

ABSTRACT
Methanosarcina acetivorans uses a variety of methylated sulfur compounds as carbon and energy sources. Previous studies implicated the mtsD, mtsF, and mtsH genes in catabolism of dimethylsulfide, but the genes required for use of other methylsulfides have yet to be established. Here, we show that a four-gene locus, designated mtpCAP-msrH, is specifically required for growth on methylmercaptopropionate (MMPA). The mtpC, mtpA, and mtpP genes encode a putative corrinoid protein, a coenzyme M (CoM) methyltransferase, and a major facilitator superfamily (MFS) transporter, respectively, while msrH encodes a putative transcriptional regulator. Mutants lacking mtpC or mtpA display a severe growth defect in MMPA medium but are unimpaired during growth on other substrates. The mtpCAP genes comprise a transcriptional unit that is highly and specifically upregulated during growth on MMPA, whereas msrH is monocistronic and constitutively expressed. Mutants lacking msrH fail to transcribe mtpCAP and grow poorly in MMPA medium, consistent with the assignment of its product as a transcriptional activator. The mtpCAP-msrH locus is conserved in numerous marine methanogens, including eight Methanosarcina species that we showed are capable of growth on MMPA. Mutants lacking the mtsD, mtsF, and mtsH genes display a 30% reduction in growth yield when grown on MMPA, suggesting that these genes play an auxiliary role in MMPA catabolism. A quadruple ΔmtpCAP ΔmtsD ΔmtsF ΔmtsH mutant strain was incapable of growth on MMPA. Reanalysis of mtsD, mtsF, and mtsH mutants suggests that the preferred substrate for MtsD is dimethylsulfide, while the preferred substrate for MtsF is methanethiol.
IMPORTANCE Methylated sulfur compounds play pivotal roles in the global sulfur and carbon cycles and contribute to global temperature homeostasis. Although the degradation of these molecules by aerobic bacteria has been well studied, relatively little is known regarding their fate in anaerobic ecosystems. In this study, we identify the genetic basis for metabolism of methylmercaptopropionate, dimethylsulfide, and methanethiol by strictly anaerobic methanogens of the genus Methanosarcina. These data will aid the development of predictive sulfur cycle models and enable molecular ecological approaches for the study of methylated sulfur metabolism in anaerobic ecosystems.
INTRODUCTION
Methylated sulfur compounds (generically known as methylsulfides), including methylmercaptopropionate (MMPA), dimethylsulfide (DMS), and methanethiol (MeSH), are abundant in many ecosystems and play pivotal roles in the global sulfur and carbon cycles (1, 2). The predominate source of methylsulfides in marine systems is believed to be dimethylsulfoniopropionate (DMSP), a compatible solute whose synthesis by marine algae is estimated to account for 1 to 10% of global primary production (3). DMSP is degraded via two distinct microbial metabolic processes: the cleavage pathway and the demethylation pathway (1). The majority of DMSP (50 to 90%) is degraded via the demethylation pathway, producing MMPA, which is subsequently converted to 3-mercaptopropionate or MeSH (4, 5). MMPA and MeSH are often assimilated by marine microorganisms, thus retaining sulfur in the marine microbial food web (5). The cleavage pathway, on the other hand, generates volatile DMS, which is the primary contributor to the ocean-atmosphere sulfur flux (6) and a precursor to aerosol particles that facilitate cloud condensation, thus contributing to the global temperature homeostasis (7).
While the aerobic catabolism of DMSP and its methylsulfide by-products has been studied extensively (1, 2), considerably less is known regarding the fate of these compounds in anaerobic ecosystems. In anoxic marine sediments, DMSP is ultimately converted to methane; however, pure cultures of methanogenic archaea are not known to use DMSP (8). Instead, it is likely that anaerobic bacteria convert DMSP into MMPA and DMS, which are then used by methanogens. This conclusion is based on the observations that DMS is produced via the cleavage pathway in certain clostridia (9) and that both sulfate-reducing and acetogenic bacteria produce MMPA via the demethylation pathway (10–12). Significantly, it was noted that MMPA was not further metabolized by these anaerobic bacteria (11). However, methylsulfides, including DMS, MMPA, and MeSH, are known to be substrates for the growth of methanogenic archaea (8, 13, 14). Several members of the order Methanosarcinales have been reported to grow on DMS (13), while Methanosarcina sp. strain MTP4 (14), Methanosarcina siciliae strain HI350, and Methanolobus taylorii strain GS-16 have been shown to grow on MeSH (15). Methanogenic catabolism of MMPA is less common, with Methanosarcina sp. strain MTP4, Methanosarcina acetivorans strain C2A, and Methanosarcina siciliae strain T4/M being the only pure cultures known to use this compound (8). Despite numerous reports demonstrating the use of methylsulfides by methanogens, mechanistic studies of methylsulfide metabolism have only been carried out in Methanosarcina barkeri and M. acetivorans.
In methylotrophic methanogens, the methyl moiety from C-1 compounds like methanol, methylamines, and methylsulfides is channeled into methanogenesis via transfer from the substrate to coenzyme M (CoM) (reviewed in reference 16). This C-1 activation reaction is catalyzed by a series of substrate-specific CoM methyltransferases. Typically, these enzymes consist of a two-subunit methyltransferase 1 (MT1) component that transfers the methyl group from the substrate to a corrinoid carrier protein and a single-subunit methyltransferase 2 (MT2), which then transfers the methyl group from the corrinoid protein to CoM (Fig. 1). A variation on this theme was discovered during the characterization of a 480-kDa corrinoid protein purified from acetate-grown Methanosarcina barkeri strain MS (17, 18). This two-subunit enzyme complex, comprised of the MtsA and MtsB proteins, is capable of methylating CoM using either DMS or MMPA as the substrate. It is believed that MtsA catalyzes the methyl transfer reaction from DMS to the corrinoid subunit, MtsB, as well as the subsequent transfer from methyl-MtsB to the CoM (17). Nevertheless, M. barkeri MS does not utilize either DMS or MMPA as a sole growth substrate (8, 18), and thus, the biological function of this interesting enzyme has yet to be established. A family of single-subunit CoM methyltransferases, encoded by the MA0859, MA4384, and MA4558 loci of M. acetivorans, has also been characterized. These proteins are comprised of an N-terminal corrinoid domain and a C-terminal MT2 domain. Genetic studies showed that these proteins are both required and sufficient for the synthesis and catabolism of DMS, leading to their designation as MtsD, MtsF, and MtsH, respectively (19, 20). It was suggested that these proteins catalyze a DMS:CoM methyltransferase reaction analogous to that catalyzed by the MtsA/B system of M. barkeri (19). However, biochemical characterization of MA4384 (MtsF) showed that the enzyme is capable of catalyzing the transfer of methyl groups from methyl-tetrahydromethanopterin (H4MPT) to CoM. DMS also served as a methyl donor, but at lower rates, prompting these authors to rename the protein CmtA (21). Thus, although the MA0859, MA4384, and MA4558 loci are clearly involved in DMS metabolism, the mechanism remains obscure.
FIG 1.
