

Abstract
Purpose
Fixed-dose rate gemcitabine plus docetaxel achieves objective response in 35% of patients with uterine leiomyosarcoma (uLMS). This study aimed to determine whether the addition of bevacizumab to gemcitabine-docetaxel increases progression-free survival (PFS) in uLMS.
Patients and Methods
In this phase III, double-blind, placebo-controlled trial, patients with chemotherapy-naive, metastatic, unresectable uLMS were randomly assigned to gemcitabine-docetaxel plus bevacizumab or gemcitabine-docetaxel plus placebo. PFS, overall survival (OS), and objective response rates (ORRs) were compared to determine superiority. Target accrual was 130 patients to detect an increase in median PFS from 4 months (gemcitabine-docetaxel plus placebo) to 6.7 months (gemcitabine-docetaxel plus bevacizumab). Treatment effects on PFS and OS were described by hazard ratios (HRs), median times to event, and 95% CIs.
Results
In all, 107 patients were accrued: gemcitabine-docetaxel plus placebo (n = 54) and gemcitabine-docetaxel plus bevacizumab (n = 53). Accrual was stopped early for futility. No statistically significant differences in grade 3 to 4 toxicities were observed. Median PFS was 6.2 months for gemcitabine-docetaxel plus placebo versus 4.2 months for gemcitabine-docetaxel plus bevacizumab (HR, 1.12; P = .58). Median OS was 26.9 months for gemcitabine-docetaxel plus placebo and 23.3 months for gemcitabine-docetaxel plus bevacizumab (HR, 1.07; P = .81). Objective responses were observed in 17 (31.5%) of 54 patients randomly assigned to gemcitabine-docetaxel plus placebo and 19 (35.8%) of 53 patients randomly assigned to gemcitabine-docetaxel plus bevacizumab. Mean duration of response was 8.6 months for gemcitabine-docetaxel plus placebo versus 8.8 months for gemcitabine-docetaxel plus bevacizumab.
Conclusion
The addition of bevacizumab to gemcitabine-docetaxel for first-line treatment of metastatic uLMS failed to improve PFS, OS, or ORR. Gemcitabine-docetaxel remains a standard first-line treatment for uLMS.
INTRODUCTION
Patients who present with advanced or recurrent uterine leiomyosarcoma (uLMS) have a poor prognosis. Few chemotherapy agents have been identified with activity against LMS. In a phase II trial, as second-line therapy, fixed-dose-rate gemcitabine-docetaxel achieved objective responses in 27% of patients with metastatic uLMS.1 In a subsequent phase II trial, as first-line therapy, fixed-dose-rate gemcitabine-docetaxel achieved objective responses in 35.8% of patients.2 Vascular endothelial growth factor (VEGF) and/or VEGF receptors are expressed in a wide variety of tumor types, including gynecologic cancers, and higher levels of vascularity have been associated with poorer prognosis. The murine parent monoclonal antibody of bevacizumab, A4.6.1, demonstrated potent growth inhibition in vivo in a variety of human cancer xenograft and metastasis models, including those for SK-LMS-1 LMS.3 In a phase IB study of gemcitabine-docetaxel plus bevacizumab for patients with chemotherapy-naive soft tissue sarcoma, objective responses were achieved in 31% of patients, with a median response duration of 6 months.4 We aimed to determine whether the addition of the vascular-targeted agent bevacizumab could increase progression-free survival (PFS) when added to fixed-dose-rate gemcitabine-docetaxel as first-line treatment for metastatic uLMS.
PATIENTS AND METHODS
Patient Eligibility
Eligible patients had advanced or recurrent uLMS with documented disease progression and measurable disease as defined by RECIST 1.1.5 Patients must not have received any prior cytotoxic chemotherapy for management of uterine sarcoma, or any prior VEGF-pathway-targeted agent, or any prior treatment with a multikinase inhibitor such as pazopanib, sorafenib, or sunitinib. Patients must not have received any prior therapy with docetaxel or gemcitabine.
