

Abstract
HIV-1 infected individuals are living longer but experiencing a prevalence rate of over 50% for HIV-1 associated neurocognitive disorders (HAND) for which no effective treatment is available. Viral and cellular factors secreted by HIV-1 infected cells leads to neuronal injury and HIV-1 Tat continues to be implicated in the pathogenesis of HAND. Here we tested the hypothesis that creatine protected against HIV-1 Tat-induced neuronal injury by preventing mitochondrial bioenergetic crisis and/or redox catastrophe. Creatine blocked HIV-1 Tat1-72-induced increases in neuron cell death and synaptic area loss. Creatine protected against HIV-1 Tat-induced decreases in ATP. Creatine and creatine plus HIV-1 Tat increased cellular levels of creatine, and creatine plus HIV-1 Tat further decreased ratios of phosphocreatine to creatine observed with creatine or HIV-1 Tat treatments alone. Additionally, creatine protected against HIV-1 Tat-induced mitochondrial hypopolarization and HIV-1 Tat-induced mitochondrial permeability transition pore opening. Thus, creatine may be a useful adjunctive therapy against HAND.
Keywords: creatine, mitochondria dysfunction, neuroprotection, HIV-1 associated neurocognitive disorders, HIV-1 Tat
Introduction
HIV-1 infection is a major global health problem, with greater than 40 million people worldwide infected [1]. Although antiretroviral therapies (ART) have increased the life span of HIV-1 infected individuals, these people are now experiencing a prevalence rate of over 50% for HIV-1 associated neurocognitive disorders (HAND) [2]. Clinically, HAND represents a set of conditions including asymptomatic neurocognitive impairment, mild neurocognitive disorder, and to a lesser extent in the ART era, HIV-1 associated dementia [3]. Currently the underlying mechanisms for HAND pathogenesis are not fully understood and no effective treatment is available. It is known that HIV-1 does not infect neurons and that HIV-1 associated neurodegenerative pathology is not proportional to viral load [4]. Thus, neuronal injury as occurs in HAND results from viral and cellular factors released from bystander HIV-1 infected cells such as infected glia [5].
Among those viral and cellular factors, HIV-1 transactivator of transcription protein (HIV-1 Tat) continues to be implicated in the pathogenesis of HAND [6–13]. HIV-1 is present in brains of HIV-1 infected individuals and levels stay elevated in the cerebrospinal fluid of people living with HIV-1 infection and/or acquired immunodeficiency syndrome even when their viral levels are immeasurable because of effective treatment with ART [14]. Others and we have shown that HIV-1 Tat disturbs neuronal calcium homeostasis [6, 15], induces mitochondrial dysfunction as determined by changes in levels of reactive oxygen species, ATP, and mitochondrial membrane polarization [7, 16–20], disrupts synaptic integrity [21, 22], and promotes neuronal cell death [8, 23].
Creatine, a normal component of meat-based diets that is produced endogenously and ingested as a dietary supplement, is present at high levels in brain [24]. Creatine is neuroprotective and in model systems has been shown to protect against Parkinson’s disease [25], Huntington’s disease [27], Alzheimer’s disease [28], and amyotrophic lateral sclerosis [29]. Mechanistically, the neuroprotective properties of creatine appear to be related mostly to stabilizing cellular energy levels by increasing mitochondrial bioenergetics and reducing free radical damage by decreasing mitochondrial redox catastrophe [26].
In the present study, we determined the extent to which creatine protects against HIV-1 Tat-induced neuronal injury in primary cultured neurons. We demonstrated that longer-term treatments with creatine protected against HIV-1 Tat-induced decreases in synaptic proteins and increases in neuronal cell death, and at shorter treatment intervals protected against changes in mitochondrial bioenergetics and redox phenomena. Thus, creatine may be a useful adjunctive therapy against HAND.
Material and Methods
Primary cortical neuron culture
Primary cultures of mouse cerebral cortical neurons were prepared using a protocol approved by the University of North Dakota Animal Care and Use Committee adherent with the Guide for the Care and Use of Laboratory Animals (NIH publication number 80-23). E-16 pups were removed aseptically from pregnant C57BL/6 mice (Charles River Laboratories), brains were isolated and placed into ice-cold sterile phosphate-buffered saline (PBS) containing 5.5 mM glucose, 1 μM ethylenediaminetetraacetic acid (EDTA) and 1 μM ethylene glycol tetraacetic acid (EGTA), and meninges were removed. Cerebral cortices were dissected, placed in fresh ice-cold buffer, minced and incubated for 15 minutes at 37°C with 5 ml of trypsin-EDTA (Gibco). Trypsin was de-activated by adding the tissue to 5 ml of heat-inactivated fetal bovine serum (Atlanta Biologicals) for 1 min at room temperature. The tissue was added to 10 ml of Neurobasal media (Gibco) containing B-27 supplement (Gibco), 0.5 mM L-glutamine, and antibiotic/antimycotic (Sigma) containing penicillin (100 units), streptomycin (0.10 mg) and amphotericin B (0.25 μg) and was triturated through a 5 ml pipette. Dissociated cells were seeded into uncoated 96-well plates (Nunc, Roskilide, Denmark) or glass bottom 35 mm culture dishes (Matek, Ashland, MA, USA) coated previously with poly-D-lysine. After 10–14 days in culture, cells which were found typically to be >95% neurons were taken for experimentation.
HIV-1 Tat
Purified (>95%) recombinant HIV-1 Tat1-72 protein from clade B was produced from the tat gene encoding for the first exon as described [30]. A deletion mutant of HIV-1 Tat was produced as described [31]; the sequence encoding amino acids 31-61 of HIV-1 Tat (mutant Tat) previously shown to contain the neurotoxic epitope [32] was removed. Both HIV-1 Tat1-72 and mutant Tat were obtained as gifts from Dr. Avindra Nath (NINDS). Low protein retention pipette tips and micro-centrifuge tubes were used to reduce the loss of HIV-1 Tat due to its adherent properties. At the time of experimental treatments, Neurobasal media was replaced with Locke’s buffer containing 156 mM NaCl, 5.6 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 3.6 mM NaHCO3, 5 mM glucose, and 5 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) in sterile water (pH 7.2) and all assays were conducted using Locke’s buffer. HIV-1 Tat once thawed was never re-frozen for later use due to degradation caused by repeat cycles of freezing and thawing. Care was also taken not to mix HIV-1 Tat vigorously to minimize oxygenation and inactivation.
