

Abstract
Cancer is a leading cause of death worldwide; due to the lack of ideal cancer biomarkers for early detection or diagnosis, most patients present with late-stage disease at the time of diagnosis, thus limiting the potential for successful treatment. Traditional cancer treatments, including surgery, chemotherapy and radiation therapy, have demonstrated very limited efficacy for patients with late-stage disease. Therefore, innovative and effective cancer treatments are urgently needed for cancer patients with late-stage and refractory disease. Cancer immunotherapy, particularly adoptive cell transfer, has shown great promise in the treatment of patients with late-stage disease, including those who are refractory to standard therapies. In this review, we will highlight recent advances and discuss future directions in adoptive cell transfer based cancer immunotherapy.
Keywords: adoptive cell transfer, cancer antigen-specific T cells, cancer immunotherapy, genetically modified T cells
Background
Cancer is a leading cause of death worldwide, and the number of cases globally continues to increase [1]. According to the World Cancer Report 2014, the global burden of cancer rose to an estimated 14 million new cancer cases in 2012, and this figure is expected to rise to 22 million annually within the next two decades. Over the same period, cancer deaths are predicted to rise from an estimated 8.2 million to 13 million per year. Most cancers can be prevented or cured if detected at an early stage and treated promptly. Unfortunately, due to the lack of ideal cancer bio-markers for early detection and diagnosis [2], most patients present with advanced-stage disease at the time of diagnosis, thus limiting the potential for successful treatment.
Traditional cancer treatments, including surgery, chemotherapy and radiation therapy, have demonstrated very limited efficacy for patients with late-stage disease. In addition, chemotherapy and radiotherapy often cause considerable side effects. Therefore, innovative and effective cancer treatments are urgently needed for cancer patients with late-stage and refractory disease. Cancer immunotherapy has emerged as a promising approach for cancer treatment [3–6]. Recent US FDA approval of immunotherapy-based vaccines/drugs sipuleucel-T (Provenge®) [7] and ipilimumab (Yervoy®) [8] represent milestones in the field of cancer immunotherapy for advanced prostate cancer and metastatic melanoma, respectively [5]. Furthermore, a Phase III clinical trial of gp100 peptide vaccine in patients with advanced melanoma has shown encouraging results [9]. Thus, cancer immunotherapy has become an important part of treating cancer patients with advanced or refractory disease. Furthermore, immunotherapy may be effective in preventing tumor recurrence following surgery. Cancer immunotherapy holds several key advantages over traditional therapies: high specificity, little or no side effects for active immunization, although adverse effects may occur in adoptive cell transfer (ACT) and good safety profile. The key point of immunotherapy is to use the patient’s own immune system to control and destroy cancer cells. Cancer immunotherapy approaches include active immunization, reversal of immunosuppression, nonspecific immune stimulation and ACT. To date, ACT has been demonstrated to be the most effective immunotherapy method for cancer treatment and has achieved very promising results in cancer clinical trials [10–14]. The first exciting clinical trial of ACT used tumor-infiltrating lymphocytes (TILs) [15]. In the TIL-based ACT approach, TILs are isolated from the tumor tissues of cancer patients, expanded in vitro using a high concentration of IL-2 (6000 U/ml), and then infused back into the patient. The feasibility of the TIL-based ACT approach was first demonstrated in melanoma [15], with a current objective response rate of 49–72% when lymphodepleting preparative regimen is performed prior to TIL infusion [4,16]. Successful TIL-based immunotherapy has promoted the rapid development of ACT. In addition to TIL-based immunotherapy, genetically modified cancer-specific T cells, such as T-cell receptor (TCR)- and chimeric antigen receptor (CAR)-transduced T cells, are being developed to augment ACT-mediated immunotherapeutic responses against various types of cancer and have already shown encouraging therapeutic effects in clinical trials [10–14].