Patients were required to have a Gynecologic Oncology Group (GOG) performance status of 0, 1, or 2; to be free of active infection; and to have recovered from effects of recent surgery or radiotherapy. Adequate bone marrow function (platelet count ≥ 100,000/μL; absolute neutrophil count ≥ 1,500/μL), renal function (creatinine ≤ 1.5× institutional upper limit of normal [ULN]), hepatic function (bilirubin within normal range; AST and alkaline phosphatase ≤ 2.5× ULN), and neurologic function (grade ≤ 1, no history of transient ischemic attack or stroke, or CNS hemorrhage within the past 6 months) were required. Baseline urine protein:creatinine ratio was required to be less than 1. International normalized ratio was required to be ≤ 1.5× the institutional ULN (or an in-therapeutic-range international normalized ratio, usually between 2 and 3, if a patient was being given a stable dose of therapeutic warfarin). Patients were excluded if they had active bleeding or pathologic conditions that carried a high risk of bleeding, including tumor that involved major vessels. Patients with brain metastases or poorly controlled seizures were excluded. Patients must not have had major surgery or significant traumatic injury within 28 days before study entry or a history of abdominal fistula or perforation within the past 12 months. Patients with a current serious nonhealing wound, ulcer, or bone fracture were excluded. Patients with hypertension were permitted on study provided their blood pressure was ≤ 140/90 mmHg. Use of blood pressure medications to achieve and maintain blood pressure control was permitted. Patients were excluded for a history of myocardial infarction or unstable angina within 6 months of the first date of bevacizumab or placebo therapy, a history of New York Heart Association grade 2 or worse congestive heart failure, significant peripheral vascular disease, a history of cerebrovascular accident, transient ischemic attack, or subarachnoid hemorrhage within 6 months of the first date of bevacizumab or placebo therapy, or a history of pulmonary embolism or deep vein thrombosis within the 6 months before enrollment. Histologic confirmation of the original primary tumor was required.
All patients signed an institutional review board (IRB) –approved informed consent and research authorization permitting release of personal health information. The protocol was reviewed and approved annually by IRBs of the participating institutions and by the National Cancer Institute Central IRB.
Study Treatment
On day 1, patients received gemcitabine 900 mg/m2 intravenously (IV) over 90 minutes, followed by bevacizumab 15 mg/kg or its placebo IV over 90 minutes (subsequent cycles of bevacizumab or placebo were permitted to be given over 60 minutes, and if tolerated, then subsequently over 30 minutes). On day 8, patients received gemcitabine 900 mg/m2 IV over 90 minutes, followed by docetaxel 75 mg/m2 IV over 60 minutes. Filgrastim 5 μg/kg was given subcutaneously on days 9 through 15, or pegfilgrastim 6 mg was given subcutaneously on day 9 or day 10. Patients with prior pelvic radiation received gemcitabine 675 mg/m2 IV over 70 to 90 minutes on days 1 and 8 and docetaxel 60 mg/m2 on day 8. Dexamethasone was given as premedication for docetaxel starting on day 7, and the use of diuretics to control fluid retention was encouraged. Blood pressure was monitored once per week during the first cycle of therapy and then on days 1 and 8 of treatment. Up to two dose reductions were permitted for significant protocol-defined toxicities (eg, febrile neutropenia, grade 4 thrombocytopenia, or grade 3 liver dysfunction).
Response and Progression Assessment
A computed tomography scan of chest, abdomen, and pelvis was required within 4 weeks of the start of treatment and was repeated for disease response approximately every 6 weeks (every other cycle). Disease progression and best response to study treatment were determined by using RECIST 1.1 criteria.
Study Design
The study was designed as a randomized, double-blind, placebo-controlled phase III trial with the primary end point of PFS. Exploratory analyses of overall survival (OS) and objective response rates (ORRs) were also planned. The randomization was stratified on prior pelvic radiation status. The target accrual was 130 patients. Assuming a constant hazard ratio (HR), this trial size had 83% power to detect a 40% reduction in the PFS event rate (HR, 0.60) as a result of treatment with bevacizumab. This HR equates to an improvement in median PFS from 4 months (expected for gemcitabine-docetaxel plus placebo) to 6.7 months (postulated for gemcitabine-docetaxel plus bevacizumab). The overall type 1 error rate was limited to 0.05. An interim analysis with futility-based stopping rules was planned when at least 70 patients experienced a PFS event. Termination of the study was to be considered if the observed PFS HR estimate fell within futility regions predefined by using an O'Brien-Fleming6 (Lan-DeMets) type 1 error spending function.
