

Abstract
Yes-associated protein (YAP) is an effector of the Hippo tumor suppressor pathway. The functional significance of YAP in prostate cancer has remained elusive. In this study, we first show that enhanced expression of YAP is able to transform immortalized prostate epithelial cells and promote migration and invasion in both immortalized and cancerous prostate cells. We found that YAP mRNA was upregulated in androgen-insensitive prostate cancer cells (LNCaP-C81 and LNCaP-C4-2 cells) compared to the level in androgen-sensitive LNCaP cells. Importantly, ectopic expression of YAP activated androgen receptor signaling and was sufficient to promote LNCaP cells from an androgen-sensitive state to an androgen-insensitive state in vitro, and YAP conferred castration resistance in vivo. Accordingly, YAP knockdown greatly reduced the rates of migration and invasion of LNCaP-C4-2 cells and under androgen deprivation conditions largely blocked cell division in LNCaP-C4-2 cells. Mechanistically, we found that extracellular signal-regulated kinase–ribosomal s6 kinase signaling was downstream of YAP for cell survival, migration, and invasion in androgen-insensitive cells. Finally, immunohistochemistry showed significant upregulation and hyperactivation of YAP in castration-resistant prostate tumors compared to their levels in hormone-responsive prostate tumors. Together, our results identify YAP to be a novel regulator in prostate cancer cell motility, invasion, and castration-resistant growth and as a potential therapeutic target for metastatic castration-resistant prostate cancer (CRPC).
INTRODUCTION
Prostate cancer is the most common malignancy and the second leading cause of cancer-related mortality among men in the United States (1). Although androgen deprivation therapy (through medical or surgical castration) is highly effective for advanced prostate cancer (1, 2), the majority of patients eventually develop resistance and progress to castration-resistant prostate cancer (CRPC). Unfortunately, most cases of CRPC are currently incurable (1). The cause of castration resistance is still not completely known. It is expected that understanding the molecular mechanisms and identifying the molecular pathways underlying the acquisition of castration resistance in prostate cancer are critical for the design of therapeutic strategies and may lead to the discovery of novel targets.
The Hippo signaling pathway, originally defined by fly geneticists, plays an important role in tumorigenesis by regulating cell proliferation and apoptosis (3–7). In mammals, protein kinases Mst1/2 (mammalian sterile 20-like 1 and 2) and Lats1/2 (large tumor suppressor 1 and 2) and the adaptor proteins WW45 (WW domain-containing protein) and Mob1 (Mps one binder 1) are the Hippo core components. These proteins form complexes to regulate their activity mainly through phosphorylation. The Hippo core is tumor suppressive and exerts its function by phosphorylating and inactivating YAP (Yes-associated protein) and its paralog, TAZ (transcriptional coactivator with a PDZ-binding motif).
Recent genetic mouse models and studies with cancer patients firmly demonstrated the critical roles of Hippo-YAP signaling in cancer development. For example, Mst1/2 and WW45 suppress the development of hepatocellular carcinoma in mice (8–11). Accordingly, the downregulation of Mst1/2 and Lats1/2 by promoter hypermethylation is often observed in various types of human cancer (12–15). Although mutations in the Hippo pathway are rare, mutation or deletion of Lats2 is significant in malignant mesothelioma (16). Furthermore, the oncoprotein YAP has been implicated in promoting the formation of several types of tumors, such as liver and skin tumors and rhabdomyosarcoma (17–21). As expected, overexpression or hyperactivation (nuclear localization) of YAP is frequently detected in several human malignancies, including liver, ovarian, breast, lung, and pancreatic cancer (18–20, 22–28). In addition to the role of Hippo-YAP signaling in cancer development, recent studies also implicate YAP in the metastatic progression of breast cancer and melanoma (29).
Accumulated evidence has shown that the Hippo-YAP pathway activity is regulated by many cues and factors, including cell adhesion, cell polarity, contact inhibition/cell density, and cytoskeleton dynamics/mechanical forces (6, 30). Recent studies have also demonstrated that YAP/TAZ activity can be regulated independently of Hippo signaling and YAP/TAZ cross talk with many other canonical signaling pathways, including Wnt/β-catenin (31–37), transforming growth factor β/Smad (38–40), and Ras–extracellular signal-regulated kinase (ERK) (28, 41, 42), in the regulation of cancer cell proliferation, survival, and tumorigenesis. Despite the role of YAP signaling in mediating these physiological processes, however, the biological significance of YAP in prostate cancer has not been previously defined.
Here, we explored the functional role of YAP in prostate cancer cell motility, invasion, and castration-resistant growth and determined the clinical relevance of YAP in CRPC. Our data identify YAP to be a critical regulator in prostate cancer, especially for CRPC, providing an alternative mechanism underlying the development of castration resistance of prostate tumor cells.
MATERIALS AND METHODS
Expression constructs.
The pcDNA-YAP expression construct has been described previously (18). Retroviral wild-type YAP and YAP mutant constructs have been described previously (43). The lentiviral YAP short hairpin RNA (shRNA) constructs and packaging vectors (psPAX2 and pMD2.G) were from Addgene (Cambridge, MA). Point mutations were generated by use of a QuikChange site-directed PCR mutagenesis kit (Stratagene, La Jolla, CA) and verified by sequencing.
Cell culture, transfection, virus packaging, and infection.
The HEK293T, HEK293GP, RWPE-1, and LNCaP cell lines and related media and supplements were purchased from the American Type Culture Collection (ATCC; Manassas, VA), and the cell lines were cultured following ATCC's instructions. The cell lines were authenticated at ATCC and were used at low (<25) passage numbers. The LNCaP-C4-2 and LNCaP-C81 sublines have been described previously (44–46). The Attractene and HiPerFect reagents (Qiagen, Valencia, CA) were used for transient overexpression and small interfering RNA (siRNA) transfections, respectively, following the manufacturer's instructions. R1881 was purchased from PerkinElmer (Waltham, MA). YAP-specific siRNA oligonucleotides were synthesized by GenePharma (Shanghai, China) on the basis of the following target sequences: 5′-CAGGTGATACTATCAACCAAA-3′ (YAP#1) and 5′-GACCAATAGCTCAGATCCTTT (YAP#2). Ectopic expression of empty vector, YAP, or the YAP S127A mutant (YAP-S127A) in the RWPE-1 and LNCaP cell lines was achieved by a retrovirus-mediated approach as described previously (47). The transduced cells were then selected with 800 μg/ml of neomycin (at 48 h postinfection) to establish cells stably expressing YAP or YAP-S127A. YAP downregulation in LNCaP-C4-2 cells was obtained by lentivirus-mediated YAP shRNA expression (48). Briefly, the YAP shRNA-expressing plasmid (2.5 μg) was cotransfected with the psPAX2 (2.0 μg) and pMD2.G (1.0 μg) genes into the virus-packaging cell line HEK293T. The medium was replaced, and HEPES (10 mM) and sodium butyrate (10 mM) were added at 16 h posttransfection. At 48 h posttransfection, the resulting lentiviral supernatant was collected and further filtered through a 0.45-μm-pore-size filter and used to infect cells in the presence of 10 μg/ml of Polybrene (Millipore, Billerica, MA). The transduced cells were then selected with puromycin (2 μg/ml) to establish cell lines in which YAP expression was stably knocked down.
Quantitative real time-PCR.