The promising results achieved with the use of genetically modified T cells to target cancer earned cancer immunotherapy being named as the Science ‘Breakthrough of the Year’ in 2013 [17]. For the first time in many years, many pharmaceutical industries are investing heavily to facilitate the development of effective genetically modified T cells to treat various cancer types. For example, the pharmaceutical giant Novartis teamed with the University of Pennsylvania in 2012 and invested $100 million to develop CAR-transduced T cells. More recently, a new biotechnology company, Juno Therapeutics Inc., has just been launched in December 2013 with an initial investment of $145 million to develop TCR- and CAR-transduced T cells. In this review, we will highlight recent advances in ACT-based cancer immunotherapy and will also briefly discuss future directions in ACT-based cancer immunotherapy.
Cancer immunotherapy
The innate and acquired immune systems play a critical role in immune surveillance and immune defense [18,19]. Therefore, the use of the immune system to eliminate cancer is a very promising approach for cancer treatment [20,21]. Indeed, immunotherapy has demonstrated great potential for cancer treatment [3–6], especially for disease refractory to traditional treatments, including surgery, chemotherapy and radiotherapy. Cancer immunotherapy approaches include active immunization, nonspecific immune stimulation and ACT. Among these strategies, ACT has achieved more exciting results in cancer clinical trials and therefore, holds the most promise for the treatment of malignant diseases [10–14]. The success of cancer immunotherapy relies largely on the identification of suitable cancer antigens for the generation of effective cancer vaccines and antigen-specific T cells. Since the first human cancer antigen MAGEA1 was identified in 1991 using in vitro expanded cancer-specific T cells from melanoma [22], a growing number of cancer antigens have been identified in different tumor types. To date, 403 cancer antigenic peptides have been included in the peptide database [23,24]. Our group has been working on cancer antigen discovery for many years and has identified many cancer antigens including TRP1, TRP2, NY-ESO-1, EBNA-1, PSGR and SATB1 [25–34]. We have also developed a novel genetic approach to identify cancer antigens recognized by CD4+ T cells [35–38], which are also believed to play an important role in antitumor immunity.
Cancer immunotherapy requires the activation and expansion of cancer-specific T cells, which kill cancer cells by recognizing antigen targets expressed on cancer cells. Over the past 20 years, studies have shown that the generation of cancer-specific immunity requires three steps (Figure 1). First, antigen-presenting cells (e.g., dendritic cells [DCs]) capture and process cancer antigens into antigenic peptides, which are presented in combination with human leukocyte antigen (HLA) molecules for recognition by TCR of T cells (signal 1) [39]. Second, T-cell activation requires the binding of the costimulatory surface molecules B7 and CD28 on antigen-presenting cells and T cells, respectively (signal 2). To achieve optimal T-cell activation, both signals 1 and 2 are required. Conversely, antigenic peptide stimulation (signal 1) in the absence of costimulation (signal 2) cannot induce full T-cell activation, thus resulting in T-cell tolerance. In addition to costimulatory molecules, there are also inhibitory molecules, such as CTLA-4 and PD-1, which induce signals to prevent T-cell activation [40]. Third, activated cancer-specific T cells arrive at tumor sites and recognize tumor antigens expressed by cancer cells, thereby killing the cancer cells. Although antigenic peptide stimulation initiates T-cell activation, the degree of T-cell activation is further determined by the balance between costimulation and cosuppression. Recent clinical trials have demonstrated that blockade of PD-1 coinhibition with anti-PD-1 or anti-PD-L1 therapy enhances T-cell-mediated anticancer responses without severe adverse events (SAE) [41,42]. Also, inhibition of CTLA-4 signaling has been shown to significantly improve the survival of patients with late-stage melanoma [8,43], leading to the FDA approval of the anti-CTLA-4 antibody ipilimumab (Yervoy) for metastatic melanoma in 2011. Besides T-cell-intrinsic regulation, T-cell activation can also be regulated by external factors (extrinsic). For example, cytokines, such as IL-2, released by CD4+ helper T cells (Th1 and Th17) can directly promote cancer-specific T cell expansion, while IL-2 may also mediate expansion of CD4+ Treg, which antagonize function of cancer-specific T cells [44]. On the other hand, myeloid-derived suppressor cells (MDSCs) and Treg cells, which are normally present in the tumor microenvironment, inhibit cancer-specific T cell function and induce immunosuppression [45,46], leading to poor immunotherapy efficacy. Therefore, overcoming Treg/MDSC-mediated immunosuppression in the tumor microenvironment might be critical for successful cancer immunotherapy. Anti-CD25 antibodies (Ontak and Daclizumab) and cyclophosphamide have been used to eliminate CD4+CD25+ Treg cells. However, anti-CD25 antibodies and cyclophosphamide are not specific for Treg cell depletion, because CD25 is also expressed on effector T cells. In an effort to overcome immune suppression and potential problems associated with current depletion strategies, we recently demonstrated that stimulation of human Toll-like receptor 8 with its ligand, Poly-G3 oligonucleotide, resulted in the functional reversal of Treg cells [45,47,48]. Our group has recently generated human Toll-like receptor 8 transgenic mice to determine whether blockade of Treg cell-mediated immune suppression by Poly-G3 can enhance antitumor immunity in vivo.