Adenine nucleotides, creatine and phosphocreatine
ATP, ADP, AMP, creatine and phosphocreatine levels were measured by high-performance liquid chromatography (HPLC) as described [33, 34]. Four hours after experimental treatments were applied, buffer was removed, cultures were washed three-times with ice-cold PBS, and cells were lyzed and proteins were precipitated with 2% trichloroacetic acid (Supelco) and three freeze/thaw cycles. A small amount of the lysate was removed for protein analysis (Bio-Rad). Lysate was added to an equal amount of dichloromethane and tri-octylamine (775:225 v:v) and samples were shaken vigorously. Aliquots (30 μl) of the aqueous phase were injected onto a LC-18-T HPLC column (Supelco) and compounds of interest were separated using a mobile phase of 0.1M KH2PO4 run isocratically at a rate of 1 ml/minute. Adenine nucleotides, creatine and phosphocreatine levels were measured spectrophotometrically at an absorbance of 254 nm for nucleotides and 210 nm for creatine and phosphocreatine. Data were measured as integrated areas under the peaks and were identified based on retention time and sample spiking. Adenlyate energy charge was calculated using the formula ( ).
Cell viability
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and trypan blue exclusion assays were conducted in 96-well plates to determine neuronal cell death [35, 36]. For the MTT assay, thiazolyl blue tetrazolium bromide (MTT, 5 mg/ml, Sigma) diluted in Neurobasal media without phenol red was added to neuronal cultures 24 h after treatments and incubated at 37°C and 5% CO2 for 3 h. Volumes of 0.1 N HCl in isopropanol equal to that of the media were added to solubilize purple formazan crystals. Absorbance was measured at 570 nm using a SpectraMax Plus 384 plate-reader (Molecular Devices). Each experiment was conducted in triplicate and experiments were repeated at least four times using different batches of cultured cells. For trypan blue exclusion, media was removed from cell cultures 24 h after treatments, 50 μl trypan blue (0.2%) diluted in PBS was added, and after 5 min at room temperature the total number of cells and the number of cells lacking trypan blue were counted; data on dead cells were expressed as a percentage of total cells. Each experiment was conducted in triplicate and experiments were repeated at least four times using different batches of cultured cells.
Mitochondrial membrane potential
Mitochondrial membrane potential was determined 4 h after treatments in cells grown on glass bottom 35 mm culture dishes by removing culture media, replacing the media with 2 ml PBS containing 1.0 μg/ml of the cationic duel emission dye tetraethylbenzimidazolylcarbocyanine iodide (JC-1) (Molecular Probes), and incubating cells at 37°C and 5% CO2 for 25 min. After incubation, cells were washed two-times with media at 37°C and fluorescence was measured using a Zeiss Axiovert 200M microscope system at excitation/emission wavelengths of 485/530 nm for the monomer and 535/570 nm for the J-aggregate. Fluorescence was quantified for each cell using average pixel intensity of both J-aggregate and JC-1 monomers using Image-J software (NIH). JC-1 is a lipophilic cationic dye that selectively enters the mitochondria and reversibly changes color. JC-1 aggregates at regions of high membrane potential and remains as a monomer in regions of low membrane potential. Data (average pixel intensity) were normalized to the ratio of J-aggregate/J-monomer in untreated control cultures (ratios for controls were set to a value of 1). Each experiment was conducted in duplicate and experiments were repeated at least four times using different batches of neurons.
Mitochondrial permeability transition pore
Mitochondrial permeability transition pore opening was determined fluorometrically [37, 38]. Following treatment of cells for 4 h, cultures were washed in 2 ml Locke’s buffer and incubated in 2 ml Locke’s buffer containing calcein-AM (1 μM) and cobalt chloride (1 mM) for 20 min at 37°C. Cells were washed with 2 ml Locke’s buffer and fluorescence was measured with a Zeiss Axiovert 200M microscope at an excitation wavelength of 488 nm and an emission wavelength of 525 nm. Following de-esterification, calcein fluorescence was visible in mitochondria, and cobalt chloride that quenches the fluorescence only enters intact mitochondria when mitochondrial permeability transition pores are open. Fluorescence was quantified using average pixel intensity using Image-J software and data were expressed as cells with open pores as a percentage of total cells. Each experiment was conducted in duplicate using 35 mm glass-bottomed plates and was repeated at least four times using different batches of cultured cells.
Reactive oxygen species
Levels of reactive oxygen species were measured in 96-well plates using the fluorescent dye dichloro-dihydrofluorecein diacetate (H2DCFDA), a cell membrane permeable dye that fluoresces in the presence of hydrogen peroxide, peroxyl radicals, peroxynitrite anions, and nitric oxide. Following treatment of cells for 4 h, culture media was replaced with 50 ml PBS containing 20 μM H2DCFDA and cells were incubated at 37°C and 5% CO2 for 45 minutes. Cells were washed once with 50 ml PBS and fluorescence was determined using a SpectraMax Plus 384 plate-reader (Molecular Devices). Each experiment was conducted in triplicate and experiments were repeated at least 4-times using different batches of cultured cells.
Immunoblotting
Neurons were lysed with RIPA buffer (Pierce) containing protease inhibitor cocktail (Sigma). After centrifugation (14,000 × g for 10 min at 4°C), supernatants were collected, and protein concentrations were determined with a DC protein assay (Bio-Rad). Equal amounts of proteins (10 μg) were separated by SDS-PAGE (12% gel), and following transfer, polyvinylidene difluoride membranes (Millipore) were incubated overnight at 4°C with anti-synaptophysin (1:1000, Sigma), and anti-β-actin (1:10000, mouse monoclonal, Abcam) antibody was used as a gel loading control. The blots were developed with enhanced chemiluminescence, and bands were visualized and analyzed by LabWorks 4.5 software on a BioSpectrum® imaging System (UVP). Quantification was performed by densitometry and the results were analyzed as total integrated densitometric volume values (arbitrary units).
Statistical analysis
All data were reported as means and SEM. Statistical comparisons were made using one-way analysis of variance (ANOVA) followed by Tukey-Kramer multiple comparison post hoc tests, unless indicated otherwise. Statistically significant differences were set at p < 0.05.