Analysis Samples
Statistical analyses for PFS, OS, and ORR were done on an intention-to-treat basis and included all patients in the study who were randomly assigned. The analyses for safety and toxicity included patients who received at least one dose of study treatment (Fig 1).
Fig 1.

CONSORT diagram. Patients who received none of the protocol therapy were excluded from the assessment of toxicity. The analysis was done on an intent-to-treat basis. Some patients were deemed to have inadequate pathology, but these patients were not excluded from any of the reported results. ORR, overall response rate; OS, overall survival; PFS, progression-free survival.
Statistical Methods
The null hypothesis of no treatment effect on PFS and OS was assessed by using a grouped log-rank test. HR (gemcitabine-docetaxel plus bevacizumab v gemcitabine-docetaxel plus placebo) estimates for treatment differences were obtained from proportional hazards models. These models were stratified by the presence of prior pelvic radiation. Median times to event and 95% CIs were estimated from Kaplan and Meier curves. Differences in continuous characteristics between the treatment groups were assessed by Wilcoxon rank sum tests. Hypotheses in contingency tables (including the ORR exploratory end point) were assessed by Fisher's exact test. 95% CIs for proportions were calculated by using the Jeffreys method. All data analyses were generated by using SAS/STAT software, Version 9.4 (SAS Institute, Cary, NC).
RESULTS
Between fourth quarter 2009 and second quarter 2013, 107 women with metastatic uLMS were enrolled onto the study. All randomly assigned patients (54 assigned to gemcitabine-docetaxel plus placebo, 53 assigned to gemcitabine-docetaxel plus bevacizumab) were included in the analyses of response and survival outcomes. There were no significant differences in the treatment arms for patient characteristics, including age, race/ethnicity, performance status, prior pelvic radiation, or prior hormonal treatment (Table 1).
Table 1.
Patient Characteristics (N = 107)
| Characteristic | Gemcitabine-Docetaxel + Placebo (n = 54) |
Gemcitabine-Docetaxel + Bevacizumab (n = 53) |
P | ||
|---|---|---|---|---|---|
| No. | % | No. | % | ||
| Age, years | .10 | ||||
| Median | 56.2 | 54.8 | |||
| Range | 44.2 to 75.6 | 28.9 to 69.1 | |||
| Race/ethnicity | .266 | ||||
| White | 46 | 85.2 | 36 | 67.9 | |
| African American | 6 | 11.1 | 12 | 22.6 | |
| Asian/American Indian/unspecified | 2 | 3.7 | 5 | 9.4 | |
| Performance status | .70 | ||||
| 0 (asymptomatic) | 38 | 70.4 | 41 | 77.4 | |
| 1 (fully ambulatory) | 15 | 27.8 | 11 | 20.8 | |
| 2 (in bed < 50% of the time) | 1 | 1.9 | 1 | 1.9 | |
| Prior hormonal treatment | .72 | ||||
| Yes | 4 | 7.4 | 3 | 5.7 | |
| No | 50 | 92.6 | 50 | 94.3 | |
| Prior pelvic radiotherapy | .96 | ||||
| Yes | 11 | 20.4 | 11 | 20.8 | |
| No | 43 | 79.6 | 42 | 79.2 | |
Toxicities Observed
Four patients were excluded from the safety analyses, leaving 51 patients assigned to gemcitabine-docetaxel plus placebo and 52 patients assigned to gemcitabine-docetaxel plus bevacizumab evaluable for toxicity. Toxicities observed are summarized in Table 2. The addition of bevacizumab to gemcitabine-docetaxel did not result in increased frequency of neutropenia, thrombocytopenia, or anemia. Four patients assigned to gemcitabine-docetaxel plus bevacizumab experienced grade 3 hypertension, and six experienced grade 2 hypertension compared with zero grade 3 and seven grade 2 hypertension events among patients assigned to gemcitabine-docetaxel plus placebo (P = .26). The frequency of thromboembolic events and lymphedema was similar in both treatment groups. Specific GI toxicities, such as bleeding or fistula formation, were no more common among patients assigned to bevacizumab than among those assigned to placebo. Similarly, the total number of grade 2 or worse GI events was not statistically different between the two groups.