Total RNA isolation, RNA reverse transcription, and quantitative real time-PCR were done as described previously (47). Other primer sequences were as follows: for TEAD1 (TEA domain-containing protein 1), CTTGAATGTGCAATGAAGCG (forward [F]) and CGAAGTTTGCCTCGGACTC (reverse [R]); for TEAD2, CTCACTCCGTAGAAGCCACC (F) and TGCCTTCTTCCTGGTCAAGT (R); for TEAD3, GCACCTTCTTCCGAGCTAGA (F) and TACGGCCGAAATGAGTTGAT (R); for TEAD4, GCTCCACTCGTTGGAGGTAA (F) and CTTAGCGCACCCATCCC (R); for YAP, ACGTTCATCTGGGACAGCAT (F) and GTTGGGAGATGGCAAAGACA (R); for TAZ, ATTCATCGCCTTCCTAGGGT (F) and GGCTGGGAGATGACCTTCAC (R); for connective tissue growth factor (CTGF), TTGGCAGGCTGATTTCTAGG (F) and GGTGCAAACATGTAACTTTTGG (R); for ITGB2 (integrin beta 2), ACTCCTGAGAGAGGACGCAC (F) and CAGGGCAGACTGGTAGCAA (R); for ANKRD1 (ankyrin repeat domain 1), GTGTAGCACCAGATCCATCG (F) and CGGTGAGACTGAACCGCTAT (R); for Cyr61 (cysteine-rich angiogenic inducer 61), CCCGTTTTGGTAGATTCTGG (F) and GCTGGAATGCAACTTCGG (R); for SOX4 (SRY [sex-determining region Y] box 4), AATGTATGTTTCCCCCTCCC (F) and TCGCTGTCGGGTCTCTAGTT (R); for survivin, CGAGGCTGGCTTCATCCACT (F) and ACGGCGCACTTTCTTCGCA (R); for prostate-specific antigen (PSA), ATATCGTAGAGCGGGTGTGG (F) and TCCTCACAGCTGCCCACT (R); for NKX3.1 (NK3 homeobox 1), CAGATAAGACCCCAAGTGCC (F) and CAGAGCCAGAGCCAGAGG (R); for KLK2 (kallikrein-2), TGTCTTCAGGCTCAAACAGG (F) and GTACAGTCATGGATGGGCAC (R); and for PGC-1 (peroxisome proliferator-activated receptor gamma coactivator 1-alpha), CTGCTAGCAAGTTTGCCTCA (F) and AGTGGTGCAGTGACCAATCA (R).
Cell fractionation assay.
Cell fractionation assays were done with NE-PER nuclear and cytoplasmic extraction reagents following the manufacturer's instructions (Thermo Scientific/Pierce, Rockford, IL).
Antibodies and Western blot analysis.
YAP antibodies from Cell Signaling Technology (catalog number 4912; Danvers, MA) and Abcam (catalog number 52771; Cambridge, MA) were used for Western blotting throughout the study. Anti-β-actin, anti-androgen receptor (anti-AR), anti-ERK1/2, anti-Akt, anti-glycogen synthase kinase 3β (anti-GSK3β), anti-β-catenin, anti-ribosomal s6 kinase 1 (anti-RSK1), and anti-RSK2 antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-Mst1, anti-Mst2, anti-Lats1, and anti-Lats2 antibodies were from Bethyl Laboratory (Montgomery, TX). Anti-phospho-YAP S127, anti-phospho-Akt T308, anti-phospho-Akt S473, anti-phospho-GSK3β S9, anti-phospho-ERK1/2, anti-phospho-Mst2 T180, anti-E-cadherin, antivimentin, and anti-poly(ADP-ribose) polymerase (anti-PARP) antibodies were from Cell Signaling Technology. Mouse monoclonal antibody against N-cadherin was provided by Keith Johnson (University of Nebraska Medical Center) (49). Anti-phospho-RSK S380 antibody was from BioLegend (San Diego, CA). Anti-neurofibromatosis 2 (anti-NF2) and anti-β-tubulin antibodies were from Sigma (St. Louis, MO). Total cell lysate preparation and Western blotting assays were done as previously described (50).
Cell proliferation and anchorage-independent growth assays.
For cell proliferation assays, 5,000 (LNCaP-C4-2) or 10,000 (LNCaP) cells were seeded in wells of a 24-well plate in triplicate. Cells were counted with a hemocytometer (Thermo, Waltham, MA), and proliferation curves were made on the basis of cell numbers of each well from three independent experiments. Soft agar colony formation assays were conducted in 6-well plates as described previously (18).
Cell migration and invasion assays.
In vitro analysis of invasion and migration was performed using a BioCoat invasion system (BD Biosciences, San Jose, CA) and a Transwell system (Corning, Corning, NY), respectively, according to the manufacturers' instructions. The cells were trypsinized and resuspended in the medium without serum or growth factors. Basal medium without serum was added to the bottom of the migration assay chamber and the BioCoat invasion chamber. The above-described cell suspension (400 μl containing 50,000 cells) was added to the insert and incubated for 18 h at 37°C. The cells were fixed with 3.7% paraformaldehyde (Sigma), and the cells inside the inserts were removed with cotton swabs. The invasive and migratory cells were stained with ProLong Gold antifade reagent with DAPI (4′,6-diamidino-2-phenylindole). The relative rates of invasion and migration were calculated as previously described (43, 51).
IHC staining.
Tissue microarray slides (TMAs) were obtained from the Prostate Cancer Biorepository Network (PCBN; New York University site). The TMA consists of 7 naive (hormone-responsive) and 13 castration-resistant prostate cancer tumors collected from 1983 to 2002 at the New York University Langone Medical Center. Slide deparaffinization, antigen retrieval, and blocking were performed as we have described previously (18). The sections were then stained with anti-YAP antibody (1:100 dilutions; catalog number 4912; Cell Signaling) using a Histostain-Plus immunohistochemistry (IHC) kit following the manufacturer's instructions (Invitrogen, Grand Island, NY). Cell nuclei were stained with hematoxylin. A Ventana iScan HT scanner (Roche) was used for slide scanning with a ×20 lens. The staining results were independently evaluated by three researchers, including two pathologists (S.M.L. and K.F.). Both the YAP staining intensity (a scale of from 0 to 3 was used, where 0 is negative, 1 is weak staining, 2 is moderate staining, and 3 is strong staining) and nuclear localization (the percentage of tumor cell nuclei stained, where 0 is no staining, 1 is ≤10% of tumor cell nuclei stained, 2 is 10 to 50% stained, and 3 is >50% stained) were scored (52).
Animal studies.
For in vivo xenograft studies, LNCaP cells expressing vector or YAP in 50% Cultrex basement membrane extract (2.0 × 106 cells for each line/0.1 ml; Trevigen, Gaithersburg, MD) were subcutaneously injected into the left flank of 3-month-old castrated male SCID mice (Charles River, Wilmington, MA). Six and nine animals were used for the control (vector) and experimental (YAP) groups, respectively. Mice were euthanized at 8 weeks postinjection, and the tumors were excised and fixed for subsequent histopathological examination and IHC analysis. The animals were housed in pathogen-free facilities. All animal experiments were approved by the University of Nebraska Medical Center Institutional Animal Care and Use Committee.
Statistical analysis.
Data were analyzed using a two-tailed, unpaired Student's t test. The Wilcoxon rank sum test was used to compare the IHC staining data between groups. A P value of <0.05 was considered to indicate statistical significance.
RESULTS
YAP transforms prostate epithelial cells and promotes cell motility and invasiveness.
Previous studies showed that YAP overexpression induces the transformation of immortalized pancreatic and mammary epithelial cells (18, 20). To investigate the biological significance of YAP overexpression/hyperactivation in prostate cancer, we first tested the role of YAP in RWPE-1 (immortalized prostate epithelial) cells. As shown in Fig. 1, ectopic expression of YAP stimulated cell proliferation and induced cellular transformation in RWPE-1 cells (Fig. 1A to D). As expected, the expression of constitutively active YAP-S127A (S127 is the main Hippo-mediated phosphorylation site of YAP) enhanced RWPE-1 cell proliferation and transformation to a greater extent than that of wild-type YAP did (Fig. 1B to D). YAP expression causes epithelial-mesenchymal transition (EMT) in mammary epithelial (MCF10A) cells (20, 26). Surprisingly, YAP transformed prostate cells without inducing an EMT, as the levels of E-cadherin (an epithelial marker) and vimentin (a mesenchymal marker) remained unchanged in the presence of YAP activation (Fig. 1E). Consistent with this observation, YAP was not sufficient to induce a full EMT in a nontransformed mammary epithelial cell line (NMuMG) (29).
FIG 1.

YAP promotes cellular transformation, motility, and invasiveness in RWPE-1 cells. (A) Establishment of RWPE-1 cell lines stably expressing YAP. (B) Expression of YAP/YAP-S127A stimulates proliferation in RWPE-1 cells. (C, D) Anchorage-independent growth (colony formation assay) in soft agar. (E) Western blotting analysis with the indicated antibodies in YAP-expressing RWPE-1 cells. ND, not detectable; N-Cad, N-cadherin; E-Cad, E-cadherin; p-S9, phosphorylated S9. (F to H) Cell migration (F) and invasion (G) assays with RWPE-1 cells expressing the vector, YAP, or YAP-S127A construct. (H) Migrating and invading cells were stained with DAPI, and images of representative fields are shown. Quantitative data are expressed as the mean ± SEM from three independent experiments. In panels A and E, numbers to the right of the blots are molecular masses (in kilodaltons). ***, P < 0.001; **, P < 0.01; *, P < 0.05 (t test).
