

Abstract
A gene encoding an enzyme similar to a pyrroloquinoline quinone (PQQ)-dependent sugar dehydrogenase from filamentous fungi, which belongs to new auxiliary activities (AA) family 12 in the CAZy database, was cloned from Pseudomonas aureofaciens. The deduced amino acid sequence of the cloned enzyme showed only low homology to previously characterized PQQ-dependent enzymes, and multiple-sequence alignment analysis showed that the enzyme lacks one of the three conserved arginine residues that function as PQQ-binding residues in known PQQ-dependent enzymes. The recombinant enzyme was heterologously expressed in an Escherichia coli expression system for further characterization. The UV-visible (UV-Vis) absorption spectrum of the oxidized form of the holoenzyme, prepared by incubating the apoenzyme with PQQ and CaCl2, revealed a broad peak at approximately 350 nm, indicating that the enzyme binds PQQ. With the addition of 2-keto-d-glucose (2KG) to the holoenzyme solution, a sharp peak appeared at 331 nm, attributed to the reduction of PQQ bound to the enzyme, whereas no effect was observed upon 2KG addition to authentic PQQ. Enzymatic assay showed that the recombinant enzyme specifically reacted with 2KG in the presence of an appropriate electron acceptor, such as 2,6-dichlorophenol indophenol, when PQQ and CaCl2 were added. 1H nuclear magnetic resonance (1H-NMR) analysis of reaction products revealed 2-keto-d-gluconic acid (2KGA) as the main product, clearly indicating that the recombinant enzyme oxidizes the C-1 position of 2KG. Therefore, the enzyme was identified as a PQQ-dependent 2KG dehydrogenase (Pa2KGDH). Considering the high substrate specificity, the physiological function of Pa2KGDH may be for production of 2KGA.
INTRODUCTION
Pyrroloquinoline quinone (PQQ) is a major cofactor in redox enzymes called quinoproteins and was first identified as a cofactor in bacterial methanol dehydrogenase (1) and glucose dehydrogenase (2), in 1979. The presence of PQQ is a defining feature of quinoprotein enzymes, which distinguishes them from nicotinamide- and flavin-dependent enzymes. In nature, PQQ-dependent quinoproteins have primarily been found as bacterial proteins, localized to the periplasm or bound to membranes, which catalyze the oxidation of various sugars and alcohols, such as glucose, methanol, and ethanol, in the presence of an appropriate electron acceptor (3). Because bacterial PQQ-dependent enzymes require cytochrome c or ubiquinone as an electron acceptor, they are believed to be involved in cellular respiration (4, 5).
Several Gram-negative bacteria, such as Pseudomonas spp. and Gluconobacter spp., possess a pathway for the oxidation of monosaccharides, known as oxidative fermentation. Among these bacteria, Pseudomonas spp. have been reported to accumulate an oxidized form of glucose in culture (6), producing 2-keto-d-gluconic acid (2KGA) from d-glucose, with d-gluconic acid produced as a metabolic intermediate. The 2KGA biosynthetic pathway proceeds through the sequential catalytic actions of two membrane-bound dehydrogenases located on the periplasmic side of the inner cytoplasmic membrane. In this pathway, a membrane-bound, PQQ-dependent d-glucose dehydrogenase catalyzes the oxidation of d-glucose to d-gluconic acid (7, 8). In a consecutive reaction, a membrane-bound, flavin adenine dinucleotide (FAD)-dependent d-gluconate dehydrogenase oxidizes d-gluconic acid to 2KGA (9, 10). Both d-gluconic acid and 2KGA can be transported into the cytoplasm (11, 12), where they are converted to 6-phosphogluconic acid and enter the Entner-Doudoroff pathway, producing pyruvate and glyceraldehyde-3-phosphate for energy metabolism (13–15). The physiological significance of glucose oxidation is thought to be a strategy for securing carbon sources, as few microorganisms are able to metabolize these oxidized substrates (16).
Recently, we identified a novel cellulose-binding hemoquinoprotein sugar dehydrogenase from the basidiomycete Coprinopsis cinerea (CcSDH) (17), which is organized into a three-domain structure consisting of an N-terminal cytochrome electron transfer domain that is similar to the cytochrome b domain, a PQQ-dependent sugar dehydrogenase catalytic domain, and a C-terminal family 1 carbohydrate-binding module (CBM1; previous called a cellulose-binding domain). The PQQ-dependent dehydrogenase domain has very low sequence homology to known quinoproteins and showed the highest activity against 2-keto-d-glucose (2KG), although the enzyme was able to catalyze the oxidation of various sugars. Using BLAST to search for homologs of the amino acid sequence of CcSDH, we identified a gene encoding a protein homologous to CcSDH in the genome databases of Pseudomonas spp. In contrast to CcSDH, the putative PQQ-dependent enzyme was predicted to lack both the N-terminal cytochrome b domain and the C-terminal CBM1. In the present study, we sought to better understand the functions of these bacterial enzymes. For this purpose, we cloned the corresponding gene from Pseudomonas aureofaciens, and the recombinant protein was successfully produced in Escherichia coli. Characterization of this protein revealed that it is a PQQ-dependent 2KG dehydrogenase (2KGDH) which has high specificity for the oxidation of 2KG to 2KGA. Considering these results with previously published work, we discuss the potential physiological function of this enzyme in the metabolic pathway of 2KGA synthesis.
MATERIALS AND METHODS
Strains.
P. aureofaciens ATCC 13985 was used as the source of genetic material for target gene cloning. E. coli strains JM109 (TaKaRa, Japan) and BL21(DE3) (TaKaRa, Japan) were used as hosts for subcloning experiments and heterologous production of recombinant proteins, respectively.
Extraction of genomic DNA from P. aureofaciens.
Genomic DNA was prepared from cells of P. aureofaciens grown in tryptone soy broth (Difco, Detroit, MI). The cells were harvested by centrifugation at 10,000 × g for 5 min. The cell pellet was resuspended in Tris-EDTA (TE) and incubated at 100°C for 10 min. Cellular debris was removed by centrifugation at 10,000 × g for 5 min, and the supernatant was collected. The genomic DNA solution was stored at −20°C until cloning.
Cloning of the gene encoding Pa2KGDH.