Results
Tat, but not mutant Tat, causes neuronal cell death
Previously, Tat1-72, but not Tat1-72 missing amino acids 31-61 (mutant Tat), was found to be neurotoxic [16, 34, 39–42]. To control for possible non-specific effects of Tat, parallel experiments were performed throughout these studies with equivalent concentrations of Tat1-72 and mutant-Tat. Primary cultures of mouse cerebral cortical neurons treated for 24 hours with Tat1-72 and mutant Tat at concentrations ranging from 10 pM to 400 nM. We found statistically significant increases in neuronal cell death by Tat1-72 treatment at 100 nM (10.8% ± 1.6) and 400 nM (17.1% ± 1.9), but not by mutant-Tat as determined by trypan blue exclusion (Figure 1A) and MTT assay (data not shown). Because of findings that Tat affects mitochondrial function and MTT assays reflect cell viability and mitochondrial function, trypan blue exclusion was used in all subsequent experiments for determination of cell viability. Time profile studies for Tat-induced neuronal death established that Tat1-72 at 100 nM increased significantly (Figure 1B) levels of neuronal cell death to comparable levels at incubation intervals of 24 (12.6% ± 1.6) and 48 h (12.6% ± 1.7). Accordingly, all subsequent studies on cell viability used Tat1-72 at a concentration of 100 nM and incubations of 24 h.
Figure 1.

Tat1-72 concentration- and time-dependently increased cortical neuron cell death. (A) Treatment of neurons with Tat1-72, but not mutant-Tat1-72, at concentrations of 100 and 400 nM significantly increased neuronal cell death as determined with trypan blue exclusion. (B) Tat at a concentration of 100 nM produced time-dependent increases in neuronal cell death; statistically significant increases were observed with 24 and 48 h incubations. Experiments (both Figures 1A and 1B) were performed in triplicate, repeated at least four times with different batches of cultured cells, and results were expressed as numbers of non-viable cells relative to total cells (*p < 0.05, ** p < 0.01, ***p < 0.001 versus untreated controls).
Creatine protects against Tat-induced neuronal cell death
To test for creatine protection against Tat1-72-induced neuronal cell death, neurons were treated with 100 nM Tat1-72 for 24 h in the absence or presence of creatine at 1, 3, 5, and 20 mM. In these studies, Tat increased significantly (p < 0.01) neuronal cell death from control levels of 4.2 ± 0.5 % to 12.7 ± 1.1 %. Creatine, at a concentration of 3 mM, significantly (p<0.05) reduced levels of Tat-induced neuronal cell death to 6.4 ± 0.5 % (Figure. 2). The neuroprotective effect afforded by creatine, however, was lost at the higher concentration (Figure. 2). Levels of neuronal cell death following incubations with 100 nM mutant Tat were indistinguishable from control levels (data not shown).
Figure 2.

Creatine concentration-dependently protected against Tat-induced neuronal cell death. Statistically significant (p<0.01) increases in neuronal cell death were observed with treatments of 100 nM Tat for 24 h as determined by trypan blue exclusion assay. Statistically significant (p<0.05) protection against Tat-induced neuronal cell death was observed with 3 mM creatine. Experiments were performed in triplicate, repeated at least four times with different batches of cultured cells, and results were expressed as numbers of non-viable cells relative to total cells (** p < 0.01 Tat versus control).
Creatine protects against Tat-induced loss of synaptic proteins
To test for creatine protection against Tat1-72-induced synaptic dysfunction, neurons were treated with 100 nM Tat1-72 for 24 h in the absence or presence of creatine at 3 mM, and protein levels of synaptophysin, a synaptic area marker, were determined by immunoblotting. In these studies, Tat decreased significantly (p < 0.05) protein levels of synaptophysin from control levels of 100 ± 25.9 % to 42 ± 9.4 %. Creatine, at a concentration of 3 mM, blocked significantly (p<0.05) Tat-induced decreases in protein levels of synaptophysin (Figure. 3). Levels of synaptophysin following incubations with 100 nM mutant Tat were indistinguishable from control levels (data not shown).
Figure 3.

Creatine protects against Tat-induced loss of synaptic proteins. Immunoblotting showed that HIV-1 Tat1-72 (100 nM) treatment for 24 hours decreased significantly (p<0.05, n=4) protein levels of synaptophysin in primary cultured neurons, and such an effect was blocked by creatine (3 mM) treatment (p<0.05, n=4)
Effects of Tat and creatine on levels of adenine nucleotides and phosphocreatine
To determine underlying mechanism(s) responsible for the neurotoxic actions of Tat1-72 and the neuroprotective effects of creatine, we measured, at a time period (4 h) that precedes the neurotoxic effects, levels of adenine nucleotides (ATP, ADP, and AMP), creatine, and phosphocreatine. Creatine (3 mM) treatment for 4 h increased significantly (p<0.01) levels of ATP from control levels of 10.3 ± 0.3 to 13.6 ± 0.6 nmol/mg protein (Figure. 4A). After treatment with Tat1-72 (100 nM, 4h), ATP levels were decreased significantly (p<0.05) to 7.3 ± 0.6 nmol/mg protein. Co-application of 3 mM creatine with 100 nM Tat1-72 for 4 h protected against Tat-induced decreases in ATP levels and returned levels to near control values of 10.0 ± 0.8 nmol/mg protein. A similar, but statistically non-significant pattern of changes to levels of ADP (Figure. 4B) and AMP (Figure. 4C) was observed for these same treatment groups. When the data for nucleotide levels were calculated to determine adenylate energy charge (Figure. 4D) or the ratio of ATP/ADP (Figure. 4E), no statistically significant differences were observed between control and treated cells.
Figure 4.

Effects of Tat, mutant-Tat, and creatine on levels of adenine nucleotides. (A) Compared with controls, creatine (Cr, 3 mM) increased significantly (p<0.01), and Tat (100 nM) but not mutant-Tat (mTat, 100 nM) decreased significantly (p<0.05) levels of ATP in cortical neurons. Creatine (3 mM) co-applied with Tat (100 nM) significantly (p<0.05) protected against Tat-induced decreases in levels of ATP. (B, C) Statistically non-significant changes to ADP (B) and AMP (C) were observed following treatments with Tat, mutant-Tat and creatine. (D, E) No statistically significant differences were observed for treatments with Tat, mutant-Tat and creatine on (D) adenylate energy charge (([ATP] + 1/2[ADP])/([ATP] + [ADP] + [AMP]) or (E) levels of ATP/ADP (* p < 0.05, ** p < 0.01 versus control).