Over 90% of cancer deaths are due to metastasis rather than to the primary tumors (53, 54). Migration and invasion are essential steps for primary tumor cells to metastasize and grow (54–56). We therefore examined the role of YAP in prostate cell motility. Interestingly, overexpression of YAP or YAP-S127A also significantly promoted cell migration (Fig. 1F) and invasion (Fig. 1G and H) in immortalized prostate epithelial cells. Next, we further explored whether enhanced expression of YAP stimulates migration and invasion in prostate cancer (LNCaP) cells. Similarly, YAP or YAP-S127A overexpression resulted in a significant increase in the number of LNCaP cells that invaded through Matrigel and migrated through filters compared to the number of vector control cells that did so (Fig. 2A to D). These data indicate that YAP activation is a positive regulator of prostate cell oncogenic activity.
FIG 2.

YAP promotes migration, invasion, and androgen-insensitive growth and AR activation in LNCaP cells. (A) Establishment of LNCaP cells expressing vector, YAP, or YAP-S127A. (B to D) Cell invasion (B, C) and migration (B, D) assays with the LNCaP cell lines described in the legend to panel A. (E) Representative photos of LNCaP cells expressing vector or YAP that have been cultured in normal medium with fetal bovine serum (FBS) or androgen deprivation medium with CSS for 3 days (medium with fetal bovine serum) or 5 days (medium with CSS). (F) Proliferation curves for various LNCaP cells. (G) Relative mRNA levels of known targets of YAP (determined by quantitative RT-PCR) in LNCaP cells expressing the vector or YAP. (H) LNCaP cells expressing the vector or YAP were cultured in normal medium with fetal bovine serum or androgen deprivation medium with CSS for 3 days. The total lysates were probed with the indicated antibodies. Numbers to the right of the blots are molecular masses (in kilodaltons). p-Akt, phosphorylated Akt; p-ERK, phosphorylated ERK. (I) Relative mRNA levels of known targets of androgen receptor (determined by quantitative RT-PCR) in LNCaP cells expressing the vector or YAP. Quantitative data are expressed as the mean ± SEM from three independent experiments. ***, P < 0.001; **, P < 0.01 (t test).
YAP promotes the androgen-insensitive growth of LNCaP cells.
Most prostate cancer patients with metastatic disease progress to CRPC. We next assessed whether YAP expression is sufficient to induce androgen-insensitive (castration-resistant) growth in LNCaP cells, which grow completely in an androgen-sensitive and -dependent manner. YAP overexpression stimulated the proliferation of LNCaP cells (Fig. 2E and F). Interestingly, the most significant change in these cells upon YAP expression was the ability to proliferate normally under androgen deprivation conditions (using charcoal-stripped serum [CSS] to deplete the medium of androgens); in contrast, the control parental cells stopped dividing without androgen (Fig. 2E and F). These data indicate that enhanced expression of YAP was sufficient to convert LNCaP cells from an androgen-sensitive to a castration-resistant (androgen-insensitive) state.
TEAD1 to TEAD4 are the major transcriptional factors of the Hippo pathway. Most of the known YAP-TEAD targets, including ANKRD1, SOX4, CTGF, and Cyr61, were induced by YAP expression in LNCaP cells (Fig. 2G), indicating that YAP signaling is on in YAP-expressing LNCaP cells. Survivin and ITGB2 were not induced by YAP-overexpressing LNCaP cells (data not shown). YAP was able to induce Akt and ERK activation in a cellular context-dependent manner (41, 42). Interestingly, we also detected moderate but reproducible increased phosphorylation of Akt on T308 (but not S473) upon YAP expression (Fig. 2H). Both Akt and ERK were strongly activated upon androgen depletion (Fig. 2H) (57), suggesting that multiple cellular pathways are involved in prostate cancer cell survival upon androgen deprivation.
We further explored whether YAP could regulate androgen signaling activity. Indeed, the androgen receptor (AR) targets PSA, NKX3.1, PGC-1, and KLK2 were all greatly induced by YAP overexpression (Fig. 2I), suggesting that YAP promotes AR activation.
Upregulation of YAP in castration-resistant prostate cancer cells.
We further assessed the extent to which YAP expression/activity is altered during progression from an androgen-sensitive to an androgen-insensitive state. For this purpose, we took advantage of a well-established prostate cancer cell model system. LNCaP cells grow slowly and completely rely on androgen, whereas the LNCaP-C81 and LNCaP-C4-2 sublines (both of which are androgen insensitive and castration resistant) grow aggressively even under androgen deprivation conditions. These cancer cell models closely represent the transition of the initial androgen-sensitive disease state to the androgen-insensitive state (58, 59). Interestingly, we found that compared to the YAP expression levels in LNCaP cells, YAP expression levels were dramatically upregulated in both LNCaP-C4-2 and LNCaP-C81 androgen-insensitive cells (Fig. 3A). The level of phosphorylation of YAP on S127 (the major phosphorylation site for the Hippo pathway) was proportionally increased. Cell fractionation assays confirmed that the cytoplasmic-nuclear localization of YAP was not significantly altered (Fig. 3B). In line with this observation, no change in the level of expression or the activity of upstream Hippo core components was detected (Fig. 3A and data not shown). Consistent with the findings of previous studies (60), AR levels were increased in LNCaP-C4-2 and LNCaP-C81 cells compared to those in parental LNCaP cells (Fig. 3A). Finally, quantitative reverse transcription-PCR (RT-PCR) showed that the levels YAP mRNA but not those of the mRNA of its paralog, TAZ, were significantly elevated in LNCaP-C4-2 and LNCaP-C81 cells, indicating that transcriptional regulation is involved in YAP upregulation (Fig. 3C and data not shown). YAP targets were consistently induced in LNCaP-C4-2 cells (Fig. 3D). TEAD4 mRNA but not TEAD1 to TEAD3 mRNA was upregulated in LNCaP-C4-2 cells (Fig. 3E). YAP protein stability was similar in both LNCaP and LNCaP-C4-2 cells (Fig. 3F). Together, these results suggest that YAP was transcriptionally upregulated during the transition of LNCaP cells to androgen-insensitive growth.
FIG 3.

YAP is upregulated in androgen-insensitive prostate cancer cells. (A) LNCaP (androgen-sensitive) and LNCaP-C81 and LNCaP-C4-2 (androgen-insensitive and castration-resistant) cell lysates were probed with the indicated antibodies. SE, short exposure; LE, long exposure; p-YAP, phosphorylated YAP; p-Mst, phosphorylated Mst. (B) Cell fractionation assay in LNCaP and LNCaP-C4-2 cells. The cells were harvested at 70 to 80% confluence. β-Tubulin and PARP served as cytoplasmic and nuclear markers, respectively. C, cytoplasmic fraction; N, nuclear fraction. (C, D) Quantitative RT-PCR of YAP and its known targets in LNCaP and castration-resistant sublines. (E) Quantitative RT-PCR of TEAD1 to TEAD4 in LNCaP and castration-resistant sublines. (F) LNCaP and LNCaP-C4-2 cells were treated with cycloheximide (CHX) at the indicated times, and the total lysates were probed with YAP and actin antibodies. (G) Quantitative RT-PCR of YAP and PSA in LNCaP cells treated with or without R1881 (1 nM) for 24 h. (H) LNCaP cells were treated with or without R1881 (1 nM) for 24 h, and the total lysates were probed with the indicated antibodies. Quantitative data were derived from three independent experiments and expressed as the mean ± SEM. In panels A, B, F, and H, numbers to the right of the blots are molecular masses (in kilodaltons). **, P < 0.01; ***, P < 0.001 (t test).
Next we investigated whether androgen signaling could affect YAP activity. Treatment with R1881 (a testosterone analog) failed to alter YAP expression (Fig. 3G) or phosphorylation (localization) (Fig. 3H). Together, these observations suggest that YAP promotes AR activation and androgen signaling fails to regulate YAP activity.
YAP promotes castration-resistant growth in vivo.