For determination of the full-length DNA sequence of 2KGDH from P. aureofaciens (Pa2KGDH), oligonucleotide primers (forward, 5′-CGACTTCGTGCATTCATAGGGAAATCAG-3′; and reverse, 5′-GGCTTGACCCTGTGCCGCAAG-3′) were designed based on the whole-genome shotgun sequence of Pseudomonas chlororaphis subsp. aureofaciens 30-84 (accession number AHHJ01000000). The primers were designed to hybridize to the 5′ and 3′ untranscribed regions, respectively, of a gene annotated an l-sorbosone dehydrogenase gene (accession number gi:397883170). The region containing the full-length DNA sequence of Pa2KGDH was amplified by PCR by using the above primers, genomic DNA from P. aureofaciens as the template, and KOD-Plus, version 2, DNA polymerase (Toyobo, Japan). The PCR product was purified, subcloned into the pGEM-T Easy vector (Promega, Madison, WI), and sequenced.
Sequence analysis.
Homology searches were performed using BLASTP (18, 19) at the National Center for Biotechnology Information website (http://www.ncbi.nlm.nih.gov/BLAST/). All searches were performed using the default settings and the BLOSUM 62 matrix. The presence of a signal peptide was predicted using the SignalP, version 4.1, server (20) on the Center for Biological Sequence Analysis website (http://www.cbs.dtu.dk/services/SignalP/). Multiple-sequence alignment analysis was performed using MAFFT, version 6.85 (21), on the European Bioinformatics Institute website (https://www.ebi.ac.uk/Tools/msa/mafft/). For phylogenetic analysis of the bacterial PQQ-dependent enzymes, complete amino acid sequences were initially aligned using MAFFT and then manually edited using SeaView (22). A phylogenetic tree was constructed from this alignment by using the neighbor-joining method (23) in ClustalX software (24), with 1,000 bootstraps.
Heterologous expression of recombinant Pa2KGDH.
The DNA fragment encoding mature Pa2KGDH was obtained by a PCR using oligonucleotide primers (forward, 5′-CATATGGGCGAAACCTCCACCCTCCAG-3′; and reverse, 5′-GCGGCCGCTTGCGCCTTGGCTGCTGACACG-3′) designed from the nucleotide sequence for cloned Pa2KGDH, with genomic DNA from P. aureofaciens as the template. The primers contain restriction endonuclease sites for NdeI and NotI (underlined) for insertion into the corresponding sites within the pET-21a(+) expression vector (Novagen, Madison, WI). The PCR product was purified and ligated into the pGEM-T Easy vector. The sequence of the cloned gene was confirmed by DNA sequencing. The target gene was digested with NdeI and NotI and then ligated into the multiple-cloning site of the pET-21a(+) vector. E. coli BL21(DE3) was transformed with the resultant plasmid. Transformants were grown in LB medium containing 100 μg/ml ampicillin with shaking (180 rpm) at 37°C until they reached an optimal optical density at 600 nm of 0.2, and expression was induced with 0.1 mM isopropyl-β-d-thiogalactopyranoside at 18°C. After an 18-h induction, cells were harvested by centrifugation at 10,000 × g for 5 min, suspended in 20 mM potassium phosphate buffer (pH 7.4) containing 0.5 M NaCl and 20 mM imidazole, and disrupted using a VP-050 ultrasonic homogenizer (Titech Co., Ltd., Japan). The sonicated cell suspension was centrifuged at 5,000 × g for 5 min to remove cellular debris and unbroken cells. The resulting supernatant was used as a cell extract for protein purification.
Purification of recombinant Pa2KGDH.
All buffers used for purification were kept at 4°C. The cell extract was applied to a HisTrap FF crude 5-ml column (GE Healthcare, Sweden). Recombinant Pa2KGDH was purified using standard immobilized-metal affinity chromatography as described by the manufacturer. After purification, the protein solution was dialyzed against 20 mM HEPES (pH 7.0). The solution was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) to confirm the purity of the preparation. The N-terminal amino acid sequence was determined using the Procise cLC 491 protein sequencing system (Applied Biosystems, Foster City, CA).
UV-Vis spectral characterization.
The UV-visible (UV-Vis) spectra of recombinant Pa2KGDH were recorded in both the oxidized and reduced states by using a UV-Vis spectrophotometer. The holoenzyme was prepared by incubating the apoenzyme at 4°C for 30 min in 50 mM HEPES (pH 7.0) with 100 μM PQQ and 100 mM CaCl2. Excess PQQ and CaCl2 were removed by ultrafiltration with a 5,000-molecular-weight (MW)-cutoff Vivaspin 500 filter (Sartorius Stedim, Germany) at 15,000 × g, followed by addition of 20 mM HEPES (pH 7.0). Ultrafiltration was repeated until all excess additives were removed completely. The reduced form of the holoenzyme was prepared by addition of 2KG.
Enzyme activity assay.
Enzyme activity was measured spectrophotometrically by reduction of 2,6-dichlorophenol indophenol (DCPIP) at 600 nm, coupled with phenazine methosulfate (PMS), at 37°C. Enzyme activity was calculated using an extinction coefficient of DCPIP of 600 nm (21.0 mM−1 cm−1). For substrate specificity testing, the reaction mixture contained the enzyme solution, 0.1 mM PQQ, 1 mM CaCl2, 50 mM HEPES (pH 7.0), 1 mM substrate, 0.7 mM PMS, and 0.1 mM DCPIP in a total volume of 500 μl. Substrates added were as follows: d-allose, l-allose, d-galactose, l-galactose, d-glucose, l-glucose, d-gulose, l-gulose, d-mannose, l-mannose, d-tallose, l-tallose, d-arabinose, l-arabinose, d-lyxose, l-lyxose, d-xylose, l-xylose, d-fructose, l-fructose, d-tagatose, l-tagatose, l-sorbose, maltose, sophorose, d-fucose, l-fucose, l-rhamnose, d-ribose, l-ribose, N-acetyl-d-glucosamine, d-glucosamine, 2KG, and cellobiose.