Table 2.
Toxicities Observed (n = 103)
| Toxicity Grade | Gemcitabine-Docetaxel + Placebo (n = 51) |
Gemcitabine-Docetaxel + Bevacizumab (n = 52) |
P | ||
|---|---|---|---|---|---|
| No. | % | No. | % | ||
| Neutropenia | .72 | ||||
| 3 | 7 | 14 | 8 | 15 | |
| 4 | 5 | 9 | 4 | 7 | |
| Thrombocytopenia | .69 | ||||
| 3 | 11 | 21 | 13 | 25 | |
| 4 | 4 | 7 | 6 | 11 | |
| Anemia | .09 | ||||
| 3 | 17 | 33 | 7 | 13 | |
| 4 | 0 | 0 | |||
| Hypertension | .26 | ||||
| 2 | 7 | 14 | 6 | 12 | |
| 3 | 0 | 4 | 8 | ||
| 4 | 0 | 0 | |||
| Thromboembolic | .71 | ||||
| 2 | 3 | 6 | 1 | 2 | |
| 3 | 3 | 6 | 3 | 6 | |
| 4 | 1 | 2 | 2 | 4 | |
| Lymphedema | .18 | ||||
| 2 | 0 | 1 | 2 | ||
| 3 | 0 | 0 | |||
| 4 | 0 | 0 | |||
| GI hemorrhage (No. of patients with event) | .79 | ||||
| 2 | 1 | 1 | |||
| 3 | 1 | 0 | |||
| 4 | 0 | 0 | |||
| Fistula (any site; No. of patients with event) | 1.0 | ||||
| 3 | 0 | 1 | |||
| 4 | 0 | 0 | |||
| Any GI event | .60 | ||||
| 2 | 13 | 14 | |||
| 3 | 12 | 7 | |||
| 4 | 1 | 0 | |||
NOTE. The number (proportion) of patients, classified by maximum grade, is provided.
PFS and OS Outcomes
PFS among patients on both treatment arms is illustrated in Figure 2 and detailed in Table 3. The median PFS was 6.2 months (95% CI, 2.9 to 9.9 months) among patients on the gemcitabine-docetaxel plus placebo arm versus 4.2 months (95% CI, 3.1 to 8.4 months) among those on the gemcitabine-docetaxel plus bevacizumab arm (P = .58). The percentage of patients alive and progression-free at 12 months from enrollment was 26.4% with gemcitabine-docetaxel plus placebo versus 25% with gemcitabine-docetaxel plus bevacizumab (HR, 1.12; 95% CI, 0.74 to 1.7). At the interim analysis, the Data Safety and Monitoring Board recommended closure of the study on the basis of the futility of gemcitabine-docetaxel plus bevacizumab relative to gemcitabine-docetaxel plus placebo.
Fig 2.