Figure 1.
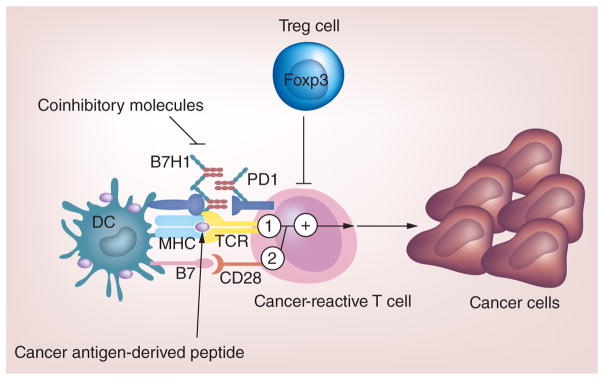
The key points in cancer immunotherapy are ‘one activation and two inhibitions’ where cancer-reactive T-cell activation by cancer antigen represents the one activation and blockade of coinhibitory molecules on T cells and reversal of Treg cell-mediated immunosuppression represent the two inhibitions.
DC: Dendritic cell.
Sipuleucel-T (Provenge) and ipilimumab (Yervoy) have been approved by the FDA for treatment of prostate cancer and metastatic melanoma, respectively [7,8], and a Phase III clinical trial of the gp100 peptide-based vaccine in patients with melanoma also produced encouraging results [9]. However, the clinical benefits reported for these agents have fallen far short of complete responses and permanent cures, although the clinical data for ipilimumab are still emerging [49]. In contrast, ACT therapy using cancer antigen-specific T cells including TILs, peptide-induced T cells and engineered T cells (TCR and CAR) has demonstrated dramatic potency in cancer treatment [10–14,50], leading to complete and durable responses in some patients with late-stage and refractory disease. Here, we will present a summary of the most exciting clinical results obtained with ACT summarized in Table 1 and also discuss future directions of ACT-based cancer immunotherapy.
Table 1.
Recent clinical trials using T-cell-based immunotherapy.