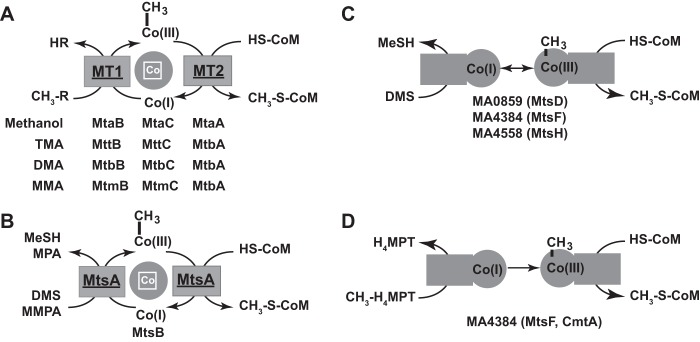
Methanosarcina enzymes involved in C-1 metabolism. (A) Schematic of the three-subunit, substrate-specific MT1/MT2 system for activation of methanol and methylamines found in all Methanosarcina species. The designations for specific MT1/MT2 enzymes and their cognate substrates are shown. (B) Schematic of the biochemically characterized two-subunit methylsulfide MT1/MT2 system from M. barkeri. (C) Schematic of the putative function of a single-subunit DMS MT1/MT2 system, based on genetic analysis of the mtsD, mtsF, and mtsH genes of M. acetivorans (19). (D) Schematic of the putative function of an H4MPT:CoM methyltransferase system encoded by the mtsD, mtsF, and mtsH genes of M. acetivorans, based on biochemical analysis of the MtsF (CmtA) protein (21).
Analysis of the M. acetivorans genome reveals numerous genes that appear to encode MT1/MT2 homologs, including 10 MT1 methyltransferase subunits, 15 MT1 corrinoid subunits, and 13 MT2 proteins (22). While most of these can be confidently assigned to known substrates, several remain unassigned. Among the known substrates used by M. acetivorans, only MMPA and MeSH have yet to be characterized at a genetic level. Thus, we suspected that some of the unassigned MT1/MT2 homologs might be involved in the use of these substrates. Here, we show that the MA4164 to MA4166 locus (which we designated the mtpCAP operon) is specifically required for MMPA metabolism and that its expression is positively regulated by MA4167 (MsrH) when MMPA is present. We also reanalyzed the phenotypes of ΔmtsD, ΔmtsF, and ΔmtsH mutants, showing that these genes contribute to the efficient metabolism of MMPA and that MtsD is the primary enzyme for DMS metabolism, while MtsF is the primary enzyme for MeSH metabolism. MtsH, however, appears to accept both substrates.
MATERIALS AND METHODS
Strains, media, and growth conditions.
The Methanosarcina strains used in the study are described in Table 1. The Methanosarcina strains were grown in single-cell morphology (23) at 37°C using high-salt (HS) broth containing one or more of the following substrates: 125 mM methanol, 50 mM trimethylamine (TMA), 50 mM dimethylamine (DMA), 50 mM monomethylamine (MMA), 120 mM acetate (24), 20 mM MMPA, 20 mM DMS, or 20 mM MeSH. Growth on medium solidified with 1.5% agar was as described previously (25). All plating manipulations were carried out under strictly anaerobic conditions in an anaerobic incubator as described previously (26). Puromycin (CalBiochem, San Diego, CA) was added from sterile, anaerobic stocks at a final concentration of 2 μg/ml for selection of Methanosarcina strains carrying the puromycin transacetylase gene (pac) (27, 28). The purine analog 8-aza-2,6-diaminopurine (Sigma, St. Louis, MO) was added from sterile, anaerobic stocks at a final concentration of 20 μg/ml for selection against the hpt gene. MMPA was prepared by alkaline hydrolysis of methyl-3-methylmercaptopropionate (Sigma-Aldrich, St. Louis, MO) (29) or purchased directly from Tokyo Chemical Industry Co. (Japan).
TABLE 1.
Strains used in the studya
| Strain | Genotype or description | Source or reference |
|---|---|---|
| M. acetivorans C2A | Wild type | DSM 2834 |
| WWM82 | Δhpt::(PmcrB-ϕC31 int-attP) | 31 |
| WWM829 | Δhpt ΔmtpCAP | This study |
| WWM830 | Δhpt ΔmtpC | This study |
| WWM831 | Δhpt ΔmtpA | This study |
| WWM832 | Δhpt ΔmtpP | This study |
| WWM833 | Δhpt ΔmsrH | This study |
| WWM898 | Δhpt::pFH009 ΔmtpC | This study |
| WWM899 | Δhpt::pFH010 ΔmtpA | This study |
| WWM900 | Δhpt::pFH013A ΔmsrH | This study |
| WWM901 | Δhpt::pFH012 ΔmtpCAP | This study |
| WWM810 | Δhpt ΔmtsD::frt | 19 |
| WWM811 | Δhpt ΔmtsF::frt | 19 |
| WWM812 | Δhpt ΔmtsH::frt | 19 |
| WWM813 | Δhpt ΔmtsD::frt ΔmtsF::frt | 19 |
| WWM814 | Δhpt ΔmtsD::frt ΔmtsH::frt | 19 |
| WWM815 | Δhpt ΔmtsF::frt ΔmtsH::frt | 19 |
| WWM816 | Δhpt ΔmtsD::frt ΔmtsF::frt ΔmtsH::frt | 19 |
| WWM897 | Δhpt ΔmtsD::frt ΔmtsF::frt ΔmtsH::frt ΔmtpCAP | This study |
| M. siciliae C2J | Wild type | Laboratory stock from K. Sowers |
| M. siciliae T4/M | Wild type | DSM 3028 |
| M. siciliae HI350 | Wild type | DSM 6564 |
| Methanosarcina sp. Naples 100 | Wild type | DSM 8689 |
| Methanosarcina sp. WH1 | Wild type | DSM 4659 |
| Methanosarcina sp. MTP4 | Wild type | DSM 6636 |
| Methanosarcina sp. WWM596 | Wild type | Laboratory stock from K. Sowers |
Plasmids and primers used for strain constructions are described in Tables S1 and S2 in the supplemental material.
Construction, verification, and complementation of mutant strains.
Plasmids used for creating the mtpC, mtpA, mtpP, msrH, and mtpCAP mutants were constructed as described in Table S1 in the supplemental material. Individual gene deletions removed most of the coding sequence, leaving behind an in-frame fusion peptide consisting of the first and last 10 amino acids, with the goal of creating nonpolar mutations. The mutated alleles were recombined onto the chromosome using the markerless genetic exchange method as described previously (30). All mutations were verified by PCR and by DNA hybridization experiments, using pFH002 as the probe against restriction endonuclease-digested genomic DNA isolated from each mutant (see Fig. S1). Plasmids used for complementation were constructed using pJK027A, which, in the strain background used, expresses the genes in question from the strong, constitutive pmcrB-tetO1 promoter (31). Each complementation plasmid was integrated in single copy into the chromosome of its respective mutant via a site-specific recombination method described previously (31).
DNA methods.
The plasmid constructions and primers used are described in Tables S1 and S2 in the supplemental material. Standard methods were used throughout for isolation and manipulation of plasmid DNA from Escherichia coli (32). Genomic DNA from M. acetivorans was isolated as described previously (33). DNA hybridizations were performed using the DIG (digoxigenin) System (Roche, Mannheim, Germany) as recommended by the manufacturer, using MagnaGraph Nylon transfer membranes (Micron Separations, Inc., Westborough, MA). DNA sequencing was performed at the W. M. Keck Center for Comparative and Functional Genomics, University of Illinois.
Phenotypic characterization.