The effect of pH on activity was examined by using 2KG as the substrate in a series of 50 mM MES (morpholineethanesulfonic acid)-NaOH buffers ranging in pH from 5.5 to 6.5, 50 mM PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)]-NaOH buffers ranging in pH from 6.5 to 7.5, and 50 mM Tris-HCl buffers ranging in pH from 7.5 to 9.0. For kinetic analyses, the activity assay was performed with 0.5 to 32 mM 2KG as the substrate, with a 50 nM enzyme solution in 50 mM PIPES-NaOH buffer (pH 7.5) containing 1 μM PQQ and 1 mM CaCl2. To investigate metal ion dependence, enzyme activity was monitored using the protocol described above, except that CaCl2 was replaced with MgCl2. The total protein concentration was determined using a Pierce bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific, Rockford, IL) according to the manufacturer's instructions, using bovine serum albumin as a standard.
Analysis of catalytic reaction products.
To acquire 1H nuclear magnetic resonance (1H-NMR) spectra of the reaction products, the reaction mixture was prepared as follows: 300 μM holoenzyme, 50 mM HEPES (pH 7.0), 0.3 mM PMS, 16.9 mM DCPIP, and 16.9 mM 2-keto-d-glucose in a total volume of 1 ml. After incubation for 12 h at 28°C, the reaction mixture was frozen by use of liquid nitrogen, lyophilized, and dissolved in D2O. 1H-NMR spectra were recorded on a JEOL ECA600 spectrometer (JEOL, Japan). The chemical shifts were referenced by setting the value for 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt to 0 ppm.
Nucleotide sequence accession number.
The nucleotide sequence of the gene encoding Pa2KGDH has been deposited in the DDBJ database under accession number AB932861.
RESULTS
Nucleotide and amino acid sequence analyses.
Using the PQQ-dependent sugar dehydrogenase CcSDH (accession number gi:397883170) as a query, a BLAST search against the NCBI database identified a homologous gene in P. aureofaciens. The gene has been annotated an l-sorbosone dehydrogenase gene, although its experimental characterization has not been reported. The corresponding gene was cloned from the genomic DNA of P. aureofaciens strain ATCC 13985. The gene consisted of a 1,317-bp open reading frame, indicating that it encodes a protein of 439 amino acids. When the nucleotide and amino acid sequences were compared with those in the NCBI database, 67 nucleotides and 9 amino acids (D59G, D78E, I108F, A147V, R184K, S186N, K236E, T344A, and N400S) were different, possibly due to strain differences.
The N-terminal 19 amino acids were predicted to form a signal peptide by the SignalP program, suggesting that the mature protein consists of 420 amino acids, with a molecular mass of 45.0 kDa and a pI of 6.1. The amino acid sequence showed the highest similarity among characterized proteins with the catalytic domain of CcSDH. However, in contrast to CcSDH, the protein consists of only the catalytic domain. Structural homology was also observed with bacterial and archaeal PQQ-dependent enzymes shown to have a six-bladed β-propeller structure by three-dimensional structural analysis. These homologous enzymes were, in order of decreasing similarity, aldose sugar dehydrogenase (Asd) from E. coli (25% identity; PDB accession number 2G8S), Asd from Streptomyces coelicolor (22% identity; PDB accession number 3DAS), Asd from Pyrobaculum aerophilum (21% identity; PDB accession number 3A9G), and glucose dehydrogenase from Acinetobacter calcoaceticus (17% identity; PDB accession number 1CRU).
The amino acid sequences of Pa2KGDH, CcSDH, and bacterial and archaeal PQQ-dependent enzymes structurally homologous to Pa2KGDH were aligned using MAFFT (Fig. 1). A histidine in Pa2KGDH (His203) was identified at the same position as a histidine predicted to be the catalytic residue in previously characterized enzymes. Three arginine residues are known to be involved in PQQ binding in bacterial and archaeal enzymes. Among them, two were conserved in Pa2KGDH (Arg259 and Arg408), whereas one was replaced with valine (Val410). In addition, Asn260, which was predicted to interact with the O-4 atom of PQQ, was found in Pa2KGDH. On the other hand, the basic amino acid residue that was predicted to make an ion pair interaction with the C-7 carboxyl group of PQQ (e.g., Lys377 in the soluble glucose dehydrogenase from Acinetobacter calcoaceticus) was not found in Pa2KGDH.
FIG 1.

Alignment of the Pa2KGDH amino acid sequence with those of known six-bladed quinoproteins. Perfect matches are boxed with a black background. Open boxes represent amino acid residues predicted to interact with PQQ via direct hydrogen bonding in known enzymes. The black arrowhead indicates the predicted catalytic histidine residue. Open arrowheads indicate conserved residues involved in PQQ binding in Pa2KGDH and known quinoproteins (27, 33, 34). 1CRU, glucose dehydrogenase from A. calcoaceticus; 3A9G, Asd from Pyrobaculum aerophilum; 3DAS, Asd from Streptomyces coelicolor; 2G8S, Asd from E. coli.
Physical and spectral properties of recombinant Pa2KGDH.
The soluble recombinant hexahistidine-tagged Pa2KGDH enzyme was heterologously expressed in E. coli and recovered from the cell extract by affinity chromatography using a Ni-nitrilotriacetic acid (Ni-NTA) column. The purity and molecular mass of the purified enzyme were confirmed by SDS-PAGE, which showed a single band of approximately 47 kDa (Fig. 2). The molecular mass observed was almost identical to that estimated from the amino acid sequence of this enzyme. The identity of the purified enzyme was confirmed by N-terminal amino acid sequencing, which determined the first 10 residues to be “GETSTLQVSD.”
FIG 2.
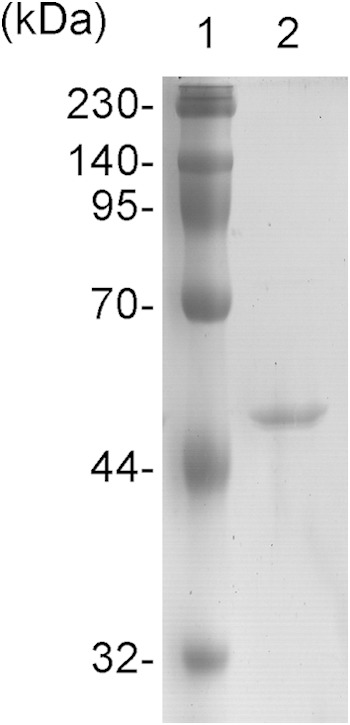
SDS-PAGE analysis of purified recombinant Pa2KGDH. Lane 1, molecular mass standard; lane 2, purified recombinant Pa2KGDH.