We next determined the effects of Tat and/or creatine on cellular levels of creatine and its associated source of tissue energetics, phosphocreatine. Incubation of cells with creatine (3 mM for 4 h) resulted in statistically significant (p<0.01) increases in cellular levels of creatine from control levels of 62.3 ± 3.2 nmol/mg protein to 86.2 ± 5.0 nmol/mg protein (Figure. 5A). Tissue levels of creatine were not significantly changed when cells were treated with 100 nM Tat (64.9 ± 3.4 nmol/mg protein) or with 100 nM mutant-Tat (60.0 ± 4.8 nmol/mg protein). Co-application of creatine with Tat resulted in statistically significant (p<0.001) increases over controls in levels of creatine to 98.0 ± 4.4 nmol/mg protein (Figure. 5A). Levels of phosphocreatine were not affected significantly by any of the treatments (Figure. 5B). The ratio of phosphocreatine to creatine (Figure. 5) of 1.19 ± 0.07 in controls was decreased significantly to 0.99 ± 0.04 by Tat (p<0.05), to 0.78 ± 0.02 by creatine (p<0.01) and to 0.72 ± 0.07 by creatine plus Tat (p<0.001). Co-application of creatine with Tat decreased the phosphocreatine/creatine ratio to a level significantly (p<0.05) less than that observed with Tat alone.
Figure 5.
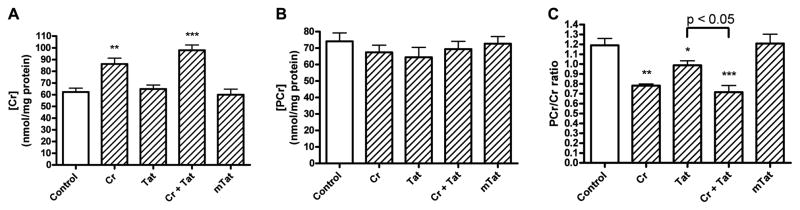
Effects of Tat, mutant-Tat and creatine on levels of creatine and phosphocreatine. (A) Creatine (Cr, 3 mM) application resulted in significantly increased cellular levels of creatine in the absence (p<0.01) or presence of Tat (p<0.001). Tat and mutant-Tat (mTat) at concentrations of 100 nM did not significantly affect levels of creatine. (B) Treatment of cortical neurons with creatine, Tat and mutant-Tat did not produce statistically significant changes in levels of phosphocreatine (PCr). (C) Ratios of phosphocreatine to creatine were decreased significantly by creatine (Cr, p<0.01), Tat (p<0.05), and creatine plus Tat (p<0.001). Co-application of creatine and Tat decreased significantly (p<0.001) the ratio of phosphocreatine to creatine to a level significantly (p<0.05) less than that observed with Tat alone (* p < 0.05, ** p < 0.01, *** p ≤ 0.001 versus control).
Creatine protects against Tat-induced decreases in mitochondrial membrane potential
Because HIV-1 Tat has been shown to affect mitochondrial function [16, 17, 43–45] and because creatine has been shown to have neuroprotective actions through mitochondrial mechanisms [16, 46, 47], we next examined mitochondrial mechanisms related to redox catastrophe. Tat (100 nM, 4 h) decreased significantly (p<0.001) the ratio of JC-1 aggregate to monomer by about 36 % (Figure. 6). This Tat-induced hypopolarization of mitochondria was blocked completely (p<0.001) with co-application of creatine (Figure 6). Creatine and mutant-Tat alone had no significant effects on mitochondrial membrane potential.
Figure 6.

Effects of Tat, mutant-Tat and creatine on mitochondrial membrane potential as determined with the fluorescent dye JC-1. Tat (100 nM), but not mutant-Tat (mTat, 100 nM), decreased significantly (p<0.001) mitochondrial membrane potential. Creatine (Cr, 3 mM) did not affect mitochondrial membrane potential, but creatine (3 mM) co-applied with Tat (100 nM) significantly (p<0.001) blocked Tat-induced mitochondrial hypopolarization (*** p < 0.001 versus control).
Creatine protects against Tat-induced increases in the opening of mitochondrial permeability transition pores
Treatment of cultured neurons with 100 nM Tat caused a significant (p<0.01) increase in the percentage of cells with open mitochondrial permeability transition pores (Figure. 7). Co-application of creatine (3 mM) with Tat resulted in a statistically significant (p<0.05) decrease in the number of cells with mitochondrial permeability transition pore opening compared to cells treated with Tat alone (Figure. 7). Mutant-Tat and creatine alone had no significant effects on mitochondrial permeability transition pore opening.
Figure 7.

Effects of Tat, mutant-Tat and creatine on opening of mitochondrial permeability transition pores. Tat (100 nM), but not mutant Tat (mTat, 100 nM), increased significantly (p<0.01) the percentage of cells with opening of mitochondrial permeability transition pores. Creatine (Cr, 3 mM) did not affect pore opening but creatine (3 mM) when co-applied with Tat (100 nM) significantly (p<0.05) blocked Tat-induced mitochondrial permeability transition pore opening (** p < 0.01 versus control).
Effects of Tat and creatine on levels of reactive oxygen species
Tat (100 nM) increased significantly (p<0.01) reactive oxygen species to levels similar to those observed with the positive control used, hydrogen peroxide (Figure. 8). Creatine (3 mM) did not itself significantly affect levels of reactive oxygen species nor did creatine significantly reduce Tat-induced increases in levels of reactive oxygen species. Mutant-Tat (100 nM) produced statistically significant (p<0.05) increases in levels of reactive oxygen species similar to those of cells treated with Tat1-72.
Figure 8.

Effects of Tat, mutant-Tat, and creatine on formation of reactive oxygen species. Tat (100 nM), creatine (3 mM) plus Tat (100 nM), mutant-Tat (100 nM) and the positive control hydrogen peroxide (H2O2, 500 mM) significantly increased reactive oxygen species. Creatine (3 mM) did not block Tat-induced increases in levels of reactive oxygen species (* p < 0.05, ** p < 0.01, *** p < 0.001 versus control).
Discussion
The present study was focused to determine the extent to which creatine protects against HIV-1 Tat-induced neurotoxicity and the involvement of mitochondria in this neuroprotection. The major findings of the present study were that creatine protected against HIV-1 Tat-induced neuronal cell death and synaptic dysfunction. Creatine also protected against HIV-1 Tat-induced decreases in ATP levels, depolarization of mitochondrial membrane potentials, and mitochondrial permeability pore opening. These findings provide further evidence for the involvement of mitochondria in the effects of HIV-1 Tat on neurons and show an ability of creatine to protect against these effects.