Growth rates were determined by monitoring the optical densities of three or more independent cultures at 600 nm using a Bausch and Lomb Spectronic 21.
Determination of metabolites.
DMS, MeSH, and methane were quantified using a Hewlett-Packard gas chromatograph (5890 series II) equipped with a stainless steel 80/120 Carbopack B column (Supelco; Sigma-Aldrich, St. Louis, MO) and a flame ionization detector. The column, injector, and detector were at 130°C, and the nitrogen flow rate was 22.5 ml/min. Under these conditions, methane, MeSH, and DMS were eluted with retention times of 0.9, 1.9, and 4.0 min, respectively. The partitioning coefficients of DMS (11.2) and MeSH (7.8) between liquid and gas phases were determined using at least six replicates in stoppered Balch tubes containing the desired substrate in 10 ml HS medium. The measured partitioning coefficients did not vary significantly over the concentrations examined, as previously observed (8, 18, 34).
RNA-seq analysis.
M. acetivorans C2A strains were adapted to different growth media for at least 30 generations prior to RNA isolation. The total RNA was isolated from early exponential-phase cultures using TRIzol (Invitrogen, Carlsbad, CA) and Zymo Direct-zol RNA miniprep kits (Zymo Research, Irvine, CA). The 16S and 23S rRNAs were subtracted using biotin-labeled 16S and 23S RNA probes as previously described (35). Briefly, 16S and 23S rRNA genes were amplified from M. acetivorans strain C2A with T7 promoter-appended primers (see Table S2 in the supplemental material) to allow in vitro synthesis of biotinylated antisense rRNA probes, which were then hybridized to total RNA prior to removal using streptavidin-coated magnetic beads. Construction and sequencing of libraries on the Illumina HiSeq2000 was performed at the W. M. Keck Center for Comparative and Functional Genomics at the University of Illinois at Urbana-Champaign. Briefly, 50 nanograms of rRNA-depleted mRNA was converted to indexed high-throughput RNA sequencing (RNA-seq) libraries with the ScriptSeq version 2 RNA-seq library preparation kit (Epicentre Biotechnologies, Madison, WI). The libraries were pooled in equimolar concentrations and quantified by quantitative PCR (qPCR) with the Illumina-compatible KAPA library quantification kit (Kapa Biosystems, Woburn, MA). The pooled libraries were sequenced for 101 cycles, plus 7 cycles for the index read, on a HiSeq2000 using TruSeq SBS version 3 reagents. The fastq files were generated and demultiplexed with Casava 1.8.2 (Illumina, San Diego, CA). Further bioinformatics processing was performed using the CLC Genomics Workbench (version 7.0; CLCbio, Aarhus, Denmark). fastq files were imported as high-throughput data and trimmed for quality (quality, 0.001; ambiguous, 2: discard, min 30, max 100) and to remove adapter sequences. After trimming, the remaining reads that mapped to stable RNAs (16S, 23S, and 5S rRNA and all tRNAs) were removed (similarity, 0.9; length, 0.85; mismatch, 2; insertion, 3; deletion, 3; global alignment and stand-alone read). The remaining reads were then mapped to the genome for RNA-seq analyses (mapping parameters, 0.9; length, 0.85; mismatch, 2; insertion, 3; deletion, 3; maximum number of hits for a read, 10). The mapped reads were normalized using the default settings, and differentially expressed genes identified using empirical analysis of differential gene expression (EDGE test) (36). Genes showing fold expression changes greater than 4 or less than −4 with P values of <0.01 were considered to be differentially expressed.
Microarray data accession number.
The raw and processed RNA-seq data have been deposited in the Gene Expression Omnibus (GEO) under the accession number GSE64349.
RESULTS
In silico analysis of the M. acetivorans mtpCAP-msrH locus.
The results of bioinformatics analyses suggested that the MA4164 to MA4167 locus, which we designated mtpCAP-msrH, is involved in the metabolism of an unknown C-1 compound. The MtpC protein (MA4164) shares 30 to 50% identity with the corrinoid protein subunits of numerous characterized Methanosarcina methyltransferase I (MT1) enzymes, being most closely related to the putative DMS methyltransferase MtsD, whereas MtpA (MA4165) is homologous (25% to 31% identity) to MT2 methyltransferase proteins, being most closely related to MtsA from M. barkeri (see Fig. S2 in the supplemental material). The MtpP (MA4166) protein is a member of the major facilitator superfamily (MFS) and is likely to be a transporter for the cognate substrate. Lastly, as noted by Reichlen et al. (37), MsrH (MA4167) is a member of the methanol-specific regulator (Msr) family, known to be involved in the transcriptional activation of numerous MT1-/MT2-encoding genes in Methanosarcina (20, 38).
Construction and characterization of mtpCAP-msrH mutants.
To address the in vivo function of the mtpCAP-msrH locus, we constructed ΔmtpCAP, ΔmtpC, ΔmtpA, ΔmtpP, and ΔmsrH mutants and examined their growth on various substrates (Fig. 2; see also Table S3 in the supplemental material). The mutants were indistinguishable from the parent when grown on methanol, acetate, methylamines, DMS, or MeSH. The parental strain and each of the mutants also transiently accumulate MeSH when grown on DMS and DMS when grown on MeSH (see Fig. S3 and S4). Therefore, the mtpCAP-msrH locus is not required for the catabolism of methanol, acetate, methylamines, DMS, or MeSH, nor is it involved in the synthesis of DMS or MeSH. In contrast, the ΔmtpCAP, ΔmtpC, ΔmtpA, and ΔmsrH mutants were severely impaired in their ability to utilize MMPA as a growth substrate, having ca. one-third of the cell yield, measured by optical density, and lag phases over 200 h longer than that of the parental strain. The ΔmtpC and ΔmtpA phenotypes were alleviated upon complementation with the respective genes; however, the defect of the ΔmsrH mutant was not recovered, possibly due to inappropriate levels of MsrH caused by the strong promoter used in the complementation plasmid (31). The ΔmtpP strain showed no observable defect during growth on MMPA.
FIG 2.

Growth of M. acetivorans strains in MMPA medium. The indicated mutants were grown in HS medium with 20 mM MMPA. The strains used were WWM82 (parental strain), WWM830 (ΔmtpC), WWM898 (complemented ΔmtpC), WWM831 (ΔmtpA), WWM899 (complemented ΔmtpA), WWM832 (ΔmtpP), WWM833 (ΔmsrH), WWM900 (complemented ΔmsrH), WWM829 (ΔmtpCAP), WWM901 (complemented ΔmtpCAP), WWM816 (ΔmtsDFH), and WWM897 (ΔmtpCAP ΔmtsDFH). Error bars represent standard deviations of the results from triplicate cultures. OD600, optical density at 600 nm.
MMPA metabolism in diverse Methanosarcina species.
Analysis of 30 sequenced Methanosarcina genomes revealed 10 strains that contain homologs of the mtpCAP-msrH locus (see Fig. S5 in the supplemental material). Seven of eight strains examined grew on MMPA, including M. acetivorans, M. siciliae C2J, M. siciliae HI350, Methanosarcina sp. strain WH1, Methanosarcina sp. strain WWM596, Methanosarcina sp. strain Naples 100, and Methanosarcina siciliae T4/M. The two M. lacustris strains were not tested due to their very poor growth in our standard medium. Surprisingly, Methanosarcina sp. MTP4, which had previously been reported to grow on MMPA (8), did not use this substrate in our experiments. We note that the Methanosarcina sp. MTP4 genome carries a duplication of the mtpCAP-msrH locus that is not found in any of the other strains, raising the possibility that rearrangement of this genomic region, which may have occurred during serial transfer following its original isolation, altered the ability to use MMPA. This result notwithstanding, these data provide strong, correlative support for the role of the mtpCAP-msrH locus in MMPA catabolism.