PQQ binding to the recombinant enzyme was examined spectrophotometrically. The UV-Vis absorption spectrum of the apo form of recombinant Pa2KGDH showed a sharp peak at around 280 nm that was attributed to the protein (Fig. 3). When the holoenzyme was prepared by incubating the apoenzyme with PQQ and CaCl2, followed by removal of excess additives, a broad peak was observed close to 350 nm, suggesting that Pa2KGDH binds to PQQ. Moreover, the addition of 2KG to the oxidized enzyme resulted in a sharp peak at 331 nm, whereas no effect was observed upon 2KG addition to authentic PQQ (Fig. 3, inset). This indicates that PQQ is reduced only when bound to the recombinant enzyme, with the addition of 2KG.
FIG 3.
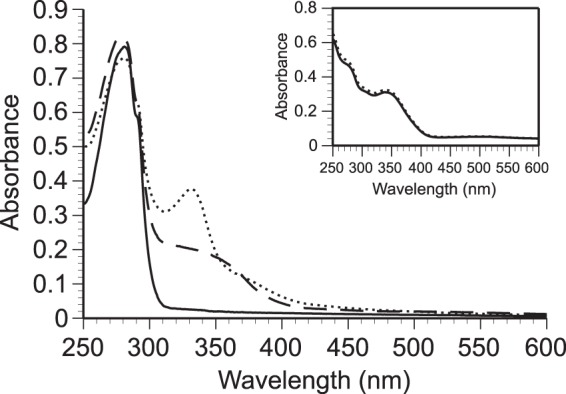
Absorption spectra of recombinant Pa2KGDH. Solid line, apoenzyme; dashed line, oxidized holoenzyme; dotted line, reduced holoenzyme. The reduced form was prepared by addition of 2-keto-d-glucose. (Inset) UV-Vis absorption spectra of authentic PQQ. Solid line, authentic PQQ; dotted line, PQQ with 2-keto-d-glucose. All determinations were conducted in 20 mM HEPES-NaOH buffer (pH 7.0) at room temperature.
Enzymatic properties of recombinant Pa2KGDH.
To investigate the role of PQQ in Pa2KGDH catalytic activity, we tested the activity of the recombinant enzyme with 2KG, using DCPIP as an electron acceptor in the presence or absence of PQQ. DCPIP was appreciably reduced when the enzyme was mixed with 2KG in the presence of PQQ and CaCl2, whereas no activity was observed in their absence (data not shown). These results clearly indicate that PQQ is necessary for the catalytic activity of Pa2KGDH. To date, the ability to oxidize 2KG has been observed for PQQ-dependent enzymes such as CcSDH (17) and l-sorbosone dehydrogenase from Ketogulonicigenium vulgare DSM 4025 (25). These enzymes demonstrated broad substrate specificities against various sugars, in addition to 2KG. Recombinant Pa2KGDH displayed significant activity against 2KG, as predicted in spectral studies, whereas no or very low activity was observed against other substrates (Table 1). These results indicate that the catalytic activity of Pa2KGDH is specific to 2KG.
TABLE 1.
Activities of recombinant Pa2KGDH toward various mono- and disaccharidesa
| Sugar | Pa2KGDH activity (μmol/min/mg) |
|---|---|
| 2-Keto-d-glucose | 1,479.45 |
| d-Glucose | 0.38 |
| l-Glucose | 0.15 |
| d-Mannose | 0.88 |
| l-Mannose | 1.03 |
| d-Galactose | 0.03 |
| l-Galactose | 2.13 |
| d-Allose | 2.55 |
| l-Allose | 6.64 |
| d-Talose | 9.83 |
| l-Talose | 0.47 |
| d-Gulose | 1.40 |
| l-Gulose | 5.88 |
| d-Xylose | ND |
| l-Xylose | 4.26 |
| d-Lyxose | 3.68 |
| l-Lyxose | 2.60 |
| d-Arabinose | 8.30 |
| l-Arabinose | 0.26 |
| d-Fucose | 0.47 |
| l-Fucose | 7.20 |
| d-Fructose | ND |
| l-Fructose | ND |
| d-Tagatose | 3.22 |
| l-Tagatose | 2.71 |
| l-Rhamnose | 2.13 |
| l-Sorbose | 2.23 |
| d-Glucosamine | 3.21 |
| N-Acetyl-d-glucosamine | 0.30 |
| Cellobiose | 0.20 |
| Maltose | ND |
| Sophorose | 0.15 |
| d-Ribose | ND |
| l-Ribose | 5.94 |
Enzyme activity was monitored by the reduction of DCPIP in 50 mM HEPES (pH 7.0) containing 1 mM substrate at 37°C. ND, not detected.
The effect of pH on recombinant Pa2KGDH activity was examined by using a series of buffers ranging in pH from 5.5 to 9.0. The recombinant enzyme displayed optimal activity at pH 7.0 to 7.5 in PIPES-NaOH buffers, while activities at pH 6.0 and 8.0 were decreased by more than half. Therefore, kinetic analysis of 2KG oxidation by recombinant Pa2KGDH was conducted in a pH 7.5 PIPES-NaOH buffer. The Km and kcat for the substrate were measured as 43.9 ± 9.9 mM and 32.0 ± 4.7 s−1, respectively. The Km values of PQQ-dependent enzymes with a six-bladed β-propeller structure are known to be high, generally above the millimolar range. For example, the Km values of the soluble glucose dehydrogenase from A. calcoaceticus for d-glucose and of Asd from E. coli for d-glucose were reported to be 22 mM and 400 mM, respectively (26, 27), similar to our results for Pa2KGDH. It is well known that PQQ-dependent enzymes require divalent metal ions, such as calcium, to bind PQQ (28). In the present study, the effects of divalent metal ions on the catalytic activity of recombinant Pa2KGDH were examined by using CaCl2 and MgCl2 in the reaction buffers. Significant activities were observed with both CaCl2 and MgCl2, although the enzymatic activity in the presence of CaCl2 was higher than that with MgCl2 (data not shown). Therefore, Pa2KGDH seems to prefer calcium for binding to PQQ, although both metal ions can be utilized.
Identification of reaction products.