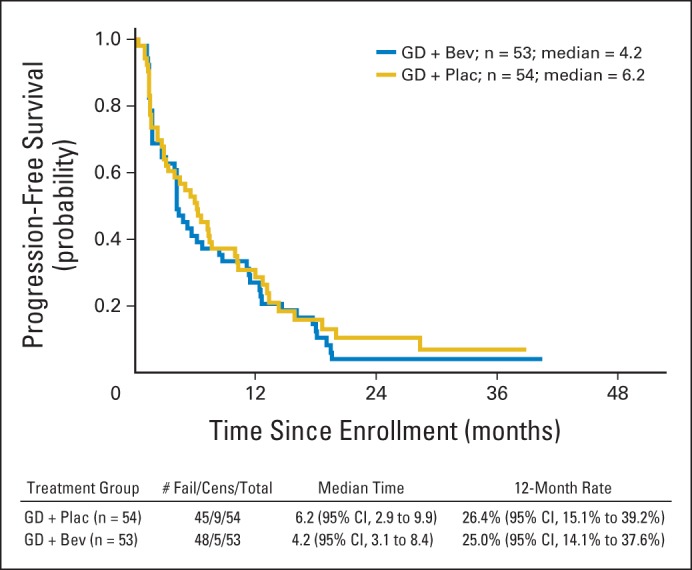
Progression-free survival among patients assigned to gemcitabine-docetaxel (GD) plus placebo (Plac; n = 54) or GD plus bevacizumab (Bev; n = 53). Cens, censored.
Table 3.
PFS and OS Among All Randomly Assigned Patients
| Therapy | Median (months) |
95% CI | % Progression Free at 12 Months | % Alive at 12 Months | 95% CI | HR | 95% CI | |
|---|---|---|---|---|---|---|---|---|
| PFS | OS | |||||||
| Gemcitabine-docetaxel + placebo | 6.2 | 2.9 to 9.9 | 26.4 | 15 to 39 | 1.12 | 0.74 to 1.7 | ||
| Gemcitabine-docetaxel + bevacizumab | 4.2 | 3.1 to 8.4 | 25 | 14 to 38 | ||||
| Gemcitabine-docetaxel + placebo | 26.9 | 15.9 to 32.1 | 74.7 | 60.4 to 84.4 | 1.07 | 0.63 to 1.81 | ||
| Gemcitabine-docetaxel + bevacizumab | 23.3 | 16.6 to 27.3 | 71 | 55.9 to 81.7 | ||||
NOTE. Assigned to gemcitabine-docetaxel + placebo, n = 54; assigned to gemcitabine-docetaxel + bevacizumab, n = 53.
Abbreviations: HR, hazard ratio; OS: overall survival; PFS: progression-free survival.
Exploratory analysis showed no OS difference between the two arms, as illustrated in Figure 3, and detailed in Table 3. Median OS was 26.9 months (range, 15.9 to 32.1 months) for patients receiving gemcitabine-docetaxel plus placebo and 23.3 months (range, 16.6 to 27.3 months) for patients receiving gemcitabine-docetaxel plus bevacizumab (HR, 1.07; 95% CI, 0.63 to 1.81; P = .81).
Fig 3.
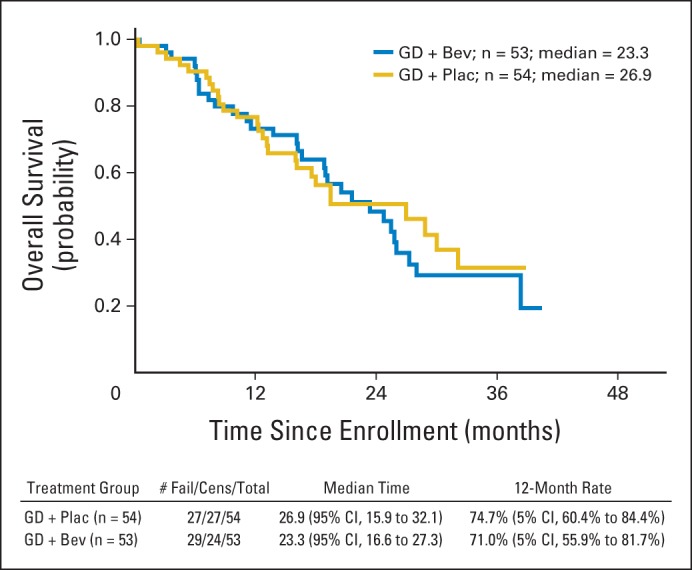
Overall survival among patients assigned to gemcitabine-docetaxel (GD) plus placebo (Plac; n = 54) or GD plus bevacizumab (Bev; n = 53). Cens, censored.