| Malignancy | Patient number | Type of T cells infused | Target antigen | Clinical outcomes | Ref. |
|---|---|---|---|---|---|
| Metastatic melanoma | 20 | TIL | Unknown | 1 CR, 10 PR, 1 MR | [15] |
| Metastatic melanoma | 13 | TIL | Unknown | 6 PR, 4 MR | [51] |
| Metastatic melanoma | 93 | TIL | Unknown | 22 CR, 34 PR | [16] |
| Metastatic melanoma | 10 | CD8+ T cells | MART1/MelanA or gp 100 | 5 SD, 1 MR, 2 minor response | [52] |
| Metastatic melanoma | 11 | CD8+ T cells | MelanA | 1 CR, 1 PR, 1 MR, 1 SD | [53] |
| Metastatic melanoma | 1 | CD4+ T cells | NY-ESO-1 | 1 CR | [54] |
| Metastatic melanoma | 14 | CD8+ T cells | MelanA | 2 CR, 4 PR, 1 SD | [55] |
| Metastatic melanoma | 11 | CD8+ T cells | MART1/Tyr/gp 100 | 1 CR, 5 SD | [56] |
| Metastatic melanoma | 17 | TCR T cells | MART1 | 2 PR, 1 MR | [57] |
| Metastatic melanoma | 36 | High affinity TCR T cells | MART1 or gp 100 | 1 CR, 8 PR | [58] |
| Metastatic melanoma | 3 | TCR T cells | CEA | 1 PR | [59] |
| Metastatic melanoma/ synovial sarcoma | 17 | TCR T cells | NY-ESO-1 | 2 CR, 7 PR | [60] |
| Metastatic melanoma | 9 | High affinity TCR T cells | MAGEA3/MAGEA12 | 1 CR, 4 PR | [61] |
| Metastatic melanoma/ myeloma | 2 | High affinity TCR T cells | MAGEA3/Titin | Not evaluable | [62,63] |
| Neuroblastoma | 19 | CAR T cells | GD2 | 3 CR | [64] |
| Renal carcinoma | 12 | CAR T cells | CAIX | Not evaluable | [65] |
| Colon cancer | 1 | CAR T cells (CD28-4-1BB-CD3ζ) | ERBB2 | Not evaluable | [66] |
| Lymphoma | 1 | CAR T cells (CD28-CD3ζ) | CD19 | 1 PR | [67] |
| Lymphoma and CLL | 8 | CAR T cells (CD28-CD3ζ) | CD19 | 1 CR, 5 PR, 1 SD | [68] |
| CLL | 3 | CAR T cells (4-1BB-CD3ζ) | CD19 | 2 CR, 1 PR | [69,70] |
| ALL | 2 | CAR T cells (4-1BB-CD3ζ) | CD19 | 2 CR | [71] |
| ALL | 5 | CAR T cells (CD28-CD3ζ) | CD19 | 5 CR | [72] |
| ALL | 16 | CAR T cells (CD28-CD3ζ) | CD19 | 14 CR | [73] |
ALL: Acute lymphoblastic leukemia; CAR: Chimeric antigen receptor; CEA: Carcinoembryonic antigen; CLL: Chronic lymphocytic leukemia; CR: Complete response; MR: Mixed response; PR: Partial response; SD: Stable disease; TCR: T-cell receptor; TIL: Tumor-infiltrating lymphocyte.
A brief history of ACT for cancer treatment
As early as in the 1980s, lymphokine-activated killer (LAK) cells were used to treat tumors in mice and humans [74]. Subsequently, in the 1990s, cytokine-induced killer (CIK) cells amplified from peripheral blood mononuclear cells (PBMCs) were evaluated for cancer therapy [75]. However, due to the lack of specificity, LAK and CIK-based therapy exhibited only limited efficacy [76,77]. With advances in immunology and new technologies, cancer antigen-specific T-cell-based therapy has shown great promise in treating cancer patients. The first cancer antigen-specific T-cell-based therapy was the use of TILs for the treatment of melanoma in 1988 [15,78]. In a recent series of three consecutive clinical trials, lymphodepletion prior to autologous TIL infusion achieved objective anticancer responses ranging from 49% to 72% [16]. Lymphodepletion may deplete Treg cells and other suppressive cells in the circulation and the tumor micro environment, thus enabling the survival and amplification of adoptively transferred TILs to achieve effective killing of cancer cells. TIL-based therapy can result in long-term remission (>5 years) and low recurrence rate [16]. Despite the clinical benefits of TIL-based therapy, there are limitations to its successful implementation. TIL-based therapy is an individualized treatment that requires surgical removal of tumor tissues for TIL cultivation. Furthermore, few medical centers worldwide provide TIL-based therapy as it requires a highly skilled medical staff to isolate and cultivate TILs. To overcome these barriers, unmodified peptide-stimulated T cells and genetically engineered T cells (e.g., TCR- and CAR-transduced T cells) that specifically recognize antigen targets expressed on cancer cells have been employed in clinical trials and have achieved promising and exciting results [10–14,50].