Reassessment of the roles of mtsD, mtsF, and mtsH in methylsulfide metabolism.
Based on the observation that MMPA is a substrate for M. barkeri MtsA/B, we suspected that the related M. acetivorans MtsD, MtsF, and MtsH proteins might be responsible for the residual MMPA-dependent growth of the mtpCAP mutants. To test this, we obtained the previously described (19) mts mutants and examined their growth on MMPA (Fig. 2 and Table 2). None of the single or double mts mutants differed significantly from the parental strain with respect to growth rate on MMPA; however, when all three genes were deleted, the growth yield was ca. one-third lower than that of the parental strain (Fig. 2). Thus, the MtsD, MtsF, and MtsH proteins play a role in MMPA metabolism in otherwise wild-type cells. The quadruple mutant lacking all three mts genes and the mtpCAP operon showed no growth in MMPA medium.
TABLE 2.
Generation times of mtsD, mtsF, and mtsH mutants on methylsulfides
| Strain genotype | Avg generation time ± SD (h) ona: |
||
|---|---|---|---|
| MMPA | DMS | MeSH | |
| C2A WT | 44.7 ± 1.9 | 20.7 ± 3.6 | 44.8 ± 5.7 |
| ΔmtsD | 35.3 ± 7.5 | No growth | 61.3 ± 0.8 |
| ΔmtsF | 39.6 ± 7.5 | 31.6 ± 6.6 | No growth |
| ΔmtsH | 40.4 ± 2.8 | 71.0 ± 5.1 | 187.3 |
| ΔmtsDF | 38.1 ± 5.2 | No growth | No growth |
| ΔmtsDH | 40.4 ± 6.2 | No growth | 28.1 ± 4.1 |
| ΔmtsFH | 45.3 ± 13.2 | 86.1 ± 8.4 | No growth |
| ΔmtsDFH | 51.0 ± 4.8 | No growth | No growth |
Growth was measured as indicated in Materials and Methods. Values represent the results from three replicates, with the exception of the ΔmtsH strain on MeSH, whose growth was measured once. MMPA, methylmercaptopropionate; DMS, dimethylsulfide; MeSH, methanethiol.
We also reexamined the phenotypes of the mts mutants during growth on DMS and MeSH (Table 2). Contrary to previously published results (19), we found that strains lacking mtsD were incapable of growth on DMS, regardless of the presence or absence of mtsF and mtsH, whereas strains lacking mtsF failed to grow on MeSH, regardless of the presence or absence of mtsD and mtsH. The mtsH gene was not required for growth on either MeSH or DMS; however, the generation times of strains lacking this gene were substantially longer on both substrates, with the exception of the ΔmtsDH mutant, which grew significantly faster than the wild-type on MeSH. Interestingly, unlike the parental strain, the ΔmtsDH mutant did not produce DMS during growth on MeSH (see Fig. S4 in the supplemental material), suggesting that the higher growth rate may be due to more efficient catabolism of the substrate. It should be noted that our DMS results differ from those obtained by Oelgeschlager et al. (19), who showed that any one of the mts genes was sufficient to allow growth on DMS. Although lower levels of DMS were used in the previous study, we obtained similar results at both 5 mM and 20 mM DMA (see Fig. S6). Thus, we believe subtle differences in medium preparation or precultivation conditions may be responsible for the discrepancy.
Transcriptional profiling of M. acetivorans grown on methylsulfides.
Transcriptional profiling of M. acetivorans via RNA-seq provided additional support for the substrate specificities assigned to the various methyltransferase genes based on the mutant phenotypes. Accordingly, the mtpCAP locus is highly and specifically upregulated during growth on MMPA relative to its transcription during growth on methanol, MeSH, or DMS (Table 3; see also Table S3 in the supplemental material). Moreover, during growth on MMPA, mtpA, mtpC, and mtpP are among the most abundant transcripts in the cell, but their expression is low during growth on the other substrates. The mtsD, mtsF, and mtsH genes are also upregulated during growth on MMPA and DMS relative to their transcription in methanol-grown cells, consistent with these genes having a role during growth on both substrates. However, only mtsF is upregulated during growth on MeSH (and only relative to its transcription during growth on methanol), consistent with the observation that mtsF mutants fail to grow on this substrate. It should be noted that MeSH is an intermediate of DMS catabolism, explaining the induction of mtsF during growth on DMS.
TABLE 3.
Relative abundances of mRNAs of wild-type M. acetivorans C2A on different substratesa
| Locus | Gene or function | Rank on indicated substrateb |
Fold change forc: |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MMPA | DMS | MeSH | MeOH | MeOH vs MMPA | DMS vs MMPA | MeSH vs MMPA | MeOH vs DMS | MeSH vs DMS | MeOH vs MeSH | ||
| MA4164 | mtpC | 7 | 1,568 | 1,574 | 2,015 | −173 | −145 | −149 | NS | NS | NS |
| MA4165 | mtpA | 4 | 1,659 | 1,121 | 736 | −153 | −188 | −161 | NS | NS | NS |
| MA4166 | mtpP | 21 | 3,310 | 3,733 | 2,995 | −57 | −65 | −82 | NS | NS | NS |
| MA4167 | msrH | 1,534 | 2,053 | 1,155 | 938 | NS | NS | NS | NS | NS | NS |
| MA1617 | mtaC3 | 8 | 251 | 92 | 2,688 | −80 | −12 | −9 | −7 | NS | −9 |
| MA1616 | mtaB3 | 13 | 289 | 144 | 2,826 | −58 | −10 | −9 | −6 | NS | −7 |
| MA0849 | ramS | 34 | 235 | 114 | 3,144 | −37 | −5 | −4 | −7 | NS | −8 |
| MA3860 | cdhA2 | 24 | 277 | 1,275 | 1,009 | −24 | −11 | −25 | NS | NS | NS |
| MA3861 | cdhB2 | 16 | 136 | 608 | 333 | −21 | −13 | −22 | NS | NS | NS |
| MA3862 | cdhC2 | 17 | 348 | 788 | 880 | −29 | −11 | −24 | −3 | NS | NS |
| MA3863 | cooC2 | 9 | 159 | 113 | 106 | −22 | −8 | −11 | −3 | NS | NS |
| MA3864 | cdhD2 | 32 | 386 | 628 | 971 | −21 | −7 | −15 | −3 | NS | NS |
| MA3865 | cdhE2 | 22 | 467 | 939 | 1,576 | −30 | −11 | −22 | −3 | NS | NS |
| MA3300 | thiS homolog | 97 | 308 | 2,774 | 4,084 | −20 | NS | −12 | −9 | −5 | NS |
| MA0859 | mtsD | 70 | 41 | 366 | 2,001 | −16 | NS | NS | −18 | NS | NS |
| MA4384 | mtsF | 44 | 16 | 4 | 1,333 | −20 | 4 | NS | −73 | NS | −62 |
| MA4558 | mtsH | 192 | 60 | 695 | 2,047 | −8 | NS | NS | −16 | −9 | NS |
| MA3302 | mreA | 25 | 424 | 1,428 | 3,577 | −42 | −6 | −22 | −7 | NS | NS |
| MA3130 | mreD | 14 | 48 | 30 | 123 | −18 | NS | NS | −9 | NS | −4 |
| MA0685 | Sulfite reductase | 37 | 166 | 53 | 572 | −16 | NS | NS | −5 | NS | −6 |
| MA3736 | mdrA | 23 | 125 | 59 | 227 | −15 | −4 | −4 | −4 | NS | −4 |
| MA3607 | pta | 35 | 266 | 335 | 229 | −11 | −6 | −9 | NS | NS | NS |
| MA3606 | ack | 39 | 178 | 288 | 256 | −11 | −3 | −8 | −4 | NS | NS |
| MA0803 | Putative regulator | 2,777 | 1 | 3,233 | 2,548 | NS | 7,277 | NS | −6,037 | −7,783 | NS |
| MA0269 | mtrH | 287 | 146 | 119 | 148 | NS | NS | NS | −3 | NS | NS |
| MA0276 | mtrE | 195 | 102 | 101 | 93 | NS | NS | NS | −3 | NS | NS |
The full RNA-seq data set is presented in Table S4 in the supplemental material.