Objective Responses and Response Duration
With a median follow-up time of 25 months (maximum 41 months), objective responses (complete response [CR] plus partial responses [PRs] by RECIST) were observed in 17 (31.5%; 95% CI, 21.2% to 43.4%) of 54 patients assigned to gemcitabine-docetaxel plus placebo versus 19 (35.8%; 95% CI, 24.9% to 48.0%) of 53 patients assigned to gemcitabine-docetaxel plus bevacizumab (P = .69). A best response of stable disease was observed in 17 patients in each arm (31% for gemcitabine-docetaxel plus placebo; 32% for gemcitabine-docetaxel plus bevacizumab). Among the 17 patients with objective responses on the gemcitabine-docetaxel plus placebo arm, the median duration of response was 8.6 months (range, 1.3 to 30.2 months). Among the 19 patients with objective responses on the gemcitabine-docetaxel plus bevacizumab arm, the median response duration was 8.8 months (range, 1.7 to 32.8 months).
A total of 24 patients (12 in each treatment arm) elected to stop study treatment for reasons other than toxicity or disease progression. These patients had achieved CR (n = 2), PR (n = 15), or stable disease (n = 7). The median number of cycles these patients had received before stopping therapy was nine (range, five to 23). These patients received follow-up with protocol-specified computed tomography imaging to assess time to progression, while undergoing no additional therapy. The median time from stopping active study treatment to progression of disease was 5.7 months (range, 2 to 19.5 months).
DISCUSSION
This randomized, double-blind, placebo-controlled phase III trial showed that the addition of bevacizumab to fixed-dose-rate gemcitabine-docetaxel failed to improve PFS, OS, ORR, or response duration among patients with metastatic uLMS undergoing first-line treatment. The addition of bevacizumab did not increase the frequency of toxicities observed. The results of this study provide several important observations for the management of women with advanced uLMS regarding docetaxel dose and duration of treatment.
The dose of docetaxel in this phase III trial was 75 mg/m2. Previous phase II studies of fixed-dose-rate gemcitabine plus docetaxel used 100 mg/m2 docetaxel. This study showed that ORRs and response duration are comparable to those seen in the previous phase II trials, GOG 87L2 (A Phase II Evaluation of Docetaxel and Gemcitabine Plus G-CSF in the First-Line Treatment of Recurrent or Advanced Leiomyosarcoma of the Uterus) and GOG 131G (A Phase II Evaluation of Docetaxel and Gemcitabine Plus G-CSF in the Second-Line Treatment of Recurrent or Advanced Leiomyosarcoma of the Uterus).1 These results suggest that docetaxel 75 mg/m2 rather than the potentially more toxic dose of 100 mg/m2 can be used in the fixed-dose-rate gemcitabine-docetaxel regimen with the expectation of similarly high ORRs. The acute and cumulative toxicities are likely to be fewer with the lower dose of docetaxel.
Twenty-four patients (23% of the study patients) elected to stop active study treatment while in CR or PR or stable disease. Among these patients, the median time from stopping active treatment until progression of disease was nearly 6 months. This observation suggests that it may be reasonable to offer patients a break from active treatment and the related toxicities.
The failure of bevacizumab to improve any clinical outcome in uLMS raises the question of whether there is any role for antivascular-directed therapy in this disease. Phase II single-agent studies of vascular-targeted agents have mostly yielded negative results. The multikinase inhibitor sunitinib achieved objective response in only two (8.7%) of 23 patients, and only 17% of patients remained progression-free at 6 months. The median PFS was 1.5 months. These results failed to meet the study's criteria for further investigation of this agent in uLMS.7 Similarly, sorafenib failed to achieve any objective responses among the patients with LMS enrolled onto a phase II trial,8 and in a separate multicohort phase II trial, objective response was observed in only one (2%) of 37 patients in the LMS cohort.9 In a phase III trial, pazopanib was compared with placebo for patients with advanced nonadipocytic soft tissue sarcomas. Although the ORR was low (6%), the median PFS was 4.6 months among patients assigned to treatment with pazopanib versus 1.6 months among those assigned to placebo. There was no statistically significant difference in OS.10 These data led to regulatory agency approval of pazopanib for patients with advanced nonadipocytic soft tissue sarcoma. It is not known whether pazopanib can be safely combined with fixed-dose-rate gemcitabine, or whether it would improve response rates or survival outcomes. Because there is no comparable study of single-agent bevacizumab versus placebo in advanced soft tissue sarcoma, it is not known whether pazopanib is truly a better antiangiogenic agent than bevacizumab in sarcomas.