TILs
Isolation and expansion of TILs for adoptive therapy was pioneered by Rosenberg et al. at the Surgery Branch, NIH [15]. Studies in mouse models showed that cancer-reactive lymphocytes are concentrated in TILs and can mediate efficient cancer regression [79]. Recently, the mining of exomic sequencing data has shown that TILs can recognize epitopes of mutated melanoma antigens [80,81], further demonstrating that tumor tissues are a rich source of cancer-reactive T cells for ACT-based therapy. For TIL preparation, cancer tissues are cut into pieces, digested and then cultured in T cell medium containing 6000 U/ml IL-2 for approximately 2 weeks. Positive wells containing cancer-reactive cells are selected for further expansion to achieve large numbers of T cells for adoptive transfer. Rosenberg et al. [15] performed the first clinical trial of TIL-based adoptive T cell transfer, which demonstrated a significant objective response in patients with metastatic melanoma. However, cancer regression was transient, which was associated with the lack of persistence of the transferred cells in vivo. Subsequently, in 2002, chemotherapy-induced lymphodepletion was introduced to improve TIL-based treatment [51]. Chemotherapy-induced lymphodepletion prior to adoptive cell infusion has been shown to dramatically enhance the persistence of the transferred cells and improve anti-cancer effects [51]. More recently, in three sequential clinical trials, 93 patients with metastatic melanoma were treated with the adoptive transfer of autologous TILs administered in conjunction with IL-2 following a lymphodepleting preparative regimen. Lymphodepleting preparative regimens (chemotherapy alone or in combination with 2 or 12 Gy irradiation) prior to TIL transfer resulted in objective response rates of 49–72% as assessed by Response Evaluation Criteria in Solid Tumors [16]. It has been clearly shown that increasing the intensity of lymphodepletion enhances the cancer treatment efficacy of adoptively transferred TILs. The mechanisms underlying the beneficial effects of lymphodepleting preparative regimens may include the following [82]: elimination of Treg cells and/or MDSCs; elimination of endogenous lymphocytes, which may remove the competition for homeostatic cytokines such as IL-7 and IL-15; and activation of antigen-presenting cells, which increases antigen presentation to T cells, thus enhancing anticancer immunity [83].
TIL-based adoptive immunotherapy has been shown to be the best available treatment for patients with metastatic melanoma compared with other treatments including antibody and small molecule therapies. However, there are still many issues that need to be resolved: to identify the basis for variation in patient response. Why do some patient respond to treatment and others do not? Why is TIL-based immunotherapy only effective against melanoma even though TILs can be also isolated from other solid tumors including colorectal cancer, breast cancer, lung cancer and ovarian cancer? Can TIL-based therapy be used to treat other solid cancers in addition to melanoma? Furthermore, it is not possible to obtain tumor tissue for TIL cultivation from every patient or successfully cultivate TILs from resected tumor tissues. Therefore, alternative immunotherapeutic approaches to TIL-based ACT are needed to treat metastatic melanoma as well as other types of solid cancers.