HIV-1 does not productively infect neurons, but neuronal injury represents a key pathological feature of HAND. Others and we have demonstrated the critical role of HIV-1 viral products and pro-inflammatory mediators released from productively infected glia in the pathogenesis of HAND [5]. One HIV-1 viral product that is neurotoxic at nanomolar concentrations is HIV-1 Tat, an RNA binding protein essential for viral replication [5, 17, 48]. HIV-1 Tat is released from infected glial cells within the CNS and it can be transported across the blood-brain barrier [49]. Importantly, HIV-1 Tat levels stay elevated in the CSF of people living with HIV-1/AIDS even when their viral levels are immeasurable because of effective treatment with ART [14]. In addition to its ability to induce neuroinflammation, others and we have demonstrated that HIV-1 Tat has direct neurotoxic effects [6, 8, 16, 23, 32, 40, 50].
Although the mechanisms are still being investigated, mitochondria have been implicated in the deleterious effects of HIV-1 Tat on neurons [62,63]. Such HIV-1 Tat-induced mitochondria dysfunction is evidenced by alterations in bioenergetics [17], mitochondrial membrane potential [17, 39], reactive oxygen species [17, 39, 51], mitochondrial permeability transition pore opening [16], mitochondrial calcium levels [62, 63], and caspase activation [16, 52]. Such mitochondrial dysfunction might result from HIV-1 Tat-induced disruption of calcium homeostasis [6, 7, 16, 53–59], because elevated intracellular calcium levels leads to mitochondrial damage [60].
We found that HIV-1 Tat1-72 opened mitochondrial permeability transition pores in cultured cortical neurons. Opening of the mitochondrial permeability transition pore results in rapid decreases in proton gradients across the inner mitochondrial membrane, and this may explain our observations that HIV-1 Tat1-72 decreases mitochondrial membrane potential, decreases cellular levels of ATP, and increases production of reactive oxygen species; all of which may result in neuronal injury through the release of pro-apoptotic factors such as cytochrome c, apoptosis inducing factor, and caspases [61]. However, others have shown that Tat can lead to mitochondrial hyperpolarization [17, 62, 63]. Although it is not clear what leads to such discrepancy, clearly there were many differences between how the studies were conducted; rat instead of mouse neurons were used, anti-oxidant free media and lower (typically 8 nM) concentrations of HIV-1 Tat were used, transient responses in minutes following Tat treatment rather than hours were measured, and Neurobasal or Leibovitz’s L15 media instead of Locke’s buffer was used. Nevertheless, similar to our findings, HIV-1 Tat was reported to cause mitochondrial hypopolarization [43, 64–66] and the loss of mitochondrial membrane potential [67]. In the present study, HIV-1 Tat decreased significantly levels of intracellular ATP. These decreases may have been due to hypopolarization of mitochondrial membrane potentials, loss of proton gradients across mitochondrial membranes, and reduced ATP synthase activity. However, the inability of HIV-1 Tat to induce changes in either the adenylate energy charge or ATP/ADP ratios suggests that the effects of HIV-1 Tat on cellular bioenergetics might be rather mild.
A promising agent that protects mitochondria function and exerts neuroprotection is creatine, a readily available dietary supplement that is present at high levels in brain [24]. Mechanistically, the neuroprotective properties of creatine appear to be related mostly to stabilizing mitochondrial bioenergetics and preventing mitochondrial redox catastrophe. In terms of stabilizing mitochondrial bioenergetics, creatine can be phosphorylated to yield phosphocreatine and the production of ATP. For the prevention of redox catastrophe, creatine can reduce the production of reactive oxygen species by acting as a free-radical scavenger [46] and preventing the opening of mitochondrial permeability transition pores [47, 68, 69].
Given robust protective effects of creatine on mitochondria, the present study tested our hypothesis that creatine protects against HIV-1 Tat induced neuronal injury. We demonstrated that creatine, at pharmacologically relevant concentrations increased levels of creatine in neurons and protected against HIV-1 Tat-induced synaptic dysfunction and neuronal cell death. Neurons have limited ability to synthesize creatine endogenously and most intraneuronal creatine results from extra-neuronal uptake [70]. This might help explain our findings that exogenously added creatine protected against HIV-1 Tat-induced decreases in ATP levels as well as HIV-1 Tat-induced mitochondrial bioenergetic crisis and redox catastrophe. This might be of relevance to the HIV-1 infected population because creatine levels are decreased in brain of HIV-1 positive individuals [71, 72]. Although we did not observe statistically significant changes in levels of phosphocreatine itself we did observe statistically significant changes in phosphocreatine: creatine ratios most likely because of the observed increases in intraneuronal creatine. These findings suggest that the creatine/phosphocreatine system may have played a role in buffering HIV-1 Tat-induced changes in high-energy phosphates.
Although creatine can act as a direct antioxidant by quenching superoxide anions, hydrogen peroxide and peroxynitrite [46], we did not observe any protective effects of creatine against HIV-1 Tat-induced increases in reactive oxygen species. Indeed, we observed statistically significant increases in reactive oxygen species with HIV-1 Tat and the deletion mutant of HIV-1 Tat. However, we caution against the interpretation that oxidative stress in not involved in HIV-1 Tat-induced neuronal injury or against the ability of creatine to act as an antioxidant because we studied this only at a single 4 h post-treatment time point.
Taken together, we demonstrated that creatine protected against HIV-1 Tat-induced neuronal injury. The mechanisms underlying creatine’s neuroprotective effects appeared to be through stabilization of mitochondrial membrane potential and keeping closed permeability transition pores. In this manner, creatine would prevent redox catastrophe and the resulting injury. However, the very tight and “U”-shaped dose-response relationship observed here and by others studying the neuroprotective effects of creatine in model systems of neurodegenerative diseases [26, 27, 29] highlights the therapeutic challenge of possibly using creatine to prevent or lessen the severity of HAND.
Acknowledgments
Supported by P30GM103329, R21AG043338, and R01MH100972.
We would like to think Dr. Avindra Nath for providing us HIV-1 Tat and mutant Tat.