The OS observed in this study among the 51 patients assigned to gemcitabine-docetaxel plus placebo was 26.9 months (range, 15.9 to 32.1 months). This result may be compared with observed OS rates in the first-line treatment setting for women with metastatic uLMS treated on recent phase II studies conducted by the GOG. For example, in GOG 87L,2 the phase II trial of fixed-dose-rate gemcitabine-docetaxel, the median OS exceeded 16 months (range, 0.4 to 41.3 months) among 39 evaluable patients. In the GOG phase II trial of trabectedin, the median OS was 26.1 months among the 20 patients enrolled, with interpretation limited by the smaller sample size and several censored observations.11
This study was specifically designed to limit eligibility to chemotherapy-naive patients with uLMS. Such a design helps ensure homogeneity of the treatment population and improves the reliability of the results. Because the study population was limited to a single histology (LMS) from one anatomic site (uterus), we cannot conclude that the observed results can be applied to all soft tissue sarcoma histologies, because different sarcoma histologies have different drug sensitivities and behaviors. In the opinion of the authors, it is likely that similar results would be observed among patients with LMS of other anatomic sites.
The timely accrual and completion of this uLMS trial demonstrates that prospective, phase III, double-blind, placebo-controlled trials are feasible in this rare disease in the cooperative group setting. The successful conduct of such studies helps to establish standards of care. This phase III trial demonstrated that bevacizumab does not improve outcomes when added to fixed-dose-rate gemcitabine-docetaxel. Therefore, gemcitabine-docetaxel remains a standard first-line treatment for advanced uLMS.
Supplementary Material
Appendix
The following gynecologic oncology institutions participated in this study: Roswell Park Cancer Institute, Duke University Medical Center, Abington Memorial Hospital, Walter Reed National Military Medical Center, University of Mississippi Medical Center, University of Colorado Cancer Center-Anschutz Cancer Pavilion, University of California at Los Angeles Health System, Fred Hutchinson Cancer Research Center, The University of Pennsylvania, University of Cincinnati, University of North Carolina at Chapel Hill, University of Iowa Hospitals and Clinics, University of Texas Southwestern Medical Center, Indiana University Hospital, University of California Medical Center at Irvine-Orange Campus, Rush University Medical Center, State University of New York Downstate Medical Center, University of New Mexico, Cleveland Clinic, Stony Brook University Medical Center, Memorial Sloan Kettering Cancer Center, Cooper Hospital-University Medical Center, Ohio State University Medical Center, MD Anderson Cancer Center, Fox Chase Cancer Center, Women's Cancer Center of Nevada, University of Oklahoma Health Sciences Center, University of Chicago, Mayo Clinic, Yale University, University of Wisconsin Hospital and Clinics, Women and Infants Hospital, The Hospital of Central Connecticut, Georgia CORE, Aurora Health Care, University of California at San Francisco-Mount Zion, and Community Clinical Oncology Program.
Footnotes
Supported by National Cancer Institute Grants No. CA27469 to the Gynecologic Oncology Group Administrative Office and No. CA37517 to the Gynecologic Oncology Group Statistical and Data Center and NRG Oncology Grant No. 1 U10 CA180822.
Clinical trial information: NCT01012297.
Authors' disclosures of potential conflicts of interest are found in the article online at www.jco.org. Author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Disclosures provided by the authors are available with this article at www.jco.org.