The reaction products of the oxidation of 2KG by recombinant Pa2KGDH were determined by 1H-NMR analysis (Fig. 4). The 1H-NMR spectrum of the reaction products was almost identical to that of 2KGA, suggesting that the enzyme oxidizes the C-1 position of 2KG. To the best of our knowledge, this is the first report of an enzyme that specifically attacks the C-1 position of 2KG to produce 2KGA.
FIG 4.
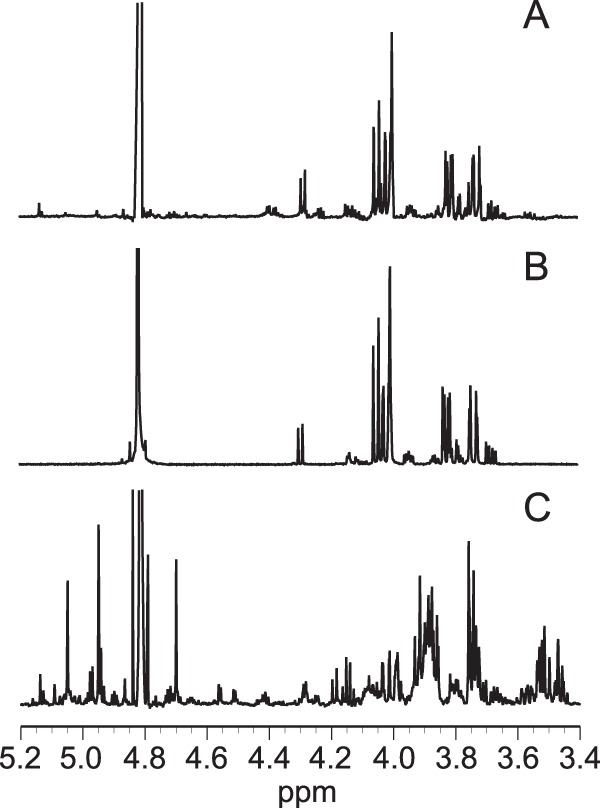
Partial 1H-NMR spectra of reaction products (A), 2-keto-d-gluconic acid (B), and 2-keto-d-glucose (C). 3-(Trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt was used as a reference (0 ppm).
DISCUSSION
PQQ-dependent quinoproteins are divided into the following two groups based on their structure: those containing an eight-bladed β-propeller where each blade consists of a four-stranded antiparallel β-sheet (29–32) and those with a six-bladed β-propeller, such as soluble glucose dehydrogenase from A. calcoaceticus and soluble Asd from E. coli and P. aerophilum (27, 33, 34). Members of these two groups do not have significant primary sequence homology. In the present study, we cloned a gene encoding a putative quinoprotein from P. aureofaciens which shows homology to known PQQ-dependent enzymes with a six-bladed β-propeller structure. The protein demonstrated no similarity to eight-bladed β-propeller-containing quinoproteins. Consistent with the primary sequence homology, UV-Vis spectral analysis and enzymatic analyses using the recombinant enzyme suggested that the gene encodes a PQQ-dependent enzyme, as PQQ was both bound to the enzyme and required for its catalytic activity. Interestingly, the enzyme lacks one of the three arginine residues that are known to be involved in PQQ binding in other PQQ-dependent quinoproteins. Several amino acid residues that are predicted to be associated with the enzyme-PQQ interaction were similarly not conserved. Together, these facts clearly indicate that Pa2KGDH is a PQQ-dependent enzyme with features distinct from those of known bacterial PQQ-dependent enzymes. Similar structural characteristics were observed for CcSDH (17), which also contains only two of the conserved arginine residues involved in PQQ binding (Fig. 1). Therefore, in the case of Pa2KGDH, other amino acid residues are likely to be involved in PQQ binding, as for CcSDH, although the three-dimensional structures of the enzymes are still unsolved.
Several bacteria, such as acetic acid bacteria and Pseudomonas spp., have been known to accumulate oxidized forms of d-glucose, including d-gluconic acid, 2KGA, and 5-keto-d-gluconic acid, in culture (6, 35). The productivity of these oxidized sugars depends on the microbial species. For example, the acetic acid bacteria Gluconobacter spp. produce 2KGA and 5-keto-d-gluconate in addition to d-gluconic acid, whereas Pseudomonas spp. are unable to synthesize 5-keto-d-gluconate. In Pseudomonas spp., d-glucose is oxidized to d-gluconic acid by a periplasmic membrane-bound, PQQ-dependent d-glucose dehydrogenase, and the resulting d-gluconic acid is further oxidized to 2KGA by a periplasmic membrane-bound, FAD-dependent d-gluconate dehydrogenase (7–10). In the present study, enzymatic analysis using recombinant Pa2KGDH demonstrated that the enzyme reacted with 2KG to produce 2KGA, as identified by 1H-NMR analysis, and that the substrate specificity was very specific. Therefore, the physiological role of this enzyme in this bacterium may be in the production of 2KGA (Fig. 5). However, this hypothesis predicts the presence of an enzyme capable of oxidizing the C-2 position of d-glucose, which is so far undiscovered in Pseudomonas spp. To date, only the FAD-dependent enzymes pyranose oxidase (POX) and pyranose dehydrogenase (PDH) have been shown to oxidize d-glucose to 2KG (36, 37). However, these enzymes have been isolated only from fungi, and a corresponding prokaryotic enzyme has not yet been identified. Therefore, the physiological role of Pa2KGDH is still unclear, and more studies will be needed to clarify the pathway related to the metabolism of 2KGA via Pa2KGDH.
FIG 5.
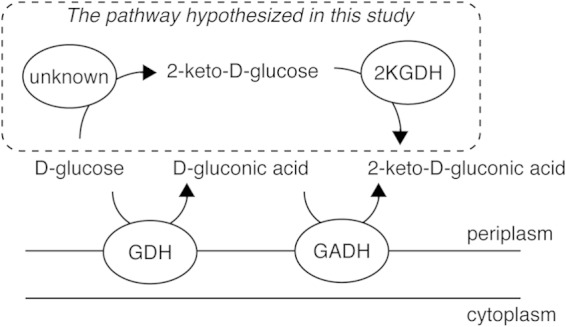
Putative pathway of 2-keto-gluconic acid synthesis in the periplasm of Pseudomonas aureofaciens. The pathway proposed in the present study is enclosed by a dashed line. GDH, d-glucose dehydrogenase; GADH, d-gluconate dehydrogenase; 2KGDH, 2-keto-glucose dehydrogenase.