We next evaluated the influence of YAP on castration resistance in animals. LNCaP cells expressing the vector control and YAP were subcutaneously inoculated into castrated male SCID mice. As expected, most of the mice (all except one) injected with LNCaP cells carrying the vector did not form palpable tumors (n = 6). However, about 67% (6/9) of mice injected with YAP-expressing LNCaP cells grew large tumors at the endpoint of the experiment (Fig. 4A and B). The tumors on the mice harboring YAP-expressing LNCaP cells were visible at 1 month postinjection (data not shown). Histopathological examination revealed extensive tumor necrosis and hemorrhage (Fig. 4C, hematoxylin and eosin [H&E] staining), which is an indicator of aggressiveness. Most of these tumor cells expressed AR and YAP (Fig. 4C). These data strongly suggest that YAP confers the castration-resistant growth of prostate cancer cells in vitro and in vivo.
FIG 4.
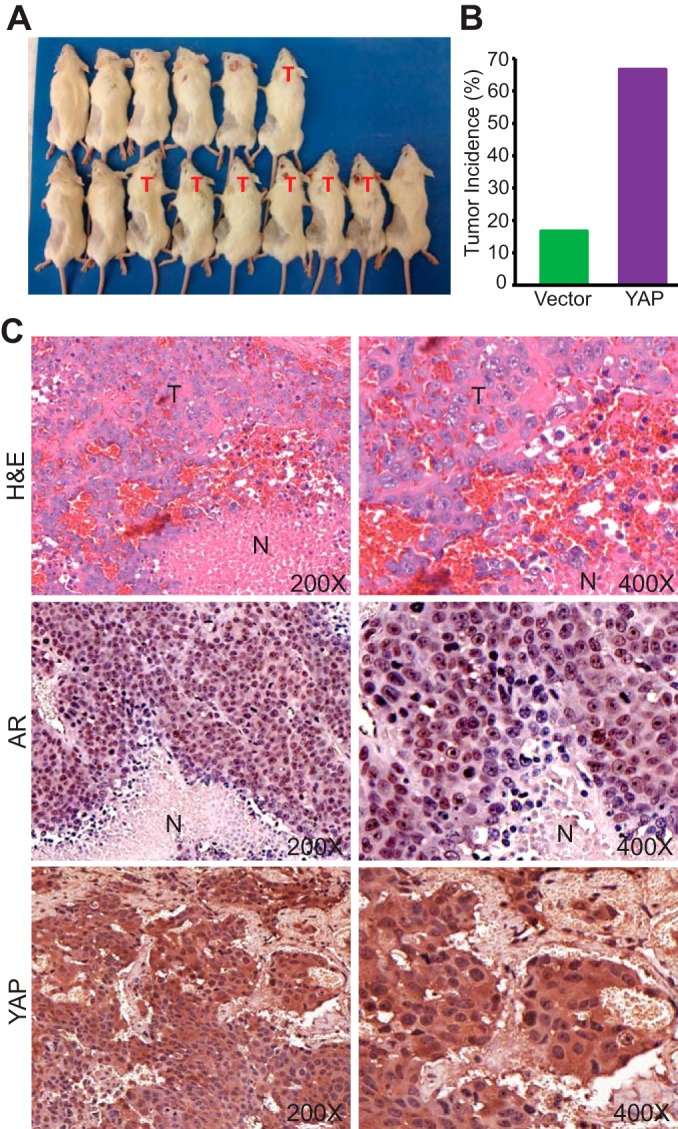
YAP confers castration resistance in vivo. (A) Castrated male SCID mice were implanted with LNCaP cells carrying the vector control (top row) or YAP-expressing LNCaP cells (bottom row) and photographed at 8 weeks postinjection. T, tumor-harboring mice. (B) Tumor incidence of the mice shown in panel A. (C) H&E and IHC staining of AR and YAP. T, tumor area; N, necrotic area.
YAP knockdown impairs migration and invasion in castration-resistant prostate cancer cells.
To explore the biological significance of YAP upregulation in castration-resistant prostate cancer cells, we reduced YAP expression by the use of shRNA (constitutive) or siRNA (transient) in LNCaP-C4-2 cells (Fig. 5A and B). Using Transwell and Matrigel assays, we demonstrated that YAP knockdown greatly impaired migration and invasion in LNCaP-C4-2 prostate cancer cells (Fig. 5C to J). These data, together with the gain of function of YAP (Fig. 1 and 2), suggest that YAP plays an important role in motility and invasion in prostate cancer cells.
FIG 5.

YAP knockdown in LNCaP-C4-2 cells impairs cell migration and invasion. (A) Establishment of cells stably expressing the shRNA control vector (shControl) and shRNAs against YAP (shYAP#1 and shYAP#2) in LNCaP-C4-2 cells. (B) LNCaP-C4-2 cells were transiently transfected with control siRNA (siControl) or siRNA targeting YAP and YAP expression (shYAP#1 and shYAP#2) and analyzed by Western blotting. Numbers to the right of the blots are molecular masses (in kilodaltons). (C to F) Cell migration and invasion assays with LNCaP-C4-2 cells established in the assay whose results are shown in panel A. (G to J) Cell migration and invasion assays with LNCaP-C4-2 cells transfected with siRNA in the assay whose results are shown in panel B. Cell migration assays with a Transwell system and invasion assays with Matrigel were performed as we previously described (43). Migrating and invading cells were stained with DAPI, and images of representative fields are shown. Quantitative data are expressed as the mean ± SEM from three independent experiments. ***, P < 0.001; **, P < 0.01; *, P < 0.05 (t test).
YAP is essential for castration-resistant growth of prostate cancer cells.
The upregulation of YAP in androgen-insensitive cell lines led us to further determine whether YAP is required for growth without androgens in these cells. Under normal growth conditions, LNCaP-C4-2 cells with YAP knockdown showed only moderately slower proliferation than control LNCaP-C4-2 cells with YAP expression (Fig. 6A, top, and B). However, while LNCaP-C4-2 cells were still able to proliferate (albeit at a lower rate) in the absence of androgens (in CSS medium), YAP-knockdown cells failed to divide under androgen deprivation conditions (Fig. 6A and B). Consistent with this observation, LNCaP-C4-2 cells with reduced amounts of YAP form colonies well in soft agar with complete serum (Fig. 6C and D); however, these cells failed to grow under conditions with CSS (Fig. 6E and F). Again, LNCaP-C4-2 control cells, but not LNCaP-C-2 cells lacking YAP, formed colonies even when androgens were removed (Fig. 6E and F). In total, these studies implicate that YAP is essential for the castration-resistant growth of prostate cancer cells.
FIG 6.

YAP is required for androgen-insensitive growth of LNCaP-C4-2 cells. (A) Representative photos of LNCaP-C4-2 cells expressing control shRNA or YAP shRNA that have been cultured in normal medium with fetal bovine serum or androgen deprivation medium with CSS for 5 days. (B) Proliferation curves for various LNCaP-C4-2 cells. (C to F) Anchorage-independent growth assay of LNCaP-C4-2 cells in soft agar under normal conditions (with fetal bovine serum) or androgen deprivation conditions (with CSS). (G) Quantitative RT-PCR of YAP, PSA, and NKX3.1 in LNCaP-C4-2 cells. Control and YAP-knockdown cells lines were cultured in serum-free medium for 24 h and treated with or without R1881 (1 nM) for an additional 24 h. Quantitative data were derived from three independent experiments and expressed as the mean ± SEM. *, P < 0.05 (t test) compared to the control; **, P < 0.01 (t test) compared to the control; ***, P < 0.001 (t test) compared to the control.
Consistent with our observations that YAP activates AR targets (Fig. 2I), YAP knockdown reduced the basal levels of PSA and NKX3.1 mRNA and partially blocked the AR targets induced by R1881 (Fig. 6G), further suggesting that YAP regulates AR signaling activity.
YAP is required for ERK-RSK signaling activation upon androgen depletion in LNCaP-C4-2 cells.
We next explored the downstream signaling of YAP in the androgen-insensitive growth of prostate cancer cells. The PTEN (phosphatase and tensin homolog)/Akt axis and MEK-ERK signaling are critical regulators in prostate tumor survival and progression (61, 62). Both Akt and MEK-ERK pathways have recently been linked with YAP activity (28, 41, 42, 63). Interestingly, we found that both Akt and ERK-RSK signaling pathways were strongly activated upon androgen depletion (Fig. 7A and B) (57), suggesting that LNCaP-C4-2 cells proliferated without androgen, at least in part, by activating these survival pathways. Importantly, ERK1/2 and downstream RSK1/2 activation (revealed by phosphorylation) was largely blocked in YAP-knockdown cells when androgens were removed (Fig. 7B). However, Akt activity was only moderately reduced when YAP was knocked down (Fig. 7A). Together, these data suggest that YAP is required for ERK-RSK activation in LNCaP-C4-2 cells under androgen depletion conditions.