Cancer antigen-induced specific T cells
Cancer antigen-specific T cells can be detected in PBMCs of cancer patients [84,85]. These antigen-specific T cells can be cultured or enriched from PBMCs or TILs following in vitro stimulation using autologous antigen-presenting cell pulsed with peptides derived from cancer antigens. Cancer antigen-induced specific T cells have been used for adoptive therapy to treat metastatic melanoma with little or no side effects [50]. A Phase I study showed that adoptive transfer of MART1/MelanA- and gp100-specific CD8+ T-cell clones did not show any serious toxicity in patients with metastatic melanoma. Furthermore, adoptively transferred T cell clones mediated the regression of individual metastases and induced minor, mixed or stable responses in eight of 10 patients with refractory metastatic disease [52]. In another clinical trial, adoptive T-cell therapy using MelanA-specific CD8+ T cells showed an antitumor response in three of 11 patients (one complete regression [CR], one partial regression and one mixed response) [53]. Interestingly, Hunder et al. reported that adoptively transferred CD4+ T cell clones with specificity for the melanoma-associated antigen NY-ESO-1 mediated a durable clinical remission and led to endogenous responses against melanoma antigens other than NY-ESO-1 in a patient with refractory metastatic melanoma who had not undergone any previous conditioning or cytokine treatment [54]. In this study, adoptively transferred CD4+ T cell clones persisted much longer in the patient’s blood (>3 months) than adoptively transferred CD8+ T cells, which survived only briefly (<20 days) in vivo in the absence of exogenous cytokine [52,53]. These results suggest that antigen-specific CD4+ T cells play a critical role in cancer treatment and may have important implications for a new generation of ACT that incorporates both antigen-specific CD4+ T cells and CD8+ T cells for the treatment of malignant diseases. Subsequently, in a Phase II study, six (43%) of 14 patients receiving ACT with PBMC-derived MelanA-specific T-cell clones experienced an objective response, with two patients achieving long-term CR (one CR for 5 years and one CR for 28 months) [55]. More recently, in another clinical trial, 11 patients with refractory, progressive, metastatic melanoma received cyclophosphamide as conditioning prior to the infusion of antigen-specific CD8+ T-cell clones followed by low-dose or high-dose IL-2. Five of 10 evaluable patients had stable disease at 8 weeks, and one of the 11 patients had a CR that continued for longer than 3 years [56].
The immunotherapeutic strategies using cancer antigen-induced specific T cells are clearly feasible and identification of potential cancer antigens recognized by autologous T cells and epitopes presented by restricting HLA alleles are critical to developing such strategies. Our group, as previously mentioned, has identified a series of cancer antigens including TRP1, TRP2, NY-ESO-1, EBNA-1, PSGR and SATB1 [25–34]. Some of these cancer antigens have been evaluated in clinical trials and have achieved encouraging results [54,86]. With identification of more and more cancer antigens expressed by different types of cancer, there are opportunities to use such antigens to generate cancer antigen-specific T cells in vitro for the treatment of various types of cancer. To avoid cancer antigen loss, cancer antigen-specific T cells targeting several cancer antigens expressed on the same cancer cell type can be obtained by in vitro stimulation of peripheral blood T cells or TILs.
ACT using cancer antigen-induced specific T cells has several advantages: cancer-reactive T cells can be obtained by in vitro peptide stimulation of PBMCs, which are easily collected from patients; a number of cancer antigens and their derived HLA-restricted epitopes have been identified, which will facilitate the development of antigen-specific T cells to treat various types of cancer; peptides are synthesized cheaply and can be easily and safely delivered to any medical center for stimulation of cancer-reactive cells; and viral vectors are not needed for gene transduction. In addition to its benefits, ACT using cancer antigen-induced specific T cells has several disadvantages: targeting of cancer antigens expressed on cancer cells requires an MHC restriction; most cancer antigens are self- antigens, which are also expressed on normal cells. Thus infusion of antigen-specific T cells can target not only antigen-positive cancer cells in vivo but also normal tissues expressing the shared tumor antigen [87]; long-term stimulation or cultivation of antigen-specific T cells in vitro may exhaust T cells, leading to shortened survival in vivo after infusion. However, a recent report suggests that exposure of T cells to IL-21 during in vitro culture may improve their in vivo persistency and expansion efficiency [88]; and the time-consuming and labor-intensive limiting dilution approach is often used for selecting antigen-specific T cells from thousands of T cell clones. Recently, CD137 has been successfully used as a marker to enrich and expand cancer-specific T cells from both PBMCs [89] and cancer tissues [90], indicating that targeting CD137 may be a useful method to rapidly achieve large numbers of cancer-specific T cells for ACT.