As discussed above, the primary sequence of Pa2KGDH is remarkably distinct from those of previously characterized prokaryotic PQQ-dependent enzymes. However, results from the BLAST search of the NCBI protein database identified many genes exhibiting significant homology to Pa2KGDH in various bacteria. As shown in Fig. 6, a phylogenetic tree demonstrates that PQQ enzymes homologous to Pa2KGDH are clearly separated from known PQQ-dependent enzymes. This indicates that these quinoproteins, including Pa2KGDH, form a new family of bacterial quinoproteins. The bacterial strains which possess genes encoding this newly identified family of enzymes belong to the phyla Proteobacteria, Bacteroidetes, Cyanobacteria, Acidobacteria, and Spirochaetes. The majority of these genes were found in the phyla Proteobacteria and Bacteroidetes, while a small number of strains of Actinobacteria, a group of Gram-positive bacteria, were found to contain homologous genes. These results suggest that this new family of PQQ-dependent enzymes is distributed mainly among Gram-negative bacteria. Features of the amino acid sequences of these enzymes predict the proteins to be membrane bound, extracellular, or intracellular. This distinct localization pattern indicates that these localization signals emerged after the separation of the enzymes into groups I and II.
FIG 6.

Phylogenetic tree for Pa2KGDH, similar putative dehydrogenases, and known PQQ-dependent enzymes. Pa2KGDH from the present study is boxed. The tree was generated from the amino acid sequences of Pa2KGDH, proteins homologous to Pa2KGDH, and known quinoproteins by using the neighbor-joining method in ClustalX (v. 2.1). 4AAH, methanol dehydrogenase from Methylophilus W3A1; 1CRU, soluble glucose dehydrogenase from A. calcoaceticus; 2G8S, soluble Asd from E. coli; 3A9G, Asd from P. aerophilum; AHE53529.1, hypothetical protein from Sphingomonas sanxanigenens; AHI25399.1, putative l-sorbosone dehydrogenase from Gluconacetobacter xylinus; AHI25551.1, l-sorbosone dehydrogenase from Gluconacetobacter xylinus; EXI83910.1, putative membrane-bound dehydrogenase domain protein from “Candidatus Accumulibacter sp.”; NP 634897.1, l-sorbosone dehydrogenase from Methanosarcina mazei; Q44091.1, l-sorbosone dehydrogenase from Gluconacetobacter liquefaciens; WP_002638173.1, sorbosone dehydrogenase from Myxococcus sp.; WP_003423316.1, l-sorbosone dehydrogenase from Pseudomonas syringae; WP_003630272.1, l-sorbosone dehydrogenase from Acetobacter pasteurianus; WP_004449305.1, l-sorbosone dehydrogenase from Acetobacter pasteurianus; WP_005046264.1, hypothetical protein from Acinetobacter calcoaceticus; WP_005246918.1, hypothetical protein from Acinetobacter sp.; WP_005284126.1, hypothetical protein from Acinetobacter sp.; WP_006117236.1, l-sorbosone dehydrogenase from Acetobacter pomorum; WP_006219208.1, sorbosone dehydrogenase from Achromobacter piechaudii; WP_006557961.1, sorbosone dehydrogenase from Acetobacter tropicalis; WP_006559625.1, l-sorbosone dehydrogenase from Acetobacter tropicalis; WP_006913414.1, sorbosone dehydrogenase from Salinisphaera shabanensis; WP_006993817.1, l-sorbosone dehydrogenase from Glaciecola mesophila; WP_007398410.1, sorbosone dehydrogenase from Gluconacetobacter sp.; WP_008850969.1, hypothetical protein from Gluconobacter morbifer; WP_009510150.1, l-sorbosone dehydrogenase from Acinetobacter baumannii; WP_009816668.1, sorbosone dehydrogenase from Roseovarius sp.; WP_010162075.1, sorbosone dehydrogenase from Sphingomonas sp.; WP_010665785.1, l-sorbosone dehydrogenase from Acetobacter aceti; WP_016418137.1, sorbosone dehydrogenase from Halomonas anticariensis; WP_016555642.1, NHL repeat-containing protein from Rhizobium grahamii; WP_017296897.1, hypothetical protein from Nodosilinea nodulosa; WP_017845324.1, sorbosone dehydrogenase from Pseudomonas veronii; WP_017902167.1, sorbosone dehydrogenase from Pseudomonas fuscovaginae; WP_018005083.1, sorbosone dehydrogenase from Cupriavidus taiwanensis; WP_018271647.1, hypothetical protein from Bradyrhizobium elkanii; WP_019087754.1, l-sorbosone dehydrogenase from Acetobacter pasteurianus; WP_019342104.1, sorbosone dehydrogenase from Pseudomonas stutzeri; WP_019495027.1, hypothetical protein from Calothrix sp.; WP_020079230.1, sorbosone dehydrogenase from Enterobacter aerogenes; WP_020090605.1, sorbosone dehydrogenase from Methylobacterium sp.; WP_020164113.1, sorbosone dehydrogenase from Methyloversatilis universalis; WP_020175448.1, hypothetical protein from Methyloferula stellata; WP_020340026.1, l-sorbosone dehydrogenase from Pseudomonas syringae; WP_020402680.1, hypothetical protein from Gracilimonas tropica; WP_021224936.1, sorbosone dehydrogenase from Sphingobium lactosutens; WP_021317704.1, sorbosone dehydrogenase from Sphingobium ummariense; WP_023096705.1, l-sorbosone dehydrogenase from Pseudomonas aeruginosa; WP_023430763.1, l-sorbosone dehydrogenase from Lutibaculum baratangense; WP_023794762.1, sorbosone dehydrogenase from Mesorhizobium sp.; YP_001236509.1, glucose/sorbosone dehydrogenase from Bradyrhizobium sp.; YP_001260163.1, PEBP family protein from Sphingomonas wittichii; YP_001600858.1, l-sorbosone dehydrogenase from Gluconacetobacter diazotrophicus; YP_001603987.1, l-sorbosone dehydrogenase from Gluconacetobacter diazotrophicus; YP_001847059.1, glucose/sorbosone dehydrogenase from Acinetobacter baumannii; YP_002275820.1, putative l-sorbosone dehydrogenase from Gluconacetobacter diazotrophicus; YP_002276996.1, putative l-sorbosone dehydrogenase from Gluconacetobacter diazotrophicus; YP_002356753.1, l-sorbosone dehydrogenase from Shewanella baltica; YP_002897724.1, putative l-sorbosone dehydrogenase homolog from Burkholderia pseudomallei; YP_003188053.1, l-sorbosone dehydrogenase from Acetobacter pasteurianus; YP_004867201.1, l-sorbosone dehydrogenase from Gluconacetobacter medellinensis; YP_005220639.1, glucose/sorbosone dehydrogenase from Rahnella aquatilis; YP_005806231.1, hypothetical protein BbuN40_0024 from Borrelia burgdorferi; YP_005919903.1, l-sorbosone dehydrogenase from Methanosaeta harundinacea; YP_008391304.1, l-sorbosone dehydrogenase from Acetobacter pasteurianus; YP_008867371.1, sorbosone dehydrogenase from Hyphomicrobium nitrativorans; YP_008881332.1, sorbosone dehydrogenase from Pandoraea sp.; YP_352796.1, putative glucose/sorbosone dehydrogenase from Rhodobacter sphaeroides; YP_363846.1, l-sorbosone dehydrogenase from Xanthomonas campestris; YP_449666.1, l-sorbosone dehydrogenase from Xanthomonas oryzae; YP_604471.1, NHL repeat-containing protein from Deinococcus geothermalis; YP_684659.1, putative l-sorbosone dehydrogenase from Methanocella arvoryzae; YP_779983.1, putative l-sorbosone dehydrogenase from Rhodopseudomonas palustris; YP_783770.1, l-sorbosone dehydrogenase from Rhodopseudomonas palustris; YP_993848.1, glucose/sorbosone dehydrogenases from Burkholderia mallei.