Rank based on the normalized reads per kilobase per million mapped reads (RPKM values) for all genes in the genome, sorted from high to low, is presented.
Statistically significant changes (P < 0.01) based on the EDGE test are shown. NS, not statistically significant (P ≥ 0.01).
Several additional classes of genes showed significant transcriptional regulation during growth on methylsulfides. The MA0849 locus, which encodes a protein with homology to the methylamine methyltransferase-activating protein RamA (39), is strongly upregulated on MMPA, DMS, and MeSH, suggesting a specific role in activation of methylsulfide methyltransferases. The mtaCB3 operon was also highly expressed during growth on all methylsulfides. Although the results of genetic experiments strongly support the idea that this operon encodes a methanol-specific MT1 enzyme, it is only expressed during growth on energetically poor substrates, such as acetate and MMA (40). Our data now add MMPA, DMS, and MeSH to the list of substrates that induce mtaCB3. Another group of highly regulated genes encompasses ones previously shown to be controlled by the global regulator MreA (37), including mreA itself, as well as the cdh-2 operon and the ack, pta, and mreD genes. Interestingly, the putative mreA regulon genes were only upregulated on MMPA. A third group involves genes with putative roles in sulfur metabolism. These include a homolog of the sulfur transfer protein ThiS (41), encoded by MA3300, the protein disulfide reductase MdrA (42), encoded by MA3736, and a putative sulfite reductase, encoded by MA0685 (43). Lastly, we observed that the protein encoded by MA0803 was strongly and specifically upregulated during growth on DMS. This protein has a C-terminal domain that is related to the Msr family transcriptional regulators but has a significantly different N-terminal DNA binding domain, suggesting that it is the founding member of a new family of archaeal transcriptional regulators involved in DMS-mediated regulation.
The RNA-seq data also support the notion that mtpCAP comprises an operon transcriptionally regulated by MsrH. Thus, continuous mapping of mRNA reads was observed across the entire operon (Fig. 3). The msrH gene is not cotranscribed with mtpCAP but instead comprises a monocistronic transcript that is constitutively expressed at low levels on all growth media (Table 3). To investigate the idea that msrH encodes a transcriptional regulator of mtpCAP, we used RNA-seq to compare the levels of mRNA abundance between the ΔmsrH strain and the parental strain. Because the ΔmsrH mutant does not grow on MMPA as the sole substrate, we grew both strains on 5 mM TMA supplemented with 20 mM MMPA. Under these conditions, mtpC, mtpA, and mtpP were the only genes that were differentially expressed between the two strains (Fig. 4). Thus, msrH appears to be a highly specific, MMPA-dependent activator of mtpCAP.
FIG 3.

RNA-seq read coverage of M. acetivorans grown on MMPA. (Top) mRNA read coverage of the entire chromosome; (middle) mRNA read coverage of the mtpCAP-msrH locus; (bottom) mRNA read coverage of the mtpA-mtpP intergenic region. Note that the coverage between genes never drops below the coverage within the genes, suggesting that mtpCAP are cotranscribed.
FIG 4.
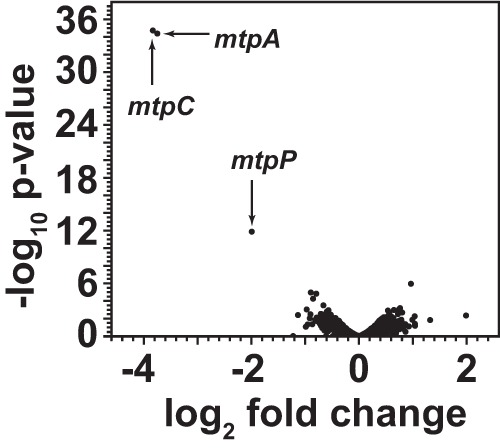
Relative mRNA abundances of the ΔmsrH mutant versus the parental strain during growth on TMA plus MMPA. Volcano plot shows the fold differences in transcript abundance between WWM833 (ΔmsrH) and WWM82 (parental strain), based on the EDGE test plotted against statistical significance. Only the three genes indicated show significant differences between the two strains.
DISCUSSION
The data presented here suggest that the products of the mtpCAP operon are the primary means for catabolism of MMPA in Methanosarcina species. These genes are strongly and specifically induced by MMPA and required for wild-type growth rates and yields on this substrate. Based on our analyses, it seems likely that MMPA is catabolized via a two-component MT1/MT2 system comprised of the methyltransferase MtpA and the corrinoid protein MtpC (Fig. 5). By analogy to the biochemically characterized MtsA/B system of M. barkeri (17), we suggest that MtpA will catalyze both the methyl transfer from MMPA to MtpC and the subsequent methyl transfer from methyl-MtpC to CoM. The highly similar chemical structures of MMPA and methyl-CoM strengthen the idea that these molecules could be substrates/products for the same enzyme. Although we found that MtpP is not required for growth on MMPA under laboratory conditions, the transporter is probably required in nature. Based on the pKa of MMPA (4.7), the ratio of protonated to unprotonated MMPA in our medium (pH 6.8) is ca. 1:100. Thus, because weak acids diffuse freely across the membrane, the transporter is probably not required when substrate concentrations are high and the pH is near neutrality. Moreover, at the marine pH of 8.3 (44), the ratio of protonated to unprotonated MMPA will be >10-fold lower than in our medium. Under these conditions, the putative transporter MtpP will almost certainly be required. The universal conservation of the mtpP gene in all methanogens that contain homologs of mtpCA supports this conclusion.
FIG 5.