Antigen-specific TCR-transduced T cells
TCRs expressed on T cells can be genetically engineered in vitro to specifically recognize and kill cancer cells [12]. Two approaches are used for TCR gene transfer. In the first approach, the antigen-specific TCR from PBMC- or TIL-derived cancer-specific T-cell clone is cloned, transduced into the peripheral blood T cells of patients using lentiviral or retroviral vectors, and subsequently amplified using the rapid expansion protocol. The second approach involves the generation of antigen-specific TCRs through the immunization of HLA-I/II transgenic mice with cancer antigen. The antigen-specific TCR is cloned and transduced into the peripheral blood T cells of the patient for immunotherapy as described above. TCRs generated in HLA-I/II transgenic mice have not been selected in the thymus against the full repertoire of human proteins; therefore, these murine TCRs usually have a higher affinity for human tumor antigens compared to those generated from human PBMCs or TILs.
TCR-transduced T cells against several tumor antigens including MART1, CEA, gp100, NY-ESO-1 and MAGEA3 have been tested in clinical trials [12]. The results from these clinical trials have shown great promise in treating various types of cancers including metastatic melanoma, metastatic colorectal cancer, metastatic synovial cell sarcoma and epithelial malignancies. Cancer regression in patients with metastatic melanoma following administration of autologous T cells genetically engineered to express a TCR against MART1 was first reported in 2006 by Rosenberg et al. at the NIH [57]. In this study, two of 15 patients experienced objective tumor responses, and none of the patients developed autoimmune toxicities. Subsequently, a TCR with greater affinity for MART1 was used in clinical trials by the same group and shown to induce partial tumor responses in six of 20 patients [58]. However, autoimmune toxicity associated with T-cell-mediated destruction of normal melanocytes occurred in 16 of the 20 patients. Transient dermatologic, ocular and otologic adverse events were observed including skin rash, uveitis and hearing loss. In another clinical trial, immunotherapy using a TCR against CEA generated by immunizing HLA-A2 transgenic mice with CEA691-699 [59] induced severe transient colitis in three patients and a partial tumor response in one patient. Importantly, more than 50% of metastatic melanoma and synovial cell sarcoma patients treated with adoptively transferred T cells expressing a TCR against the cancer/testis antigen (CTA) NY-ESO-1 [60] achieved objective tumor responses without the induction of autoimmune toxicities. Therefore, it seems that TCR gene therapy targeting a CTA can mediate tumor regression without triggering autoimmune responses in normal tissues. However, a recent study showed that four of nine patients receiving ACT with autologous T cells transduced with a high affinity TCR against the CTA MAGEA3112-120 [61] manifested severe neurological toxicities due to an unexpected cross-reactivity against a nonidentical HLA-A2-restricted epitope present in MAGEA12 that is expressed in normal brain tissue. Surprisingly, in another clinical trial, two patients who received autologous T cells transduced to express an affinity-enhanced human TCR against an HLA-A1-restricted epitope (EVDPIGHLY) of MAGEA3 experienced fatal cardiac toxicity associated with myocardial T-cell infiltration and elevated levels of genetically engineered cells in myocardial tissue [62,63]. Using an amino acid scanning approach, a peptide from the muscle protein Titin (ESDPIVAQY) was identified as an alternative target for the MAGEA3-derived peptide-specific TCR-transduced T cells and the most likely cause of in vivo toxicity.
In addition to antigen-specific TCR-transduced T cells, a novel class of recombinant TCRs, immune-mobilizing monoclonal TCRs against cancer (ImmTACs), has been recently described [91]. These molecules combine high-affinity TCR-based antigen recognition with the immune-activating potential of an anti-CD3 antibody fragment to potently redirect T cells to kill tumor cells. ImmTACs have been demonstrated to be capable of eradicating cancer cells in vitro and in vivo [92,93]. An advantage of ImmTAC is that it can be formulated as an ‘off-the-shelf’ drug for administration to patients with the relevant HLA allele and antigen-positive cancer, whereas drawbacks include the need for repeated injections and the lack of persistence of cancer-specific memory T cells in the body. The ImmTAC drug IMCgp100 is currently in clinical trials in melanoma patients.