Footnotes
Conflict of Interest: The authors declare no conflict of interest
References
- 1.Saxena SK, Tiwari S, Nair MP. A global perspective on HIV/AIDS. Science. 2012;337:798. doi: 10.1126/science.337.6096.798. [DOI] [PubMed] [Google Scholar]
- 2.Heaton RK, Clifford DB, Franklin DR, Jr, et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology. 2010;75:2087–2096. doi: 10.1212/WNL.0b013e318200d727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan P, Brew BJ. HIV associated neurocognitive disorders in the modern antiviral treatment era: prevalence, characteristics, biomarkers, and effects of treatment. Curr HIV/AIDS Rep. 2014;11:317–324. doi: 10.1007/s11904-014-0221-0. [DOI] [PubMed] [Google Scholar]
- 4.van de Bovenkamp M, Nottet HS, Pereira CF. Interactions of human immunodeficiency virus-1 proteins with neurons: possible role in the development of human immunodeficiency virus-1-associated dementia. Eur J Clin Invest. 2002;32:619–627. doi: 10.1046/j.1365-2362.2002.01029.x. [DOI] [PubMed] [Google Scholar]
- 5.Nath A, Geiger J. Neurobiological aspects of human immunodeficiency virus infection: neurotoxic mechanisms. Prog Neurobiol. 1998;54:19–33. doi: 10.1016/s0301-0082(97)00053-1. [DOI] [PubMed] [Google Scholar]
- 6.Haughey NJ, Holden CP, Nath A, et al. Involvement of inositol 1,4,5-trisphosphate-regulated stores of intracellular calcium in calcium dysregulation and neuron cell death caused by HIV-1 protein tat. J Neurochem. 1999;73:1363–1374. doi: 10.1046/j.1471-4159.1999.0731363.x. [DOI] [PubMed] [Google Scholar]
- 7.Nath A, Haughey NJ, Jones M, et al. Synergistic neurotoxicity by human immunodeficiency virus proteins Tat and gp120: protection by memantine. Ann Neurol. 2000;47:186–194. [PubMed] [Google Scholar]
- 8.Buscemi L, Ramonet D, Geiger JD. Human immunodeficiency virus type-1 protein Tat induces tumor necrosis factor-alpha-mediated neurotoxicity. Neurobiol Dis. 2007;26:661–670. doi: 10.1016/j.nbd.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sabatier JM, Vives E, Mabrouk K, et al. Evidence for neurotoxic activity of tat from human immunodeficiency virus type 1. J Virol. 1991;65:961–967. doi: 10.1128/jvi.65.2.961-967.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weeks BS, Lieberman DM, Johnson B, et al. Neurotoxicity of the human immunodeficiency virus type 1 tat transactivator to PC12 cells requires the Tat amino acid 49-58 basic domain. J Neurosci Res. 1995;42:34–40. doi: 10.1002/jnr.490420105. [DOI] [PubMed] [Google Scholar]
- 11.Perez A, Probert AW, Wang KK, et al. Evaluation of HIV-1 Tat induced neurotoxicity in rat cortical cell culture. J Neurovirol. 2001;7:1–10. doi: 10.1080/135502801300069575. [DOI] [PubMed] [Google Scholar]
- 12.King JE, Eugenin EA, Buckner CM, et al. HIV tat and neurotoxicity. Microbes Infect. 2006;8:1347–1357. doi: 10.1016/j.micinf.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 13.Agrawal L, Louboutin JP, Reyes BA, et al. HIV-1 Tat neurotoxicity: A model of acute and chronic exposure, and neuroprotection by gene delivery of antioxidant enzymes. Neurobiol Dis. 2011 doi: 10.1016/j.nbd.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 14.Johnson TP, Patel K, Johnson KR, et al. Induction of IL-17 and nonclassical T-cell activation by HIV-Tat protein. Proc Natl Acad Sci U S A. 2013;110:13588–13593. doi: 10.1073/pnas.1308673110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haughey NJ, Mattson MP. Calcium dysregulation and neuronal apoptosis by the HIV-1 proteins Tat and gp120. J Acquir Immune Defic Syndr. 2002;31(Suppl 2):S55–61. doi: 10.1097/00126334-200210012-00005. [DOI] [PubMed] [Google Scholar]
- 16.Kruman II, Nath A, Mattson MP. HIV-1 protein Tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Exp Neurol. 1998;154:276–288. doi: 10.1006/exnr.1998.6958. [DOI] [PubMed] [Google Scholar]
- 17.Perry SW, Norman JP, Litzburg A, et al. HIV-1 transactivator of transcription protein induces mitochondrial hyperpolarization and synaptic stress leading to apoptosis. J Immunol. 2005;174:4333–4344. doi: 10.4049/jimmunol.174.7.4333. [DOI] [PubMed] [Google Scholar]
- 18.Price TO, Ercal N, Nakaoke R, et al. HIV-1 viral proteins gp120 and Tat induce oxidative stress in brain endothelial cells. Brain Res. 2005;1045:57–63. doi: 10.1016/j.brainres.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 19.Rappaport J, Joseph J, Croul S, et al. Molecular pathway involved in HIV-1-induced CNS pathology: role of viral regulatory protein, Tat. J Leukoc Biol. 1999;65:458–465. doi: 10.1002/jlb.65.4.458. [DOI] [PubMed] [Google Scholar]
- 20.Zou W, Kim BO, Zhou BY, et al. Protection against human immunodeficiency virus type 1 Tat neurotoxicity by Ginkgo biloba extract EGb 761 involving glial fibrillary acidic protein. Am J Pathol. 2007;171:1923–1935. doi: 10.2353/ajpath.2007.070333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim HJ, Martemyanov KA, Thayer SA. Human immunodeficiency virus protein Tat induces synapse loss via a reversible process that is distinct from cell death. J Neurosci. 2008;28:12604–12613. doi: 10.1523/JNEUROSCI.2958-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fitting S, Xu R, Bull C, et al. Interactive comorbidity between opioid drug abuse and HIV-1 Tat: chronic exposure augments spine loss and sublethal dendritic pathology in striatal neurons. Am J Pathol. 2010;177:1397–1410. doi: 10.2353/ajpath.2010.090945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hui L, Chen X, Bhatt D, et al. Ketone bodies protection against HIV-1 Tat-induced neurotoxicity. J Neurochem. 2012 doi: 10.1111/j.1471-4159.2012.07764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mujika I, Padilla S. Creatine supplementation as an ergogenic aid for sports performance in highly trained athletes: a critical review. Int J Sports Med. 1997;18:491–496. doi: 10.1055/s-2007-972670. [DOI] [PubMed] [Google Scholar]