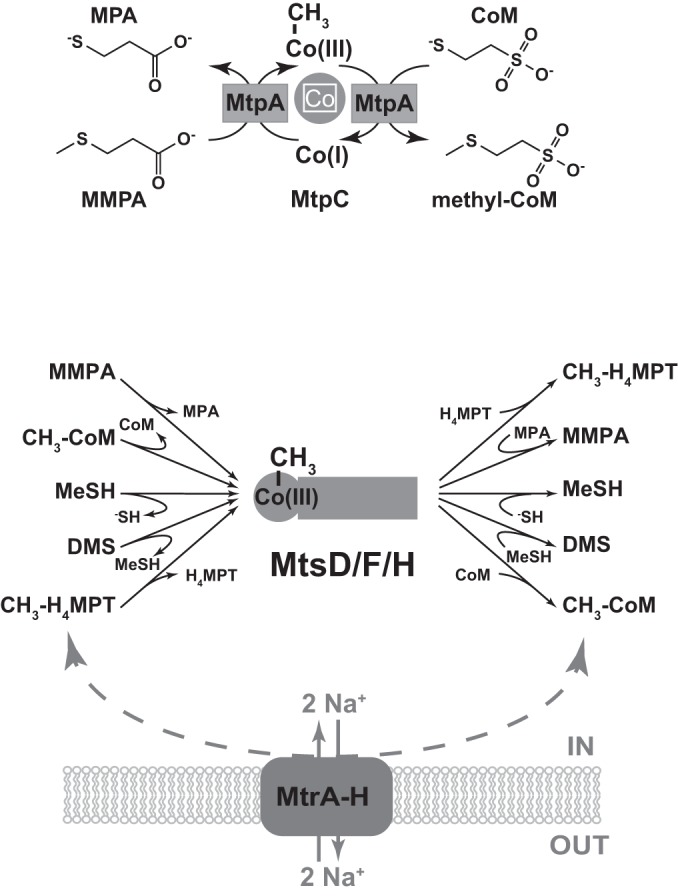
Putative roles for MtpCA and MtsD/-F/-H in methylsulfide metabolism. (Top) Schematic of a proposed bifunctional MT1/MT2 enzyme, in which MtpA catalyzes transfer of the methyl group from MMPA to the corrinoid protein MtpC and subsequent methyl transfer from methyl-MtpC to CoM. (Bottom) Schematic of a proposal for broad substrate specificity in the MtsD/-F/-H proteins. These putative multifunctional enzymes would allow bypass of the normal energy-conserving/-dependent Mtr H4MPT:CoM methyltransferase while providing a mechanism to introduce methyl groups from methylsulfides into both the reductive and the oxidative branch of the methylotrophic pathway of methanogenesis. Note that the direction of methyl transfer between CH3-H4MPT and CH3-CoM determines whether it is exergonic or endergonic and, thus, whether sodium ions are extruded or consumed.
The observation that the mtpCAP mutant showed partial growth on MMPA indicates the presence of an alternate, albeit inefficient pathway for catabolism of this compound by M. acetivorans. If MtpA/MtpC is indeed the primary MT1/MT2 system for this substrate, then the observed residual growth of the mtpCAP mutant must depend on an alternate activating system to generate methyl-CoM from MMPA. The loss of residual growth upon deletion of the three mts genes suggests that MtsF, MtsD, or MtsH acting as single-subunit MT1/MT2 enzymes for MMPA can provide this function. Our genetic data, along with the previously published results of Oelgeschlager et al. (19), show that the Mts proteins also use MeSH and DMS as substrates, with different substrate preferences for individual isozymes. Biochemical evidence for the use of multiple methylsulfide substrates has previously been demonstrated for the MtsA/B system of M. barkeri, which uses both DMS and MMPA as substrates (17).
Intriguingly, we observed that the ΔmtsDFH mutant reached a lower cell density on MMPA than did otherwise wild-type cells, suggesting that the proteins encoded by these genes play an additional role beyond substrate activation. While this phenotype is more difficult to explain, it may be related to the CoM:H4MPT methyltransferase activity of this family of proteins (21). In reporting this novel biochemical activity, Vepachedu and Ferry (21) suggested that MtsF, and probably the homologous MtsD and MtsH proteins, would act to bypass the membrane-bound, energy-conserving Mtr CoM:H4MPT methyltransferase, providing an advantage during growth on low-energy substrates, such as carbon monoxide. Assuming that both the genetic and biochemical data are correct, the Mts family of proteins would have an exceptionally broad substrate range, including methylated and unmethylated sulfides and H4MPT (Fig. 5). The ability to recognize numerous sulfide and methyl-sulfide substrates in addition to H4MPT and methyl-H4MPT would explain all available data. These multifunctional enzymes would allow bypass of the ion-pumping Mtr H4MPT:CoM methyltransferase, as well as provide a mechanism to introduce methyl groups from methylsulfides into both the reductive and oxidative branches of the methylotrophic methanogenesis pathway. Thus, the energetic advantage of bypassing Mtr would apply to growth on any of these substrates. Accordingly, the ΔmtsDFH mutants would lack this energetic advantage and would achieve a lower cell density, as seen in our data. Moreover, the broad specificity of Mts isozymes could also explain the synthesis of DMS and MeSH during growth on CO (19, 45), because the methylated corrinoid cofactor can donate the methyl moiety to sulfide or MeSH in a reaction that is thermodynamically equivalent to transfer to CoM. There is, however, one major caveat to this model. The possession of CoM:H4MPT methyltransferase activity by both Mts and Mtr introduces the possibility of a futile cycle that would deplete the ion gradient across the cell membrane, essentially preventing energy conservation. If this model, shown in Fig. 5, is correct, then the cell must possess a mechanism to prevent this from happening.
Given the dramatic differences between the aceticlastic and methylotrophic pathways for methanogenesis (reviewed in references 16 and 46), we were surprised to note the striking similarity between the transcriptional profiles of MMPA- and acetate-grown cells. This shared phenotype may involve the global regulator MreA, although the intricacies for this regulation remain murky at this point. Based on a combination of the transcriptomic profiles of acetate- versus methanol-grown cells and those of mreA mutants, Reichlen et al. (37) proposed that MreA mediates repression of methylotrophic and activation of aceticlastic genes. Our data are consistent with this interpretation but suggest that the regulation is considerably more complex. Thus, the aceticlastic genes encoding acetate kinase (ack), phosphotransacetylase (pta), and acetyl coenzyme A (acetyl-CoA) synthase (cdh-2) are highly upregulated on both MMPA and acetate, as is the mreA gene itself. However, deletion of mreA leads to increases of ca. 40- to 130-fold in mtpCAP expression while concomitantly reducing the expression of pta, ack, and cdh by 5-fold (37). This effect is likely to be indirect, probably mediated via msrH, which is required for mtpCAP expression and is significantly upregulated in the mreA mutant. The question then remains, what is the common regulatory signal that is sensed during growth on MMPA and acetate? The simplest explanation is that MMPA catabolism generates acetate; however, it has previously been shown that growth on MMPA results in nearly stoichiometric conversion of the substrate to mercaptopropionate (8). Nonetheless, it is conceivable that some mercaptopropionate is metabolized to acetate, triggering an aceticlastic transcriptional response. Alternatively, mercaptopropionate may be sufficiently similar to acetate to trigger this response without catabolism. We do not favor this idea because mreA mutants recapitulate the regulatory phenotype in the absence of MMPA. Further clues come from the observation that mtpCAP, along with mtsD, mtsF, and mtsH, are also highly upregulated upon deletion of the alternate heterodisulfide reductase encoded by hdrABC (47). Previous studies suggested that loss of HdrABC slows the regeneration of the free coenzymes M and B (CoM and CoB), which are needed for the terminal step in methanogenesis. Loss of HdrABC also slows the regeneration of oxidized ferredoxin, which is the central electron carrier in aceticlastic methanogenesis. Interestingly, hdrABC expression is highly downregulated during growth on acetate (47). Taken together, these data suggest a highly intertwined and complex mechanism for the regulation of energy-conserving metabolism in Methanosarcina that may involve direct sensing of intermediates in the methanogenic pathway.