In conclusion, the present study demonstrated that Pa2KGDH is a novel PQQ-dependent enzyme with structural features that are distinct from those of known bacterial PQQ-dependent enzymes. Pa2KGDH specifically preferred 2KG as a substrate and oxidized the C-1 position of 2KG, indicating that the enzyme is a 2KGDH. In addition, a homology search revealed the presence of genes similar to the Pa2KGDH gene in various bacteria. This PQQ-dependent enzyme provides novel insight into the pathway of oxidized sugar metabolism in bacteria.
ACKNOWLEDGMENTS
This research was financially supported by a grant-in-aid for JSPS fellows (grant 26-8518 to K. Umezawa) from the Japan Society for the Promotion of Science and by grants-in-aid for innovative areas (grants 24114001 and 24114008 to K. Igarashi) from the Japanese Ministry of Education, Culture, Sports, and Technology (MEXT).
REFERENCES
- 1.Salisbury SA, Forrest HS, Cruse WBT, Kennard O. 1979. A novel coenzyme from bacterial primary alcohol dehydrogenases. Nature 280:843–844. doi: 10.1038/280843a0. [DOI] [PubMed] [Google Scholar]
- 2.Duine JA, Frank JJ, van Zeeland JK. 1979. Glucose dehydrogenase from Acinetobacter calcoaceticus: a ‘quinoprotein.’ FEBS Lett 108:443–446. doi: 10.1016/0014-5793(79)80584-0. [DOI] [PubMed] [Google Scholar]
- 3.Matsushita K, Toyama H, Yamada M, Adachi O. 2002. Quinoproteins: structure, function, and biotechnological applications. Appl Microbiol Biotechnol 58:13–22. doi: 10.1007/s00253-001-0851-1. [DOI] [PubMed] [Google Scholar]
- 4.Matsushita K, Yakushi T, Toyama H, Shinagawa E, Adachi O. 1996. Function of multiple heme c moieties in intramolecular electron transport and ubiquinone reduction in the quinohemoprotein alcohol dehydrogenase-cytochrome c complex of Gluconobacter suboxydans. J Biol Chem 271:4850–4857. doi: 10.1074/jbc.271.9.4850. [DOI] [PubMed] [Google Scholar]
- 5.Matsushita K, Toyama H, Adachi O. 2004. Respiratory chains in acetic acid bacteria: membrane-bound periplasmic sugar and alcohol respirations. Adv Photosynth Respir 16:81–99. doi: 10.1007/978-1-4020-3163-2_4. [DOI] [Google Scholar]
- 6.Lockwood LB, Tabenkin B, Ward GE. 1941. The production of gluconic acid and 2-keto-gluconic acid from glucose by species of Pseudomonas and Phytomonas. J Bacteriol 42:51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsushita K, Ohno Y, Shinagawa E, Adachi O, Ameyama M. 1980. Membrane-bound d-glucose dehydrogenase from Pseudomonas sp.: solubilization, purification and characterization. Agric Biol Chem 44:1505–1512. doi: 10.1271/bbb1961.44.1505. [DOI] [Google Scholar]
- 8.Ameyama M, Matsushita K, Ohno Y, Shinagawa E, Adachi O. 1981. Existence of a novel prosthetic group, PQQ, in membrane-bound, electron transport chain-linked, primary dehydrogenases of oxidative bacteria. FEBS Lett 130:179–183. doi: 10.1016/0014-5793(81)81114-3. [DOI] [PubMed] [Google Scholar]
- 9.Matsushita K, Shinagawa E, Adachi O, Ameyama M. 1979. Membrane-bound d-gluconate dehydrogenase from Pseudomonas aeruginosa. Purification and structure of cytochrome-binding form. J Biochem 85:1173–1181. [PubMed] [Google Scholar]
- 10.Matsushita K, Shinagawa E, Adachi O, Ameyama M. 1979. Membrane-bound d-gluconate dehydrogenase from Pseudomonas aeruginosa. Its kinetic properties and a reconstitution of gluconate oxidase. J Biochem 86:249–256. [PubMed] [Google Scholar]
- 11.Guymon LF, Eagon RG. 1974. Transport of glucose, gluconate, and methyl α-d-glucoside by Pseudomonas aeruginosa. J Bacteriol 117:1261–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Agbanyo F, Taylor NF. 1985. The active transport of 2-keto-d-gluconate in vesicles prepared from Pseudomonas purida. J Biochem 228:257–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Narrod SA, Wood WA. 1956. Carbohydrate oxidation by Pseudomonas fluorescens. V. Evidence for gluconokinase and 2-ketogluconokinase. J Biol Chem 220:45–55. [PubMed] [Google Scholar]
- 14.Roberts BK, Midgley M, Dawes EA. 1973. The metabolism of 2-oxogluconate by Pseudomonas aeruginosa. J Gen Microbiol 78:319–329. doi: 10.1099/00221287-78-2-319. [DOI] [PubMed] [Google Scholar]
- 15.Frampton EW, Wood WA. 1961. Carbohydrate oxidation by Pseudomonas fluorescens. VI. Conversion of 2-keto-6-phosphogluconate to pyruvate. J Biol Chem 236:2571–2577. [PubMed] [Google Scholar]
- 16.Whiting PH, Midgley M, Dawes EA. 1976. The role of glucose limitation in the regulation of the transport of glucose, gluconate and 2-oxogluconate, and of glucose metabolism in Pseudomonas aeruginosa. J Gen Microbiol 92:304–310. doi: 10.1099/00221287-92-2-304. [DOI] [PubMed] [Google Scholar]