AUTHOR CONTRIBUTIONS
Conception and design: Martee L. Hensley, Robert S. Mannel, Helen Michael
Provision of study materials or patients: Robert S. Mannel
Collection and assembly of data: Martee L. Hensley, David M. O'Malley, Kian Behbakht, Jamie N. Bakkum-Gamez, Helen Michael
Data analysis and interpretation: Martee L. Hensley, Austin Miller, Jamie N. Bakkum-Gamez
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Randomized Phase III Trial of Gemcitabine Plus Docetaxel Plus Bevacizumab or Placebo As First-Line Treatment for Metastatic Uterine Leiomyosarcoma: An NRG Oncology/Gynecologic Oncology Group Study
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or jco.ascopubs.org/site/ifc.
Martee L. Hensley
Employment: sanofi-aventis (I)
Consulting or Advisory Role: GlaxoSmithKline, INSYS Therapeutics, Arno Therapeutics
Research Funding: Johnson & Johnson
Patents, Royalties, Other Intellectual Property: UpToDate
Austin Miller
No relationship to disclose
David M. O'Malley
Honoraria: Genentech, Clovis Oncology
Research Funding: Amgen, VentiRx Pharmaceuticals, AstraZeneca, Genentech, Regeneron Pharmaceuticals, ImmunoGen, Array BioPharma, Janssen R&D, Clovis Oncology, EMD Serono, Ergomed, Ajinomoto
Robert S. Mannel
Consulting or Advisory Role: Genentech, Endocyte, Eisai, Amgen, Advaxis, MedImmune, AstraZeneca
Kian Behbakht
No relationship to disclose
Jamie N. Bakkum-Gamez
No relationship to disclose
Helen Michael
No relationship to disclose
REFERENCES
- 1.Hensley ML, Blessing J, Degeest K, et al. Fixed-dose rate gemcitabine plus docetaxel as second-line therapy for metastatic uterine leiomyosarcoma: A Gynecologic Oncology Group phase II study. Gynecol Oncol. 2008;109:323–328. doi: 10.1016/j.ygyno.2008.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hensley ML, Blessing JA, Mannel R, et al. Fixed-dose rate gemcitabine plus docetaxel as first-line therapy for metastatic uterine leiomyosarcoma: A Gynecologic Oncology Group phase II trial. Gynecol Oncol. 2008;109:329–334. doi: 10.1016/j.ygyno.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim KJ, Li B, Winer J, et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. 1993;362:841–844. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- 4.Verschraegen CF, Arias-Pulido H, Lee SJ, et al. Phase IB study of the combination of docetaxel, gemcitabine, and bevacizumab in patients with advanced or recurrent soft tissue sarcoma: The Axtell regimen. Ann Oncol. 2012;23:785–790. doi: 10.1093/annonc/mdr299. [DOI] [PubMed] [Google Scholar]
- 5.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 6.Lan KK, DeMets DL. Discrete sequential boundaries for clinical trials. Biometrika. 1983;70:659–663. [Google Scholar]
- 7.Hensley ML, Sill MW, Scribner DR, Jr, et al. Sunitinib malate in the treatment of recurrent or persistent uterine leiomyosarcoma: A Gynecologic Oncology Group phase II study. Gynecol Oncol. 2009;115:460–465. doi: 10.1016/j.ygyno.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.von Mehren M, Rankin C, Goldblum JR, et al. Phase 2 Southwest Oncology Group-directed intergroup trial (S0505) of sorafenib in advanced soft tissue sarcomas. Cancer. 2012;118:770–776. doi: 10.1002/cncr.26334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maki RG, D'Adamo DR, Keohan ML, et al. Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J Clin Oncol. 2009;27:3133–3140. doi: 10.1200/JCO.2008.20.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van der Graaf WT, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379:1879–1886. doi: 10.1016/S0140-6736(12)60651-5. [DOI] [PubMed] [Google Scholar]
- 11.Monk BJ, Blessing JA, Street DG, et al. A phase II evaluation of trabectedin in the treatment of advanced, persistent, or recurrent uterine leiomyosarcoma: A Gynecologic Oncology Group study. Gynecol Oncol. 2012;124:48–52. doi: 10.1016/j.ygyno.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.