- 25.Klivenyi P, Gardian G, Calingasan NY, et al. Additive neuroprotective effects of creatine and a cyclooxygenase 2 inhibitor against dopamine depletion in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson’s disease. J Mol Neurosci. 2003;21:191–198. doi: 10.1385/jmn:21:3:191. [DOI] [PubMed] [Google Scholar]
- 26.Seidl SE, Potashkin JA. The promise of neuroprotective agents in Parkinson’s disease. Front Neurol. 2011;2:68. doi: 10.3389/fneur.2011.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matthews RT, Yang L, Jenkins BG, et al. Neuroprotective effects of creatine and cyclocreatine in animal models of Huntington’s disease. J Neurosci. 1998;18:156–163. doi: 10.1523/JNEUROSCI.18-01-00156.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burklen TS, Schlattner U, Homayouni R, et al. The Creatine Kinase/Creatine Connection to Alzheimer’s Disease: CK-Inactivation, APP-CK Complexes and Focal Creatine Deposits. J Biomed Biotechnol. 2006;2006:35936. doi: 10.1155/JBB/2006/35936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosenfeld J, King RM, Jackson CE, et al. Creatine monohydrate in ALS: effects on strength, fatigue, respiratory status and ALSFRS. Amyotroph Lateral Scler. 2008;9:266–272. doi: 10.1080/17482960802028890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma M, Nath A. Molecular determinants for cellular uptake of Tat protein of human immunodeficiency virus type 1 in brain cells. J Virol. 1997;71:2495–2499. doi: 10.1128/jvi.71.3.2495-2499.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prendergast MA, Rogers DT, Mulholland PJ, et al. Neurotoxic effects of the human immunodeficiency virus type-1 transcription factor Tat require function of a polyamine sensitive-site on the N-methyl-D-aspartate receptor. Brain Res. 2002;954:300–307. doi: 10.1016/s0006-8993(02)03360-7. [DOI] [PubMed] [Google Scholar]
- 32.Nath A, Psooy K, Martin C, et al. Identification of a human immunodeficiency virus type 1 Tat epitope that is neuroexcitatory and neurotoxic. J Virol. 1996;70:1475–1480. doi: 10.1128/jvi.70.3.1475-1480.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bough KJ, Wetherington J, Hassel B, et al. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann Neurol. 2006;60:223–235. doi: 10.1002/ana.20899. [DOI] [PubMed] [Google Scholar]
- 34.Hui L, Chen X, Bhatt D, et al. Ketone bodies protection against HIV-1 Tat-induced neurotoxicity. J Neurochem. 2012;122:382–391. doi: 10.1111/j.1471-4159.2012.07764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mattson MP, Barger SW, Begley JG, et al. Calcium, free radicals, and excitotoxic neuronal death in primary cell culture. Methods Cell Biol. 1995;46:187–216. doi: 10.1016/s0091-679x(08)61930-5. [DOI] [PubMed] [Google Scholar]
- 36.Wang X, Mori T, Sumii T, et al. Hemoglobin-induced cytotoxicity in rat cerebral cortical neurons: caspase activation and oxidative stress. Stroke. 2002;33:1882–1888. doi: 10.1161/01.str.0000020121.41527.5d. [DOI] [PubMed] [Google Scholar]
- 37.Petronilli V, Miotto G, Canton M, et al. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys J. 1999;76:725–734. doi: 10.1016/S0006-3495(99)77239-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rama Rao KV, Jayakumar AR, Norenberg MD. Role of oxidative stress in the ammonia-induced mitochondrial permeability transition in cultured astrocytes. Neurochem Int. 2005;47:31–38. doi: 10.1016/j.neuint.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 39.Aksenov MY, Aksenova MV, Nath A, et al. Cocaine-mediated enhancement of Tat toxicity in rat hippocampal cell cultures: the role of oxidative stress and D1 dopamine receptor. Neurotoxicology. 2006;27:217–228. doi: 10.1016/j.neuro.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 40.Gavriil ES, Cooney R, Weeks BS. Tat mediates apoptosis in vivo in the rat central nervous system. Biochem Biophys Res Commun. 2000;267:252–256. doi: 10.1006/bbrc.1999.1894. [DOI] [PubMed] [Google Scholar]
- 41.New DR, Maggirwar SB, Epstein LG, et al. HIV-1 Tat induces neuronal death via tumor necrosis factor-alpha and activation of non-N-methyl-D-aspartate receptors by a NFkappaB-independent mechanism. J Biol Chem. 1998;273:17852–17858. doi: 10.1074/jbc.273.28.17852. [DOI] [PubMed] [Google Scholar]
- 42.Shi B, Raina J, Lorenzo A, et al. Neuronal apoptosis induced by HIV-1 Tat protein and TNF-alpha: potentiation of neurotoxicity mediated by oxidative stress and implications for HIV-1 dementia. J Neurovirol. 1998;4:281–290. doi: 10.3109/13550289809114529. [DOI] [PubMed] [Google Scholar]
- 43.Langford D, Grigorian A, Hurford R, et al. The role of mitochondrial alterations in the combined toxic effects of human immunodeficiency virus Tat protein and methamphetamine on calbindin positive-neurons. J Neurovirol. 2004;10:327–337. doi: 10.1080/13550280490520961. [DOI] [PubMed] [Google Scholar]
- 44.Raidel SM, Haase C, Jansen NR, et al. Targeted myocardial transgenic expression of HIV Tat causes cardiomyopathy and mitochondrial damage. Am J Physiol Heart Circ Physiol. 2002;282:H1672–1678. doi: 10.1152/ajpheart.00955.2001. [DOI] [PubMed] [Google Scholar]
- 45.Darbinian N, Khalili K, Amini S. Neuroprotective activity of pDING in response to HIV-1 Tat. J Cell Physiol. 2014;229:153–161. doi: 10.1002/jcp.24392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lawler JM, Barnes WS, Wu G, et al. Direct antioxidant properties of creatine. Biochem Biophys Res Commun. 2002;290:47–52. doi: 10.1006/bbrc.2001.6164. [DOI] [PubMed] [Google Scholar]
- 47.O’Gorman E, Beutner G, Dolder M, et al. The role of creatine kinase in inhibition of mitochondrial permeability transition. FEBS Lett. 1997;414:253–257. doi: 10.1016/s0014-5793(97)01045-4. [DOI] [PubMed] [Google Scholar]
- 48.Sabatier JM, Vives E, Mabrouk K, et al. Evidence for neurotoxic activity of tat from human immunodeficiency virus type 1. J Virol. 1991;65:961–967. doi: 10.1128/jvi.65.2.961-967.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Banks WA, Robinson SM, Nath A. Permeability of the blood-brain barrier to HIV-1 Tat. Exp Neurol. 2005;193:218–227. doi: 10.1016/j.expneurol.2004.11.019. [DOI] [PubMed] [Google Scholar]