FIG 7.

YAP is required for ERK-RSK activation upon androgen depletion in LNCaP-C4-2 cells. (A, B) Cells were harvested at day 3 under normal conditions (with fetal bovine serum) or androgen-depleted conditions (with CSS), and the total lysates were probed with the indicated antibodies. Numbers to the right of the blots are molecular masses (in kilodaltons). p-RSK, phosphorylated RSK. (C to F) Cell migration (C, D) and invasion (E, F) assays under normal conditions (with fetal bovine serum) and androgen deprivation conditions (with CSS) with or without the MEK-ERK inhibitor U0126. ***, P < 0.001 (t test) compared to the control; **, P < 0.01 (t test) compared to the control. (G) A model for YAP signaling in castration-resistant prostate cancer.
To determine the functional role of ERK activation upon androgen depletion, we inhibited MEK-ERK signaling with the inhibitor U0126 and analyzed migratory and invasive activity in LNCaP-C4-2 cells. ERK inhibition partially suppressed migration under normal conditions and to a greater extent in medium without androgens (Fig. 7C and D). Interestingly, treatment with U0126 had no effect on invasion in complete medium; however, U0126 greatly impaired the invasive ability of LNCaP-C4-2 cells under androgen deprivation conditions (Fig. 7E and F). As expected, knockdown of YAP significantly reduced the levels of migration and invasion in LNCaP-C4-2 cells (Fig. 7C to F). Taken together, our data indicate that for LNCaP-C4-2 cells ERK activation (probably downstream YAP) is essential to promote survival and migration/invasion under androgen depletion conditions.
Upregulation and activation of YAP in castration-resistant tumors.
YAP is overexpressed and/or hyperactivated (as shown by nuclear localization) in prostate primary tumor samples (18, 26). However, it is not known to what extent YAP activity/expression correlates with castration resistance. Having established the role of YAP in androgen-insensitive prostate cancer growth in cell culture models, we further determined the functional relevance of YAP in CRPC in the clinical setting. For this purpose, we obtained tissue microarrays containing naive (hormone-responsive) and castration-resistant prostate tumors and performed IHC staining. Immunostaining demonstrated that overall YAP expression was relatively weak in naive prostate tumors (n = 7) (Fig. 8A to A″), and no single case was scored moderate or strong for YAP staining (see Materials and Methods). Importantly, we observed the dramatic upregulation of YAP in most hormonal therapy-resistant tumor samples (n = 13) (Fig. 8B to E). Nine of the resistant tumors showed moderate or strong staining, and 4 of them had weak staining (whereas all of the naive tumors showed weak to no staining) (P = 0.003 for resistant versus naive tumors). Furthermore, strong nucleus-localized (hyperactive) YAP staining was detected in 5 of the resistant tumors (Fig. 8B to B″, to D″, and F) (P = 0.001 for resistant versus naive tumors). Publically available data further confirmed that the amount of YAP mRNA was significantly higher in CRPC or metastatic prostate tumors than primary tumors (Fig. 8G and H). These data, together with the findings from our cell culture and animal models (Fig. 2 to 7), identify YAP to be a critical regulator in the castration-resistant growth of prostate cancer.
FIG 8.

Upregulation and activation of YAP in castration-resistant prostate tumors. (A to A″) Representative photos of immunostaining for YAP in naive (hormone-responsive) prostate tumors. (B to B″, C to C″, and D to D″) Representative photos of YAP IHC staining in hormonal therapy-resistant prostate tumors. (E) Quantification of YAP IHC staining intensity in naive and castration-resistant prostate tumors. Four resistant cases (3254108156, 8322079241, 8842201759, 6756440716) were scored low, five resistant cases (4024863604, 5962887148, 3698735602, 9182583214, 2667199309) were scored moderate, and four resistant cases (8063595154, 4976472144, 4729101711, 7346843168) were scored high for YAP staining. All naive tumor samples (4481786650, 2667199309, 8743000808, 4599423355, 8365207463, 8315345132, 2667199309) had no to low YAP staining. (F) Quantification of YAP nuclear localization based on IHC staining in naive and castration-resistant prostate tumors. Four resistant cases (3254108156, 8322079241, 7346843168, 6756440716) were scored low, four resistant cases (8842201759, 4024863604, 4729101711, 2667199309) were scored moderate, and five resistant cases (5962887148, 3698735602, 8063595154, 9182583214, 4976472144) were scored high for YAP nuclear staining. All naive tumors samples had no to low nuclear YAP staining. ***, P < 0.001; **, P < 0.01. (G) Relative YAP mRNA levels in localized (primary) tumors (n = 60) and CRPC (resistant; n = 36). Quantitative data were mined from Grasso et al. (78). ***, P < 0.001 (t test). (H) Relative YAP mRNA levels in primary (n = 65) and metastatic (Met; n = 25) tumors (most metastases are castration resistant). Data were retrieved from accession number GDS2545 of the Gene Expression Omnibus database (82). ***, P < 0.001 (t test).
DISCUSSION
Androgen deprivation therapy initially decreases the volume of both primary and metastatic lesions; however, most men experience eventual relapse. Recurring prostate cancer is typically castration resistant, since removal of testicular androgen by chemical or surgical castration does not affect tumor growth or metastasis. Ultimately, in the vast majority of cases, CRPC is lethal. Thus, there is an urgent need to identify drug targets and develop new therapeutic strategies to treat CRPC. Although the underlying mechanisms of castration resistance are not fully understood, both androgen receptor-dependent and -independent signaling pathways are known to be involved (64). Androgen receptor overexpression, activation, and androgen secretion are the major contributors to androgen receptor-dependent CRPC (64–66). For example, androgen receptor selectively upregulates M-phase cell cycle genes to promote CRPC (67). Interestingly, a recent study found that a gain-of-function mutation in dihydrotestosterone (the most potent androgen) synthesis partially accounts for castration resistance (68). However, some other studies have challenged the androgen receptor-dependent mechanism, as castration induces the activation of many kinases (69) and increases the expression of antiapoptotic genes independently of the androgen receptor (70). Furthermore, prostate cancer stem cells have been proposed to be the origin of prostate cancer progression, and they may not express androgen receptor (71). Our current study implicates YAP as a potent regulator for CRPC in vitro and in vivo (Fig. 2 to 4, 6, and 7) and in clinical samples (Fig. 8), thus identifying YAP to be a potential alternative regulator/pathway for the acquisition of castration resistance by prostate tumor cells.
Hippo-YAP signaling is often deregulated in cancer and is a potential target for cancer therapy (5–7). Among the components, the YAP-TEAD complex represents the most attractive target for several reasons. First, TEAD transcription factors are required for YAP's oncogenic activity both in cell culture and in vivo (48, 72). Second, TEAD is largely dispensable during normal tissue growth in the mouse liver (72) and in Drosophila (73) (i.e., TEAD becomes critical only when YAP is hyperactivated/overexpressed). Thus, there is a strong rationale for developing YAP-TEAD complex-disrupting agents as anticancer therapeutics against YAP-driven oncogenesis. Indeed, Liu-Chittenden et al. screened a small-molecule library (consisting of 3,300 FDA-approved drugs) for agents that inhibit YAP-TEAD activity in a cell-based assay (72). Verteporfin was identified to be a compound effective at preventing hepatic tumorigenesis driven by YAP overexpression (72) and the growth of xenograft tumors in immunodeficient mice (52, 74). Thus, administration of verteporfin is an effective pharmacologic approach to inhibit YAP signaling, and these studies strongly support the feasibility of targeting YAP in human cancer in which Hippo-YAP is deregulated. Importantly, the current study showed that depletion of YAP could cause castration-resistant prostate cancer cells to stop growing and become androgen sensitive (Fig. 6 and 7). Therefore, inhibition of YAP (e.g., by verteporfin) combined with hormonal therapy is a potential novel therapeutic strategy for prostate cancer patients with CRPC (Fig. 7G).
