [DOI] [PubMed] [Google Scholar]
- 10.Morris CA, Mervis CB, Osborne LR. 2011. Frequency of the 7q11.23 inversion polymorphism in transmitting parents of children with Williams syndrome and in the general population does not differ between North America and Europe. Mol Cytogenet 4:7. doi: 10.1186/1755-8166-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frangiskakis JM, Ewart AK, Morris CA, Mervis CB, Bertrand J, Robinson BF, Klein BP, Ensing GJ, Everett LA, Green ED, Pröschel C, Gutowski NJ, Noble M, Atkinson DL, Odelberg SJ, Keating MT. 1996. LIM-kinase1 hemizygosity implicated in impaired visuospatial constructive cognition. Cell 86:59–69. doi: 10.1016/S0092-8674(00)80077-X. [DOI] [PubMed] [Google Scholar]
- 12.Gray V, Karmiloff-Smith A, Funnell E, Tassabehji M. 2006. In-depth analysis of spatial cognition in Williams syndrome: a critical assessment of the role of the LIMK1 gene. Neuropsychologia 44:679–685. doi: 10.1016/j.neuropsychologia.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 13.Morris CA, Mervis CB, Hobart HH, Gregg RG, Bertrand J, Ensing GJ, Sommer A, Moore CA, Hopkin RJ, Spallone PA, Keating MT, Osborne L, Kimberley KW, Stock AD. 2003. GTF2I hemizygosity implicated in mental retardation in Williams syndrome: genotype-phenotype analysis of five families with deletions in the Williams syndrome region. Am J Med Genet A 123A:45–59. doi: 10.1002/ajmg.a.20496. [DOI] [PubMed] [Google Scholar]
- 14.Meng Y, Zhang Y, Tregoubov V, Janus C, Cruz L, Jackson M, Lu WY, MacDonald JF, Wang JY, Falls DL, Jia Z. 2002. Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron 35:121–133. doi: 10.1016/S0896-6273(02)00758-4. [DOI] [PubMed] [Google Scholar]
- 15.Jia Z, Todorovski Z, Meng Y, Asrar S, Wang L.-Y. 2009. LIM kinase and actin regulation of spines, p 467–472. In Squire LR. (ed), Encyclopedia of neuroscience. Academic Press, Oxford, United Kingdom. [Google Scholar]
- 16.Osbrone LR, Jia Z. 2013. Mouse models of Williams syndrome. In Handbook of molecular genetic techniques for brain and behavior research. Elsevier, Amsterdam, Netherlands. [Google Scholar]
- 17.Bernard O, Ganiatsas S, Kannourakis G, Dringen R. 1994. Kiz-1, a protein with LIM zinc finger and kinase domains, is expressed mainly in neurons. Cell Growth Differ 5:1159–1171. [PubMed] [Google Scholar]
- 18.Mizuno K, Okano I, Ohashi K, Nunoue K, Kuma K, Miyata T, Nakamura T. 1994. Identification of a human cDNA encoding a novel protein kinase with two repeats of the LIM/double zinc finger motif. Oncogene 9:1605–1612. [PubMed] [Google Scholar]
- 19.Pröschel C, Blouin MJ, Gutowski NJ, Ludwig R, Noble M. 1995. Limk1 is predominantly expressed in neural tissues and phosphorylates serine, threonine and tyrosine residues in vitro. Oncogene 11:1271–1281. [PubMed] [Google Scholar]
- 20.Wang JY, Frenzel KE, Wen D, Falls DL. 1998. Transmembrane neuregulins interact with LIM kinase 1, a cytoplasmic protein kinase implicated in development of visuospatial cognition. J Biol Chem 273:20525–20534. doi: 10.1074/jbc.273.32.20525. [DOI] [PubMed] [Google Scholar]
- 21.Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P. 1998. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 393:805–809. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- 22.Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, Nishida E, Mizuno K. 1998. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 393:809–812. doi: 10.1038/31735. [DOI] [PubMed] [Google Scholar]
- 23.Bamburg JR. 1999. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu Rev Cell Dev Biol 15:185–230. doi: 10.1146/annurev.cellbio.15.1.185. [DOI] [PubMed] [Google Scholar]