- 50.Cheng J, Nath A, Knudsen B, et al. Neuronal excitatory properties of human immunodeficiency virus type 1 Tat protein. Neuroscience. 1998;82:97–106. doi: 10.1016/s0306-4522(97)00174-7. [DOI] [PubMed] [Google Scholar]
- 51.Wallace DR, Dodson SL, Nath A, et al. Delta opioid agonists attenuate TAT(1-72)-induced oxidative stress in SK-N-SH cells. Neurotoxicology. 2006;27:101–107. doi: 10.1016/j.neuro.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 52.Singh IN, El-Hage N, Campbell ME, et al. Differential involvement of p38 and JNK MAP kinases in HIV-1 Tat and gp120-induced apoptosis and neurite degeneration in striatal neurons. Neuroscience. 2005;135:781–790. doi: 10.1016/j.neuroscience.2005.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bonavia R, Bajetto A, Barbero S, et al. HIV-1 Tat causes apoptotic death and calcium homeostasis alterations in rat neurons. Biochem Biophys Res Commun. 2001;288:301–308. doi: 10.1006/bbrc.2001.5743. [DOI] [PubMed] [Google Scholar]
- 54.Eugenin EA, D’Aversa TG, Lopez L, et al. MCP-1 (CCL2) protects human neurons and astrocytes from NMDA or HIV-tat-induced apoptosis. J Neurochem. 2003;85:1299–1311. doi: 10.1046/j.1471-4159.2003.01775.x. [DOI] [PubMed] [Google Scholar]
- 55.Haughey NJ, Nath A, Mattson MP, et al. HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J Neurochem. 2001;78:457–467. doi: 10.1046/j.1471-4159.2001.00396.x. [DOI] [PubMed] [Google Scholar]
- 56.Self RL, Mulholland PJ, Nath A, et al. The human immunodeficiency virus type-1 transcription factor Tat produces elevations in intracellular Ca2+ that require function of an N-methyl-D-aspartate receptor polyamine-sensitive site. Brain Res. 2004;995:39–45. doi: 10.1016/j.brainres.2003.09.052. [DOI] [PubMed] [Google Scholar]
- 57.Caporello E, Nath A, Slevin J, et al. The immunophilin ligand GPI1046 protects neurons from the lethal effects of the HIV-1 proteins gp120 and Tat by modulating endoplasmic reticulum calcium load. J Neurochem. 2006;98:146–155. doi: 10.1111/j.1471-4159.2006.03863.x. [DOI] [PubMed] [Google Scholar]
- 58.Zhu X, Yao H, Peng F, et al. PDGF-mediated protection of SH-SY5Y cells against Tat toxin involves regulation of extracellular glutamate and intracellular calcium. Toxicol Appl Pharmacol. 2009;240:286–291. doi: 10.1016/j.taap.2009.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wallace DR. HIV neurotoxicity: potential therapeutic interventions. J Biomed Biotechnol. 2006;2006:65741. doi: 10.1155/JBB/2006/65741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dong Z, Saikumar P, Weinberg JM, et al. Calcium in cell injury and death. Annu Rev Pathol. 2006;1:405–434. doi: 10.1146/annurev.pathol.1.110304.100218. [DOI] [PubMed] [Google Scholar]
- 61.Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]
- 62.Norman JP, Perry SW, Kasischke KA, et al. HIV-1 trans activator of transcription protein elicits mitochondrial hyperpolarization and respiratory deficit, with dysregulation of complex IV and nicotinamide adenine dinucleotide homeostasis in cortical neurons. J Immunol. 2007;178:869–876. doi: 10.4049/jimmunol.178.2.869. [DOI] [PubMed] [Google Scholar]
- 63.Norman JP, Perry SW, Reynolds HM, et al. HIV-1 Tat activates neuronal ryanodine receptors with rapid induction of the unfolded protein response and mitochondrial hyperpolarization. PLoS One. 2008;3:e3731. doi: 10.1371/journal.pone.0003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maragos WF, Young KL, Turchan JT, et al. Human immunodeficiency virus-1 Tat protein and methamphetamine interact synergistically to impair striatal dopaminergic function. J Neurochem. 2002;83:955–963. doi: 10.1046/j.1471-4159.2002.01212.x. [DOI] [PubMed] [Google Scholar]
- 65.Rumbaugh JA, Li G, Rothstein J, et al. Ceftriaxone protects against the neurotoxicity of human immunodeficiency virus proteins. J Neurovirol. 2007;13:168–172. doi: 10.1080/13550280601178218. [DOI] [PubMed] [Google Scholar]
- 66.Rumbaugh J, Turchan-Cholewo J, Galey D, et al. Interaction of HIV Tat and matrix metalloproteinase in HIV neuropathogenesis: a new host defense mechanism. FASEB J. 2006;20:1736–1738. doi: 10.1096/fj.05-5619fje. [DOI] [PubMed] [Google Scholar]
- 67.Ferri KF, Jacotot E, Blanco J, et al. Mitochondrial control of cell death induced by HIV-1-encoded proteins. Ann N Y Acad Sci. 2000;926:149–164. doi: 10.1111/j.1749-6632.2000.tb05609.x. [DOI] [PubMed] [Google Scholar]
- 68.Dolder M, Walzel B, Speer O, et al. Inhibition of the mitochondrial permeability transition by creatine kinase substrates. Requirement for microcompartmentation. J Biol Chem. 2003;278:17760–17766. doi: 10.1074/jbc.M208705200. [DOI] [PubMed] [Google Scholar]
- 69.Sullivan PG, Geiger JD, Mattson MP, et al. Dietary supplement creatine protects against traumatic brain injury. Ann Neurol. 2000;48:723–729. [PubMed] [Google Scholar]
- 70.Carducci C, Carducci C, Santagata S, et al. In vitro study of uptake and synthesis of creatine and its precursors by cerebellar granule cells and astrocytes suggests some hypotheses on the physiopathology of the inherited disorders of creatine metabolism. Neurosci. 2012;26:13–41. doi: 10.1186/1471-2202-13-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chang L, Eernst T, Witt MD, et al. Relationships among brain metabolites, cognitive function, and viral loads in antiretroviral-naïve HIV patients. Neuroimage. 2002;17:1638–1648. doi: 10.1006/nimg.2002.1254. [DOI] [PubMed] [Google Scholar]
- 72.Ernst T, Itti E, Itti L, Chang L. Changes in cerebral metabolism are detected prior to perfusion changes in early HIV-CMC: A coregistered (1)H MRS and SPECT study. J Magn Reson Imaging. 2000;12:859–865. doi: 10.1002/1522-2586(200012)12:6<859::aid-jmri8>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]