Previous studies, including ours, demonstrated that YAP is overexpressed or hyperactivated in prostate tumor samples (18, 26). Furthermore, Lats2 expression is significantly lower in metastatic prostate tissues than normal prostate tissue samples (75). Interestingly, Lats2 and MstI have been shown to be associated with androgen receptor and regulate its activity (76, 77). These reports suggest that the Hippo-YAP pathway plays a role in the pathogenesis of prostate cancer. This study adds further evidence showing that the Hippo effector YAP regulates cell motility, invasion, and the castration-resistant growth of prostate cancer cells. Together, these studies demonstrated the biological significance of the Hippo-YAP signaling in prostate cancer. There are several questions that need to be addressed. How is YAP upregulated in androgen-insensitive prostate cancer cells? Our observations suggest that the upregulation of YAP is androgen receptor independent (Fig. 3G) and methylation is dispensable for YAP transcription in LNCaP and LNCaP-C4-2 cells (L. Zhang and J. Dong, unpublished observations). Large-scale studies failed to identify YAP amplification and mutation in CRPC (78). Therefore, future studies are needed to address the underlying mechanisms of YAP upregulation in CRPC. Furthermore, how is Hippo-YAP deregulated, and what are the clinical outcomes? Answers to and understanding from answers to these questions may provide additional insights into the pathogenesis of prostate cancer.
Genetically engineered mouse alleles of most Hippo components are available, and these animal models have provided compelling evidence showing the importance of Hippo-YAP signaling in human malignancies (8–11, 17–21, 79). However, no single such model has been developed for the prostate. Such genetic models are expected to further provide evidence demonstrating the significance of Hippo-YAP signaling in prostate cancer. Since PTEN is an important tumor suppressor in prostate cancer and specific deletion of PTEN in the prostate leads to metastatic prostate cancer and castration resistance (80, 81), one might consider use of a combination of the PTEN alleles with Hippo (loss-of-function)-YAP (gain-of-function) signaling when these animal models are developed. Interestingly, a recent report showed that MstI/2 deletion or YAP activation could downregulate PTEN, suggesting a potential link between the Hippo-YAP pathway and PTEN signaling (63).
ACKNOWLEDGMENTS
We thank Ming-Fong Lin (University of Nebraska Medical Center) for providing us the LNCaP-C33 (equivalent to LNCaP) and LNCaP-C81 cell lines. We are grateful to the Prostate Cancer Biorepository Network (PCBN; New York University site) for providing the hormone-resistant tissue array. We also thank Joyce Solheim for critical reading of and comments on the manuscript.
PCBN is supported by award no. W81XWH-10-2-0056 and W81XWH-10-2-0046 from the U.S. Department of Defense Prostate Cancer Research Program. Research in the J. Dong laboratory is supported in part by grants P30 GM106397 and R01 GM109066 from the National Institutes of Health and W81XWH-14-1-0150 from the U.S. Department of Defense Health Program through the Prostate Cancer Research Program.
REFERENCES
- 1.Amaral TM, Macedo D, Fernandes I, Costa L. 2012. Castration-resistant prostate cancer: mechanisms, targets, and treatment. Prostate Cancer 2012:327253. doi: 10.1155/2012/327253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen Y, Sawyers CL, Scher HI. 2008. Targeting the androgen receptor pathway in prostate cancer. Curr Opin Pharmacol 8:440–448. doi: 10.1016/j.coph.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao B, Li L, Lei Q, Guan KL. 2010. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev 24:862–874. doi: 10.1101/gad.1909210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pan D. 2010. The Hippo signaling pathway in development and cancer. Dev Cell 19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harvey KF, Zhang X, Thomas DM. 2013. The Hippo pathway and human cancer. Nat Rev Cancer 13:246–257. doi: 10.1038/nrc3458. [DOI] [PubMed] [Google Scholar]
- 6.Park HW, Guan KL. 2013. Regulation of the Hippo pathway and implications for anticancer drug development. Trends Pharmacol Sci 34:581–589. doi: 10.1016/j.tips.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson R, Halder G. 2014. The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat Rev Drug Discov 13:63–79. doi: 10.1038/nrd4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee KP, Lee JH, Kim TS, Kim TH, Park HD, Byun JS, Kim MC, Jeong WI, Calvisi DF, Kim JM, Lim DS. 2010. The Hippo-Salvador pathway restrains hepatic oval cell proliferation, liver size, and liver tumorigenesis. Proc Natl Acad Sci U S A 107:8248–8253. doi: 10.1073/pnas.0912203107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu L, Li Y, Kim SM, Bossuyt W, Liu P, Qiu Q, Wang Y, Halder G, Finegold MJ, Lee JS, Johnson RL. 2010. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc Natl Acad Sci U S A 107:1437–1442. doi: 10.1073/pnas.0911427107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song H, Mak KK, Topol L, Yun K, Hu J, Garrett L, Chen Y, Park O, Chang J, Simpson RM, Wang CY, Gao B, Jiang J, Yang Y. 2010. Mammalian MstI and Mst2 kinases play essential roles in organ size control and tumor suppression. Proc Natl Acad Sci U S A 107:1431–1436. doi: 10.1073/pnas.0911409107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, Thasler W, Lee JT, Avruch J, Bardeesy N. 2009. MstI and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 16:425–438. doi: 10.1016/j.ccr.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang Z, Li X, Hu J, Zhou W, Jiang Y, Li G, Lu D. 2006. Promoter hypermethylation-mediated down-regulation of LATS1 and LATS2 in human astrocytoma. Neurosci Res 56:450–458. doi: 10.1016/j.neures.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 13.Li H, Wolfe A, Septer S, Edwards G, Zhong X, Abdulkarim AB, Ranganathan S, Apte U. 2012. Deregulation of Hippo kinase signalling in human hepatic malignancies. Liver Int 32:38–47. doi: 10.1111/j.1478-3231.2011.02646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seidel C, Schagdarsurengin U, Blumke K, Wurl P, Pfeifer GP, Hauptmann S, Taubert H, Dammann R. 2007. Frequent hypermethylation of MST1 and MST2 in soft tissue sarcoma. Mol Carcinog 46:865–871. doi: 10.1002/mc.20317. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi Y, Miyoshi Y, Takahata C, Irahara N, Taguchi T, Tamaki Y, Noguchi S. 2005. Down-regulation of LATS1 and LATS2 mRNA expression by promoter hypermethylation and its association with biologically aggressive phenotype in human breast cancers. Clin Cancer Res 11:1380–1385. doi: 10.1158/1078-0432.CCR-04-1773. [DOI] [PubMed] [Google Scholar]
- 16.Murakami H, Mizuno T, Taniguchi T, Fujii M, Ishiguro F, Fukui T, Akatsuka S, Horio Y, Hida T, Kondo Y, Toyokuni S, Osada H, Sekido Y. 2011. LATS2 is a tumor suppressor gene of malignant mesothelioma. Cancer Res 71:873–883. doi: 10.1158/0008-5472.CAN-10-2164. [DOI] [PubMed] [Google Scholar]
- 17.Schlegelmilch K, Mohseni M, Kirak O, Pruszak J, Rodriguez JR, Zhou D, Kreger BT, Vasioukhin V, Avruch J, Brummelkamp TR, Camargo FD. 2011. Yap1 acts downstream of alpha-catenin to control epidermal proliferation. Cell 144:782–795. doi: 10.1016/j.cell.2011.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. 2007. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 130:1120–1133. doi: 10.1016/j.cell.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Camargo FD, Gokhale S, Johnnidis JB, Fu D, Bell GW, Jaenisch R, Brummelkamp TR. 2007. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol 17:2054–2060. doi: 10.1016/j.cub.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 20.Overholtzer M, Zhang J, Smolen GA, Muir B, Li W, Sgroi DC, Deng CX, Brugge JS, Haber DA. 2006. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci U S A 103:12405–12410. doi: 10.1073/pnas.0605579103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tremblay AM, Missiaglia E, Galli GG, Hettmer S, Urcia R, Carrara M, Judson RN, Thway K, Nadal G, Selfe JL, Murray G, Calogero RA, De Bari C, Zammit PS, Delorenzi M, Wagers AJ, Shipley J, Wackerhage H, Camargo FD. 2014. The Hippo transducer YAP1 transforms activated satellite cells and is a potent effector of embryonal rhabdomyosarcoma formation. Cancer Cell 26:273–287. doi: 10.1016/j.ccr.2014.05.029. [DOI] [PubMed] [Google Scholar]