- 24.Bernard O. 2007. Lim kinases, regulators of actin dynamics. Int J Biochem Cell Biol 39:1071–1076. doi: 10.1016/j.biocel.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 25.Meng Y, Takahashi H, Meng J, Zhang Y, Lu G, Asrar S, Nakamura T, Jia Z. 2004. Regulation of ADF/cofilin phosphorylation and synaptic function by LIM-kinase. Neuropharmacology 47:746–754. doi: 10.1016/j.neuropharm.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 26.Kuroda S, Tokunaga C, Kiyohara Y, Higuchi O, Konishi H, Mizuno K, Gill GN, Kikkawa U. 1996. Protein-protein interaction of zinc finger LIM domains with protein kinase C. J Biol Chem 271:31029–31032. doi: 10.1074/jbc.271.49.31029. [DOI] [PubMed] [Google Scholar]
- 27.Yin JC, Tully T. 1996. CREB and the formation of long-term memory. Curr Opin Neurobiol 6:264–268. doi: 10.1016/S0959-4388(96)80082-1. [DOI] [PubMed] [Google Scholar]
- 28.Pittenger C, Kandel E. 1998. A genetic switch for long-term memory. C R Acad Sci III 321:91–96. doi: 10.1016/S0764-4469(97)89807-1. [DOI] [PubMed] [Google Scholar]
- 29.Huang Y-Y, Kandel ER. 2005. Theta frequency stimulation induces a local form of late phase LTP in the CA1 region of the hippocampus. Learn Mem 12:587–593. doi: 10.1101/lm.98905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou Y, Won J, Karlsson MG, Zhou M, Rogerson T, Balaji J, Neve R, Poirazi P, Silva AJ. 2009. CREB regulates excitability and the allocation of memory to subsets of neurons in the amygdala. Nat Neurosci 12:1438–1443. doi: 10.1038/nn.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meyer-Lindenberg A, Mervis CB, Sarpal D, Koch P, Steele S, Kohn P, Marenco S, Morris CA, Das S, Kippenhan S, Mattay VS, Weinberger DR, Berman KF. 2005. Functional, structural, and metabolic abnormalities of the hippocampal formation in Williams syndrome. J Clin Invest 115:1888–1895. doi: 10.1172/JCI24892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jarrold C, Baddeley AD, Phillips C. 2007. Long-term memory for verbal and visual information in Down syndrome and Williams syndrome: performance on the Doors and People test. Cortex 43:233–247. doi: 10.1016/S0010-9452(08)70478-7. [DOI] [PubMed] [Google Scholar]
- 33.Rhodes SM, Riby DM, Fraser E, Campbell LE. 2011. The extent of working memory deficits associated with Williams syndrome: exploration of verbal and spatial domains and executively controlled processes. Brain Cogn 77:208–214. doi: 10.1016/j.bandc.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 34.Asrar S, Zhou Z, Ren W, Jia Z. 2009. Ca2+ permeable AMPA receptor induced long-term potentiation requires PI3/MAP kinases but not Ca/CaM-dependent kinase II. PLoS One 4:e4339. doi: 10.1371/journal.pone.0004339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meng J, Meng Y, Hanna A, Janus C, Jia Z. 2005. Abnormal long-lasting synaptic plasticity and cognition in mice lacking the mental retardation gene Pak3. J Neurosci 25:6641–6650. doi: 10.1523/JNEUROSCI.0028-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bliss TV, Collingridge GL. 1993. A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 37.Mayford M. 2012. Navigating uncertain waters. Nat Neurosci 15:1056–1057. doi: 10.1038/nn.3174. [DOI] [PubMed] [Google Scholar]