Finally, our results shed additional light on the fate of methylated sulfur compounds in marine environments. The aerobic flux of DMSP into either the cleavage pathway to generate DMS or the demethylation pathway to produce MMPA has attracted considerable attention due to the significant consequences for climate regulation (48). Our data establishing the genetic basis for both DMS and MMPA metabolism in marine methanogens will empower molecular microbial ecology approaches to assess the fate of methylsulfides in anaerobic marine ecosystems as well.
Supplementary Material
ACKNOWLEDGMENTS
We thank Joel Cioni for help with the preparation of MMPA, Kou-San Ju for constructive discussions, and Madeline M. López Muñoz and Petra Kohler for all their help with RNA-seq analysis. We also thank Michael Rother at Germany for sharing the mtsD, mtsF, and mtsH mutant strains.
This work was supported by National Science Foundation grant MCB-1022462 to W.W.M.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02605-14.
REFERENCES
- 1.Reisch CR, Moran MA, Whitman WB. 2011. Bacterial catabolism of dimethylsulfoniopropionate (DMSP). Front Microbiol 2:172. doi: 10.3389/fmicb.2011.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Curson AR, Todd JD, Sullivan MJ, Johnston AW. 2011. Catabolism of dimethylsulphoniopropionate: microorganisms, enzymes and genes. Nat Rev Microbiol 9:849–859. doi: 10.1038/nrmicro2653. [DOI] [PubMed] [Google Scholar]
- 3.Howard EC, Henriksen JR, Buchan A, Reisch CR, Buergmann H, Welsh R, Ye WY, Gonzalez JM, Mace K, Joye SB, Kiene RP, Whitman WB, Moran MA. 2006. Bacterial taxa that limit sulfur flux from the ocean. Science 314:649–652. doi: 10.1126/science.1130657. [DOI] [PubMed] [Google Scholar]
- 4.Kiene RP, Linn LJ, Gonzalez J, Moran MA, Bruton JA. 1999. Dimethylsulfoniopropionate and methanethiol are important precursors of methionine and protein-sulfur in marine bacterioplankton. Appl Environ Microbiol 65:4549–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reisch CR, Stoudemayer MJ, Varaljay VA, Amster IJ, Moran MA, Whitman WB. 2011. Novel pathway for assimilation of dimethylsulphoniopropionate widespread in marine bacteria. Nature 473:208–211. doi: 10.1038/nature10078. [DOI] [PubMed] [Google Scholar]
- 6.Lovelock JE, Maggs RJ, Rasmusse RA. 1972. Atmospheric dimethyl sulfide and natural sulfur cycle. Nature 237:452–453. doi: 10.1038/237452a0. [DOI] [Google Scholar]
- 7.Charlson RJ, Lovelock JE, Andreae MO, Warren SG. 1987. Oceanic phytoplankton, atmospheric sulfur, cloud albedo and climate. Nature 326:655–661. doi: 10.1038/326655a0. [DOI] [Google Scholar]
- 8.van der Maarel M, Jansen M, Hansen TA. 1995. Methanogenic conversion of 3-S-methylmercaptopropionate to 3-mercaptopropionate. Appl Environ Microbiol 61:48–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wagner C, Stadtman ER. 1962. Bacterial fermentation of dimethyl-beta-propiothetin. Arch Biochem Biophys 98:331–336. doi: 10.1016/0003-9861(62)90191-1. [DOI] [PubMed] [Google Scholar]
- 10.van der Maarel MJEC, Quist P, Dijkhuizen L, Hansen TA. 1993. Anaerobic degradation of dimethylsulfoniopropionate to 3-S-methylmercaptopropionate by a marine Desulfobacterium strain. Arch Microbiol 160:411–412. doi: 10.1007/BF00252230. [DOI] [Google Scholar]
- 11.van der Maarel MJ, Jansen M, Haanstra R, Meijer WG, Hansen TA. 1996. Demethylation of dimethylsulfoniopropionate to 3-S-methylmercaptopropionate by marine sulfate-reducing bacteria. Appl Environ Microbiol 62:3978–3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jansen M, Hansen TA. 2001. Non-growth-associated demethylation of dimethylsulfoniopropionate by (homo)acetogenic bacteria. Appl Environ Microbiol 67:300–306. doi: 10.1128/AEM.67.1.300-306.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lomans BP, van der Drift C, Pol A, Op den Camp HJM. 2002. Microbial cycling of volatile organic sulfur compounds. Cell Mol Life Sci 59:575–588. doi: 10.1007/s00018-002-8450-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finster K, Tanimoto Y, Bak F. 1992. Fermentation of methanethiol and dimethylsulfide by a newly isolated methanogenic bacterium. Arch Microbiol 157:425–430. doi: 10.1007/BF00249099. [DOI] [Google Scholar]
- 15.Ni S, Boone DR. 1993. Catabolism of dimethylsulfide and methane thiol by methylotrophic methanogens, p 796–810. In Oremland RS. (ed), Biogeochemistry of global change: radiatively active trace gases. Selected papers from the Tenth International Symposium on Environmental Biogeochemistry, San Francisco, August 19-24, 1991. Chapman & Hall, Inc., New York, NY. [Google Scholar]
- 16.Keltjens JT, Vogels GD. 1993. Conversion of methanol and methylamines to methane and carbon dioxide, p 253–303. In Ferry JG. (ed), Methanogenesis: ecology, physiology, biochemistry & genetics. Chapman & Hall, New York, NY. [Google Scholar]
- 17.Tallant TC, Paul L, Krzycki JA. 2001. The MtsA subunit of the methylthiol: coenzyme M methyltransferase of Methanosarcina barkeri catalyses both half-reactions of corrinoid-dependent dimethylsulfide:coenzyme M methyl transfer. J Biol Chem 276:4485–4493. doi: 10.1074/jbc.M007514200. [DOI] [PubMed] [Google Scholar]
- 18.Tallant TC, Krzycki JA. 1997. Methylthiol:coenzyme M methyltransferase from Methanosarcina barkeri, an enzyme of methanogenesis from dimethylsulfide and methylmercaptopropionate. J Bacteriol 179:6902–6911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oelgeschlager E, Rother M. 2009. In vivo role of three fused corrinoid/methyl transfer proteins in Methanosarcina acetivorans. Mol Microbiol 72:1260–1272. doi: 10.1111/j.1365-2958.2009.06723.x. [DOI] [PubMed] [Google Scholar]
- 20.Bose A, Kulkarni G, Metcalf WW. 2009. Regulation of putative methyl-sulphide methyltransferases in Methanosarcina acetivorans C2A. Mol Microbiol 74:227–238. doi: 10.1111/j.1365-2958.2009.06864.x. [DOI] [PubMed] [Google Scholar]