- 22.Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, Fan ST, Luk JM, Wigler M, Hannon GJ, Mu D, Lucito R, Powers S, Lowe SW. 2006. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 125:1253–1267. doi: 10.1016/j.cell.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Steinhardt AA, Gayyed MF, Klein AP, Dong J, Maitra A, Pan D, Montgomery EA, Anders RA. 2008. Expression of Yes-associated protein in common solid tumors. Hum Pathol 39:1582–1589. doi: 10.1016/j.humpath.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fernandez-L A, Northcott PA, Dalton J, Fraga C, Ellison D, Angers S, Taylor MD, Kenney AM. 2009. YAP1 is amplified and up-regulated in hedgehog-associated medulloblastomas and mediates Sonic hedgehog-driven neural precursor proliferation. Genes Dev 23:2729–2741. doi: 10.1101/gad.1824509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu MZ, Yao TJ, Lee NP, Ng IO, Chan YT, Zender L, Lowe SW, Poon RT, Luk JM. 2009. Yes-associated protein is an independent prognostic marker in hepatocellular carcinoma. Cancer 115:4576–4585. doi: 10.1002/cncr.24495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, Zheng P, Ye K, Chinnaiyan A, Halder G, Lai ZC, Guan KL. 2007. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 21:2747–2761. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang X, George J, Deb S, Degoutin JL, Takano EA, Fox SB, AOCS Study Group, Bowtell DD, Harvey KF . 2011. The Hippo pathway transcriptional co-activator, YAP, is an ovarian cancer oncogene. Oncogene 30:2810–2822. doi: 10.1038/onc.2011.8. [DOI] [PubMed] [Google Scholar]
- 28.Zhang W, Nandakumar N, Shi Y, Manzano M, Smith A, Graham G, Gupta S, Vietsch EE, Laughlin SZ, Wadhwa M, Chetram M, Joshi M, Wang F, Kallakury B, Toretsky J, Wellstein A, Yi C. 2014. Downstream of mutant KRAS, the transcription regulator YAP is essential for neoplastic progression to pancreatic ductal adenocarcinoma. Sci Signal 7:ra42. doi: 10.1126/scisignal.2005049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lamar JM, Stern P, Liu H, Schindler JW, Jiang ZG, Hynes RO. 2012. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc Natl Acad Sci U S A 109:E2441–E2450. doi: 10.1073/pnas.1212021109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu FX, Guan KL. 2013. The Hippo pathway: regulators and regulations. Genes Dev 27:355–371. doi: 10.1101/gad.210773.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosenbluh J, Nijhawan D, Cox AG, Li X, Neal JT, Schafer EJ, Zack TI, Wang X, Tsherniak A, Schinzel AC, Shao DD, Schumacher SE, Weir BA, Vazquez F, Cowley GS, Root DE, Mesirov JP, Beroukhim R, Kuo CJ, Goessling W, Hahn WC. 2012. Beta-catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 151:1457–1473. doi: 10.1016/j.cell.2012.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Varelas X, Miller BW, Sopko R, Song S, Gregorieff A, Fellouse FA, Sakuma R, Pawson T, Hunziker W, McNeill H, Wrana JL, Attisano L. 2010. The Hippo pathway regulates Wnt/beta-catenin signaling. Dev Cell 18:579–591. doi: 10.1016/j.devcel.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 33.Imajo M, Miyatake K, Iimura A, Miyamoto A, Nishida E. 2012. A molecular mechanism that links Hippo signalling to the inhibition of Wnt/beta-catenin signalling. EMBO J 31:1109–1122. doi: 10.1038/emboj.2011.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, Martin JF. 2011. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 332:458–461. doi: 10.1126/science.1199010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Konsavage WM Jr, Kyler SL, Rennoll SA, Jin G, Yochum GS. 2012. Wnt/beta-catenin signaling regulates Yes-associated protein (YAP) gene expression in colorectal carcinoma cells. J Biol Chem 287:11730–11739. doi: 10.1074/jbc.M111.327767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Azzolin L, Zanconato F, Bresolin S, Forcato M, Basso G, Bicciato S, Cordenonsi M, Piccolo S. 2012. Role of TAZ as mediator of Wnt signaling. Cell 151:1443–1456. doi: 10.1016/j.cell.2012.11.027. [DOI] [PubMed] [Google Scholar]
- 37.Azzolin L, Panciera T, Soligo S, Enzo E, Bicciato S, Dupont S, Bresolin S, Frasson C, Basso G, Guzzardo V, Fassina A, Cordenonsi M, Piccolo S. 2014. YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell 158:157–170. doi: 10.1016/j.cell.2014.06.013. [DOI] [PubMed] [Google Scholar]
- 38.Alarcon C, Zaromytidou AI, Xi Q, Gao S, Yu J, Fujisawa S, Barlas A, Miller AN, Manova-Todorova K, Macias MJ, Sapkota G, Pan D, Massague J. 2009. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell 139:757–769. doi: 10.1016/j.cell.2009.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferrigno O, Lallemand F, Verrecchia F, L'Hoste S, Camonis J, Atfi A, Mauviel A. 2002. Yes-associated protein (YAP65) interacts with Smad7 and potentiates its inhibitory activity against TGF-beta/Smad signaling. Oncogene 21:4879–4884. doi: 10.1038/sj.onc.1205623. [DOI] [PubMed] [Google Scholar]
- 40.Varelas X, Sakuma R, Samavarchi-Tehrani P, Peerani R, Rao BM, Dembowy J, Yaffe MB, Zandstra PW, Wrana JL. 2008. TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat Cell Biol 10:837–848. doi: 10.1038/ncb1748. [DOI] [PubMed] [Google Scholar]
- 41.Xin M, Kim Y, Sutherland LB, Qi X, McAnally J, Schwartz RJ, Richardson JA, Bassel-Duby R, Olson EN. 2011. Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci Signal 4:ra70. doi: 10.1126/scisignal.2002278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu MZ, Chan SW, Liu AM, Wong KF, Fan ST, Chen J, Poon RT, Zender L, Lowe SW, Hong W, Luk JM. 2011. AXL receptor kinase is a mediator of YAP-dependent oncogenic functions in hepatocellular carcinoma. Oncogene 30:1229–1240. doi: 10.1038/onc.2010.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang S, Zhang L, Liu M, Chong R, Ding SJ, Chen Y, Dong J. 2013. CDK1 phosphorylation of YAP promotes mitotic defects and cell motility and is essential for neoplastic transformation. Cancer Res 73:6722–6733. doi: 10.1158/0008-5472.CAN-13-2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thalmann GN, Anezinis PE, Chang SM, Zhau HE, Kim EE, Hopwood VL, Pathak S, von Eschenbach AC, Chung LW. 1994. Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Res 54:2577–2581. doi: 10.1126/scisignal.2002278. [DOI] [PubMed] [Google Scholar]
- 45.Lin MF, Meng TC, Rao PS, Chang C, Schonthal AH, Lin FF. 1998. Expression of human prostatic acid phosphatase correlates with androgen-stimulated cell proliferation in prostate cancer cell lines. J Biol Chem 273:5939–5947. doi: 10.1074/jbc.273.10.5939. [DOI] [PubMed] [Google Scholar]
- 46.Igawa T, Lin FF, Lee MS, Karan D, Batra SK, Lin MF. 2002. Establishment and characterization of androgen-independent human prostate cancer LNCaP cell model. Prostate 50:222–235. doi: 10.1002/pros.10054. [DOI] [PubMed] [Google Scholar]
- 47.Xiao L, Chen Y, Ji M, Dong J. 2011. KIBRA regulates Hippo signaling activity via interactions with large tumor suppressor kinases. J Biol Chem 286:7788–7796. doi: 10.1074/jbc.M110.173468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM, Lai ZC, Guan KL. 2008. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev 22:1962–1971. doi: 10.1101/gad.1664408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johnson KR, Lewis JE, Li D, Wahl J, Soler AP, Knudsen KA, Wheelock MJ. 1993. P- and E-cadherin are in separate complexes in cells expressing both cadherins. Exp Cell Res 207:252–260. doi: 10.1006/excr.1993.1191. [DOI] [PubMed] [Google Scholar]