- 38.Alarcon JM, Hodgman R, Theis M, Huang Y-S, Kandel ER, Richter JD. 2004. Selective modulation of some forms of Schaffer collateral-CA1 synaptic plasticity in mice with a disruption of the CPEB-1. Gene Learn Mem 11:318–327. doi: 10.1101/lm.72704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang TY, DerMardirossian C, Bokoch GM. 2006. Cofilin phosphatases and regulation of actin dynamics. Curr Opin Cell Biol 18:26–31. doi: 10.1016/j.ceb.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 40.Cingolani LA, Thalhammer A, Yu LMY, Catalano M, Ramos T, Colicos MA, Goda Y. 2008. Activity-dependent regulation of synaptic AMPA receptor composition and abundance by beta3 integrins. Neuron 58:749–762. doi: 10.1016/j.neuron.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang W, Zhou Z, Asrar S, Henkelman M, Xie W, Jia Z. 2011. p21-activated kinases 1 and 3 control brain size through coordinating neuronal complexity and synaptic properties. Mol Cell Biol 31:388–403. doi: 10.1128/MCB.00969-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou Z, Hu J, Passafaro M, Xie W, Jia Z. 2011. GluA2 (GluR2) regulates metabotropic glutamate receptor-dependent long-term depression through N-cadherin-dependent and cofilin-mediated actin reorganization. J Neurosci 31:819–833. doi: 10.1523/JNEUROSCI.3869-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gass P, Wolfer DP, Balschun D, Rudolph D, Frey U, Lipp H-P, Schütz G. 1998. Deficits in memory tasks of mice with CREB mutations depend on gene dosage. Learn Mem 5:274–288. [PMC free article] [PubMed] [Google Scholar]
- 44.Carlezon WA Jr, Duman RS, Nestler EJ. 2005. The many faces of CREB. Trends Neurosci 28:436–445. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 45.Yang EJ, Yoon J-H, Min DS, Chung KC. 2004. LIM kinase 1 activates cAMP-responsive element-binding protein during the neuronal differentiation of immortalized hippocampal progenitor cells. J Biol Chem 279:8903–8910. doi: 10.1074/jbc.M311913200. [DOI] [PubMed] [Google Scholar]
- 46.Asanuma M, Nishibayashi S, Iwata E, Kondo Y, Nakanishi T, Vargas MG, Ogawa N. 1996. Alterations of cAMP response element-binding activity in the aged rat brain in response to administration of rolipram, a cAMP-specific phosphodiesterase inhibitor. Brain Res Mol Brain Res 41:210–215. doi: 10.1016/0169-328X(96)00098-8. [DOI] [PubMed] [Google Scholar]
- 47.Monti B, Berteotti C, Contestabile A. 2006. Subchronic rolipram delivery activates hippocampal CREB and Arc, enhances retention and slows down extinction of conditioned fear. Neuropsychopharmacology 31:278–286. doi: 10.1038/sj.npp.1300813. [DOI] [PubMed] [Google Scholar]
- 48.Otmakhov N, Khibnik L, Otmakhova N, Carpenter S, Riahi S, Asrican B, Lisman J. 2004. Forskolin-induced LTP in the CA1 hippocampal region is NMDA receptor dependent. J Neurophysiol 91:1955–1962. doi: 10.1152/jn.00941.2003. [DOI] [PubMed] [Google Scholar]
- 49.Stinton C, Elison S, Howlin P. 2010. Mental health problems in adults with Williams syndrome. Am J Intellect Dev Disabil 115:3–18. doi: 10.1352/1944-7558-115.1.3. [DOI] [PubMed] [Google Scholar]
- 50.Li HH, Roy M, Kuscuoglu U, Spencer CM, Halm B, Harrison KC, Bayle JH, Splendore A, Ding F, Meltzer LA, Wright E, Paylor R, Deisseroth K, Francke U. 2009. Induced chromosome deletions cause hypersociability and other features of Williams-Beuren syndrome in mice. EMBO Mol Med 1:50–65. doi: 10.1002/emmm.200900003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pang PT, Lu B. 2004. Regulation of late-phase LTP and long-term memory in normal and aging hippocampus: role of secreted proteins tPA and BDNF. Ageing Res Rev 3:407–430. doi: 10.1016/j.arr.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 52.Lamprecht R, LeDoux J. 2004. Structural plasticity and memory. Nat Rev Neurosci 5:45–54. doi: 10.1038/nrn1301. [DOI] [PubMed] [Google Scholar]