- 21.Vepachedu VR, Ferry JG. 2012. Role of the fused corrinoid/methyl transfer protein CmtA during CO-dependent growth of Methanosarcina acetivorans. J Bacteriol 194:4161–4168. doi: 10.1128/JB.00593-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galagan JE, Nusbaum C, Roy A, Endrizzi MG, Macdonald P, FitzHugh W, Calvo S, Engels R, Smirnov S, Atnoor D, Brown A, Allen N, Naylor J, Stange-Thomann N, DeArellano K, Johnson R, Linton L, McEwan P, McKernan K, Talamas J, Tirrell A, Ye WJ, Zimmer A, Barber RD, Cann I, Graham DE, Grahame DA, Guss AM, Hedderich R, Ingram-Smith C, Kuettner HC, Krzycki JA, Leigh JA, Li WX, Liu JF, Mukhopadhyay B, Reeve JN, Smith K, Springer TA, Umayam LA, White O, White RH, de Macario EC, Ferry JG, Jarrell KF, Jing H, Macario AJL, Paulsen I, Pritchett M, Sowers KR, et al. 2002. The genome of M. acetivorans reveals extensive metabolic and physiological diversity. Genome Res 12:532–542. doi: 10.1101/gr.223902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sowers KR, Boone JE, Gunsalus RP. 1993. Disaggregation of Methanosarcina spp. and growth as single sells at elevated osmolarity. Appl Environ Microbiol 59:3832–3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Metcalf WW, Zhang JK, Shi X, Wolfe RS. 1996. Molecular, genetic, and biochemical characterization of the serC gene of Methanosarcina barkeri fusaro. J Bacteriol 178:5797–5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boccazzi P, Zang JK, Metcalf WW. 2000. Generation of dominant selectable markers for resistance to pseudomonic acid by cloning and mutagenesis of the ileS gene from the archaeon Methanosarcina barkeri Fusaro. J Bacteriol 182:2611–2618. doi: 10.1128/JB.182.9.2611-2618.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Metcalf WW, Zhang JK, Wolfe RS. 1998. An anaerobic, intrachamber incubator for growth of Methanosarcina spp. on methanol-containing solid media. Appl Environ Microbiol 64:768–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gernhardt P, Possot O, Foglino M, Sibold L, Klein A. 1990. Construction of an integration vector for use in the archaebacterium Methanococcus voltae and expression of a eubacterial resistance gene. Mol Gen Genet 221:273–279. [DOI] [PubMed] [Google Scholar]
- 28.Metcalf WW, Zhang JK, Apolinario E, Sowers KR, Wolfe RS. 1997. A genetic system for Archaea of the genus Methanosarcina: liposome-mediated transformation and construction of shuttle vectors. Proc Natl Acad Sci U S A 94:2626–2631. doi: 10.1073/pnas.94.6.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wackett LP, Honek JF, Begley TP, Wallace V, Ormejohnson WH, Walsh CT. 1987. Substrate-analogs as mechanistic probes of methyl-S-coenzyme-M reductase. Biochemistry 26:6012–6018. doi: 10.1021/bi00393a010. [DOI] [PubMed] [Google Scholar]
- 30.Pritchett MA, Zhang JK, Metcalf WW. 2004. Development of a markerless genetic exchange method for Methanosarcina acetivorans C2A and its use in construction of new genetic tools for methanogenic archaea. Appl Environ Microbiol 70:1425–1433. doi: 10.1128/AEM.70.3.1425-1433.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guss AM, Rother M, Zhang JK, Kulkarni G, Metcalf WW. 2008. New methods for tightly regulated gene expression and highly efficient chromosomal integration of cloned genes for Methanosarcina species. Archaea 2:193–203. doi: 10.1155/2008/534081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ausubel FM. 1992. Short protocols in molecular biology: a compendium of methods from current protocols in molecular biology, 2nd ed Wiley, New York, NY. [Google Scholar]
- 33.Pritchett MA, Metcalf WW. 2005. Genetic, physiological and biochemical characterization of multiple methanol methyltransferase isozymes in Methanosarcina acetivorans C2A. Mol Microbiol 56:1183–1194. doi: 10.1111/j.1365-2958.2005.04616.x. [DOI] [PubMed] [Google Scholar]
- 34.Kiene RP, Visscher PT. 1987. Production and fate of methylated sulfur compounds from methionine and dimethylsulfoniopropionate in anoxic salt marsh sediments. Appl Environ Microbiol 53:2426–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stewart FJ, Ottesen EA, DeLong EF. 2010. Development and quantitative analyses of a universal rRNA-subtraction protocol for microbial metatranscriptomics. ISME J 4:896–907. doi: 10.1038/ismej.2010.18. [DOI] [PubMed] [Google Scholar]
- 36.Robinson MD, Smyth GK. 2008. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics 9:321–332. doi: 10.1093/biostatistics/kxm030. [DOI] [PubMed] [Google Scholar]
- 37.Reichlen MJ, Vepachedu VR, Murakami KS, Ferry JG. 2012. MreA functions in the global regulation of methanogenic pathways in Methanosarcina acetivorans. mBio 3(4):e00189-12. doi: 10.1128/mBio.00189-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bose A, Metcalf WW. 2008. Distinct regulators control the expression of methanol methyltransferase isozymes in Methanosarcina acetivorans C2A. Mol Microbiol 67:649–661. doi: 10.1111/j.1365-2958.2007.06075.x. [DOI] [PubMed] [Google Scholar]
- 39.Ferguson T, Soares JA, Lienard T, Gottschalk G, Krzycki JA. 2009. RamA, a protein required for reductive activation of corrinoid-dependent methylamine methyltransferase reactions in methanogenic archaea. J Biol Chem 284:2285–2295. doi: 10.1074/jbc.M807392200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bose A, Pritchett MA, Rother M, Metcalf WW. 2006. Differential regulation of the three methanol methyltransferase isozymes in Methanosarcina acetivorans C2A. J Bacteriol 188:7274–7283. doi: 10.1128/JB.00535-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lehmann C, Begley TP, Ealick SE. 2006. Structure of the Escherichia coli ThiS-ThiF complex, a key component of the sulfur transfer system in thiamin biosynthesis. Biochemistry 45:11–19. doi: 10.1021/bi051502y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lessner DJ, Ferry JG. 2007. The archaeon Methanosarcina acetivorans contains a protein disulfide reductase with an iron-sulfur cluster. J Bacteriol 189:7475–7484. doi: 10.1128/JB.00891-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Susanti D, Mukhopadhyay B. 2012. An intertwined evolutionary history of methanogenic archaea and sulfate reduction. PLoS One 7:e45313. doi: 10.1371/journal.pone.0045313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gruber N, Hauri C, Lachkar Z, Loher D, Frolicher TL, Plattner GK. 2012. Rapid progression of ocean acidification in the California Current System. Science 337:220–223. doi: 10.1126/science.1216773. [DOI] [PubMed] [Google Scholar]
- 45.Moran JJ, House CH, Vrentas JM, Freeman KH. 2008. Methyl sulfide production by a novel carbon monoxide metabolism in Methanosarcina acetivorans. Appl Environ Microbiol 74:540–542. doi: 10.1128/AEM.01750-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferry JG. 1997. Enzymology of the fermentation of acetate to methane by Methanosarcina thermophila. Biofactors 6:25–35. doi: 10.1002/biof.5520060104. [DOI] [PubMed] [Google Scholar]
- 47.Buan NR, Metcalf WW. 2010. Methanogenesis by Methanosarcina acetivorans involves two structurally and functionally distinct classes of heterodisulfide reductase. Mol Microbiol 75:843–853. doi: 10.1111/j.1365-2958.2009.06990.x. [DOI] [PubMed] [Google Scholar]
- 48.Simo R. 2001. Production of atmospheric sulfur by oceanic plankton: biogeochemical, ecological and evolutionary links. Trends Ecol Evol 16:287–294. doi: 10.1016/S0169-5347(01)02152-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.