- 50.Xiao L, Chen Y, Ji M, Volle DJ, Lewis RE, Tsai MY, Dong J. 2011. KIBRA protein phosphorylation is regulated by mitotic kinase aurora and protein phosphatase 1. J Biol Chem 286:36304–36315. doi: 10.1074/jbc.M111.246850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang S, Ji M, Zhang L, Chen Y, Wennmann DO, Kremerskothen J, Dong J. 2014. Phosphorylation of KIBRA by the extracellular signal-regulated kinase (ERK)-ribosomal S6 kinase (RSK) cascade modulates cell proliferation and migration. Cell Signal 26:343–351. doi: 10.1016/j.cellsig.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Song S, Ajani JA, Honjo S, Maru DM, Chen Q, Scott AW, Heallen TR, Xiao L, Hofstetter WL, Weston B, Lee JH, Wadhwa R, Sudo K, Stroehlein JR, Martin JF, Hung MC, Johnson RL. 2014. Hippo coactivator YAP1 upregulates SOX9 and endows esophageal cancer cells with stem-like properties. Cancer Res 74:4170–4182. doi: 10.1158/0008-5472.CAN-13-3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 54.Chambers AF, Groom AC, MacDonald IC. 2002. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 55.Chaffer CL, Weinberg RA. 2011. A perspective on cancer cell metastasis. Science 331:1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 56.Nguyen DX, Bos PD, Massague J. 2009. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer 9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 57.Hong SK, Jeong JH, Chan AM, Park JI. 2013. AKT upregulates B-Raf Ser445 phosphorylation and ERK1/2 activation in prostate cancer cells in response to androgen depletion. Exp Cell Res 319:1732–1743. doi: 10.1016/j.yexcr.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Karan D, Schmied BM, Dave BJ, Wittel UA, Lin MF, Batra SK. 2001. Decreased androgen-responsive growth of human prostate cancer is associated with increased genetic alterations. Clin Cancer Res 7:3472–3480. [PubMed] [Google Scholar]
- 59.Karan D, Kelly DL, Rizzino A, Lin MF, Batra SK. 2002. Expression profile of differentially-regulated genes during progression of androgen-independent growth in human prostate cancer cells. Carcinogenesis 23:967–975. doi: 10.1093/carcin/23.6.967. [DOI] [PubMed] [Google Scholar]
- 60.Qin J, Xie Y, Wang B, Hoshino M, Wolff DW, Zhao J, Scofield MA, Dowd FJ, Lin MF, Tu Y. 2009. Upregulation of PIP3-dependent Rac exchanger 1 (P-Rex1) promotes prostate cancer metastasis. Oncogene 28:1853–1863. doi: 10.1038/onc.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mulholland DJ, Dedhar S, Wu H, Nelson CC. 2006. PTEN and GSK3beta: key regulators of progression to androgen-independent prostate cancer. Oncogene 25:329–337. doi: 10.1038/sj.onc.1209020. [DOI] [PubMed] [Google Scholar]
- 62.Rodriguez-Berriguete G, Fraile B, Martinez-Onsurbe P, Olmedilla G, Paniagua R, Royuela M. 2012. MAP kinases and prostate cancer. J Signal Transduct 2012:169170. doi: 10.1155/2012/169170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tumaneng K, Schlegelmilch K, Russell RC, Yimlamai D, Basnet H, Mahadevan N, Fitamant J, Bardeesy N, Camargo FD, Guan KL. 2012. YAP mediates crosstalk between the Hippo and PI(3)K-TOR pathways by suppressing PTEN via miR-29. Nat Cell Biol 14:1322–1329. doi: 10.1038/ncb2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tanaka H, Kono E, Tran CP, Miyazaki H, Yamashiro J, Shimomura T, Fazli L, Wada R, Huang J, Vessella RL, An J, Horvath S, Gleave M, Rettig MB, Wainberg ZA, Reiter RE. 2010. Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat Med 16:1414–1420. doi: 10.1038/nm.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. 2004. Molecular determinants of resistance to antiandrogen therapy. Nat Med 10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 66.Harris WP, Mostaghel EA, Nelson PS, Montgomery B. 2009. Androgen deprivation therapy: progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat Clin Pract Urol 6:76–85. doi: 10.1038/ncpuro1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, Chen Z, Beroukhim R, Wang H, Lupien M, Wu T, Regan MM, Meyer CA, Carroll JS, Manrai AK, Janne OA, Balk SP, Mehra R, Han B, Chinnaiyan AM, Rubin MA, True L, Fiorentino M, Fiore C, Loda M, Kantoff PW, Liu XS, Brown M. 2009. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 138:245–256. doi: 10.1016/j.cell.2009.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chang KH, Li R, Kuri B, Lotan Y, Roehrborn CG, Liu J, Vessella R, Nelson PS, Kapur P, Guo X, Mirzaei H, Auchus RJ, Sharifi N. 2013. A gain-of-function mutation in DHT synthesis in castration-resistant prostate cancer. Cell 154:1074–1084. doi: 10.1016/j.cell.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Drake JM, Graham NA, Lee JK, Stoyanova T, Faltermeier CM, Sud S, Titz B, Huang J, Pienta KJ, Graeber TG, Witte ON. 2013. Metastatic castration-resistant prostate cancer reveals intrapatient similarity and interpatient heterogeneity of therapeutic kinase targets. Proc Natl Acad Sci U S A 110:E4762–E4769. doi: 10.1073/pnas.1319948110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gleave M, Miyake H, Chi K. 2005. Beyond simple castration: targeting the molecular basis of treatment resistance in advanced prostate cancer. Cancer Chemother Pharmacol 56(Suppl 1):S47–S57. doi: 10.1007/s00280-005-0098-0. [DOI] [PubMed] [Google Scholar]
- 71.Sharifi N, Kawasaki BT, Hurt EM, Farrar WL. 2006. Stem cells in prostate cancer: resolving the castrate-resistant conundrum and implications for hormonal therapy. Cancer Biol Ther 5:901–906. doi: 10.4161/cbt.5.8.2949. [DOI] [PubMed] [Google Scholar]
- 72.Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, Liu JO, Pan D. 2012. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev 26:1300–1305. doi: 10.1101/gad.192856.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wu S, Liu Y, Zheng Y, Dong J, Pan D. 2008. The TEAD/TEF family protein Scalloped mediates transcriptional output of the Hippo growth-regulatory pathway. Dev Cell 14:388–398. doi: 10.1016/j.devcel.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 74.Yu FX, Luo J, Mo JS, Liu G, Kim YC, Meng Z, Zhao L, Peyman G, Ouyang H, Jiang W, Zhao J, Chen X, Zhang L, Wang CY, Bastian BC, Zhang K, Guan KL. 2014. Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell 25:822–830. doi: 10.1016/j.ccr.2014.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhao B, Li L, Wang L, Wang CY, Yu J, Guan KL. 2012. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev 26:54–68. doi: 10.1101/gad.173435.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cinar B, Collak FK, Lopez D, Akgul S, Mukhopadhyay NK, Kilicarslan M, Gioeli DG, Freeman MR. 2011. MST1 is a multifunctional caspase-independent inhibitor of androgenic signaling. Cancer Res 71:4303–4313. doi: 10.1158/0008-5472.CAN-10-4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Powzaniuk M, McElwee-Witmer S, Vogel RL, Hayami T, Rutledge SJ, Chen F, Harada S, Schmidt A, Rodan GA, Freedman LP, Bai C. 2004. The LATS2/KPM tumor suppressor is a negative regulator of the androgen receptor. Mol Endocrinol 18:2011–2023. doi: 10.1210/me.2004-0065. [DOI] [PubMed] [Google Scholar]
- 78.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, Asangani IA, Ateeq B, Chun SY, Siddiqui J, Sam L, Anstett M, Mehra R, Prensner JR, Palanisamy N, Ryslik GA, Vandin F, Raphael BJ, Kunju LP, Rhodes DR, Pienta KJ, Chinnaiyan AM, Tomlins SA. 2012. The mutational landscape of lethal castration-resistant prostate cancer. Nature 487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cai J, Zhang N, Zheng Y, de Wilde RF, Maitra A, Pan D. 2010. The Hippo signaling pathway restricts the oncogenic potential of an intestinal regeneration program. Genes Dev 24:2383–2388. doi: 10.1101/gad.1978810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, Wang S, Plaisier S, Garraway IP, Huang J, Graeber TG, Wu H. 2011. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell 19:792–804. doi: 10.1016/j.ccr.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy-Burman P, Nelson PS, Liu X, Wu H. 2003. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 4:209–221. doi: 10.1016/S1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- 82.Chandran UR, Ma C, Dhir R, Bisceglia M, Lyons-Weiler M, Liang W, Michalopoulos G, Becich M, Monzon FA. 2007. Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer 7:64. doi: 10.1186/1471-2407-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]