- 17.Matsumura H, Umezawa K, Takeda K, Sugimoto N, Ishida T, Samejima M, Ohno H, Yoshida M, Igarashi K, Nakamura N. 2014. New family of eukaryotic pyrroloquinoline quinone-dependent oxidoreductases. PLoS One 9:e104851. doi: 10.1371/journal.pone.0104851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 19.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 21.Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galtier N, Gouy M, Gautier C. 1996. SEAVIEW and PHYLO_WIN: two graphic tools for sequence alignment and molecular phylogeny. Comput Appl Biosci 12:543–548. doi: 10.1093/bioinformatics/12.6.543. [DOI] [PubMed] [Google Scholar]
- 23.Saitou N, Nei M. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425. [DOI] [PubMed] [Google Scholar]
- 24.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. 1997. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyazaki T, Sugisawa T, Hoshino T. 2006. Pyrroloquinoline quinone-dependent dehydrogenases from Ketogulonicigenium vulgare catalyze the direct conversion of l-sorbosone to l-ascorbic acid. Appl Environ Microbiol 72:1487–1495. doi: 10.1128/AEM.72.2.1487-1495.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dokter P, Frank J, Duine JA. 1986. Purification and characterization of quinoprotein glucose dehydrogenase from Acinetobacter calcoaceticus L.M.D. 79.41. Biochem J 239:163–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Southall SM, Doel JJ, Richardson DJ, Oubrie A. 2006. Soluble aldose sugar dehydrogenase from Escherichia coli: a highly exposed active site conferring broad substrate specificity. J Biol Chem 281:30650–30659. doi: 10.1074/jbc.M601783200. [DOI] [PubMed] [Google Scholar]
- 28.Matsushita K, Toyama H, Ameyama M, Adachi O, Dewanti A, Duine JA. 1995. Soluble and membrane-bound quinoprotein d-glucose dehydrogenases of the Acinetobacter calcoaceticus: the binding process of PQQ to the apoenzymes. Biosci Biotechnol Biochem 59:1548–1555. doi: 10.1271/bbb.59.1548. [DOI] [Google Scholar]
- 29.Ghosh M, Anthony C, Harlos K, Goodwin MG, Blake C. 1995. The refined structure of the quinoprotein methanol dehydrogenase from Methylobacterium extorquens at 1.94 A. Structure 3:177–187. doi: 10.1016/S0969-2126(01)00148-4. [DOI] [PubMed] [Google Scholar]
- 30.Keitel T, Diehl A, Knaute T, Stezowski JJ, Höhne W, Görisch H. 2000. X-ray structure of the quinoprotein ethanol dehydrogenase from Pseudomonas aeruginosa: basis of substrate specificity. J Mol Biol 297:961–974. doi: 10.1006/jmbi.2000.3603. [DOI] [PubMed] [Google Scholar]
- 31.Oubrie A, Rozeboom HJ, Kalk KH, Huizinga EG, Dijkstra BW. 2002. Crystal structure of quinohemoprotein alcohol dehydrogenase from Comamonas testosteroni: structural basis for substrate oxidation and electron transfer. J Biol Chem 277:3727–3732. doi: 10.1074/jbc.M109403200. [DOI] [PubMed] [Google Scholar]
- 32.Chen ZW, Matsushita K, Yamashita T, Fujii TA, Toyama H, Adachi O, Bellamy HD, Mathews FS. 2002. Structure at 1.9 A resolution of a quinohemoprotein alcohol dehydrogenase from Pseudomonas putida HK5. Structure 10:837–849. doi: 10.1016/S0969-2126(02)00774-8. [DOI] [PubMed] [Google Scholar]
- 33.Oubrie A, Rozeboom HJ, Dijkstra BW. 1999. Active-site structure of the soluble quinoprotein glucose dehydrogenase complexed with methylhydrazine: a covalent cofactor-inhibitor complex. Proc Natl Acad Sci U S A 96:11787–11791. doi: 10.1073/pnas.96.21.11787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakuraba H, Yokono K, Yoneda K, Watanabe A, Asada Y, Satomura T, Yabutani T, Motonaka J, Ohshima T. 2010. Catalytic properties and crystal structure of quinoprotein aldose sugar dehydrogenase from hyperthermophilic archaeon Pyrobaculum aerophilum. Arch Biochem Biophys 502:81–88. doi: 10.1016/j.abb.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 35.Shinagawa E, Matsushita K, Adachi O, Ameyama M. 1983. Selective production of 5-keto-d-gluconate by Gluconobacter strains. J Ferment Technol 61:359–363. [Google Scholar]
- 36.Janssen FW, Ruelius HW. 1968. Carbohydrate oxidase, a novel enzyme from Polyporus obtusus. II. Specificity and characterization of reaction products. Biochim Biophys Acta 167:501–510. doi: 10.1016/0005-2744(68)90040-5. [DOI] [PubMed] [Google Scholar]
- 37.Volc J, Kubatova E, Wood DA, Daniel G. 1997. Pyranose 2-dehydrogenase, a novel sugar oxidoreductase from the basidiomycete fungus Agaricus bisporus. Arch Microbiol 167:119–125. doi: 10.1007/s002030050424. [DOI] [PubMed] [Google Scholar]