An obstacle to cancer immunotherapy is that the binding affinity of natural TCRs for cancer antigens expressed in tumors is generally low. Thus, modified high-affinity TCRs are being evaluated in various clinical trials. However, lessons from these clinical trials suggest that a TCR with intermediate and not high affinity might be a better choice to avoid the occurrence of unacceptable SAE due to cross-reactivity with cancer antigens expressed at low levels in vital organs. In addition, it is of great importance to identify ideal cancer antigen targets with tumor-restricted expression (e.g., NY-ESO-1). In summary, these clinical trials have demonstrated the considerable effector functions of affinity-enhanced TCR-transduced T cells in vivo and highlight the potential safety concerns for TCR-engineered T cells. Strategies to minimize the risk of developing SAEs due to cross-activity or ‘on-target/off-tumor’ effects should be included in future clinical trials.
CAR-transduced T cells
The concept of CAR was firstly introduced by Eshhar et al. in 1989 [94]. The current CAR structure is composed of an extracellular single-chain variable fragment of an antibody, an extracellular hinge and spacer element, a transmembrane domain, and intracellular signaling domains derived from molecules involved in T-cell signaling. The ectodomain can specifically recognize and bind to cancer cell surface antigens (e.g., CD19) and deliver activating signals to T cells through CD3ξ or the Fc receptor-γ chain, which in turn trigger T-cell effector functions for cancer cell elimination. As shown in Figure 2, CAR is generally classified into three generations according to the number of signaling domains [10]. First-generation CARs only contain the CD3ζ chain signaling domain. Second-generation CARs incorporate an additional signaling domain from costimulatory or accessory molecules, such as CD28, 4-1BB (CD137), OX-40, CD244, CD27, or ICOS. Second-generation CAR T cells have been shown to enhance T-cell expansion and antitumor activity compared with first-generation CARs. To further improve T-cell expansion, cytotoxicity and in vivo persistence, third-generation CARs, which contain an additional costimulatory domain, have been developed. Recently, fourth-generation CARs have been described [14,95] in which the gene construct contains a vector encoding a CAR and a CAR-responsive promoter, which responds upon successful signaling of the CAR by the transgenic production of cytokines such as IL-12 (Figure 2). Fourth-generation CAR T cells have shown enhanced in vivo anticancer efficacy in preclinical studies [96,97]. CAR-transduced T cell therapy has the following advantages: use of autologous T cells to reduce the risk of rejection; the same CAR configuration can be used for many patients; a variety of cell surface molecules including proteins with varying glycosylation and nonprotein structures such as gangliosides and carbohydrate antigens can be targeted; and CAR T cell function is unaffected by tumor escape mechanisms related to HLA downregulation and altered processing because antigen presentation is not MHC restricted.
Figure 2. Four generations of chimeric antigen receptor structures.

First-generation chimeric antigen receptor (CAR) consists of a single signaling domain derived from the CD3ζ chain. Extracellular domain scFv is an antigen-binding site, which is derived from monoclonal antibodies. The CAR is tethered to the plasma membrane via a transmembrane domain; second-generation CAR is added with an additional intracellular signaling domain to the basic first-generation receptor configuration that provides a costimulatory signal (e.g., CD28 or 4-1BB); third-generation CAR is added with two costimulatory domains in series (e.g., CD28 and 4-1BB) to the T-cell activatory signaling domain. Fourth-generation CAR employs a vector encoding a CAR and a CAR-responsive promoter that responds upon successful signaling of the CAR by the transgenic production of cytokines. Either a first-, second- or third-generation CAR may be used in this configuration.
scFv: Single-chain variable fragment; TM: Transmembrane.
First-generation CAR T cells targeting neuroblastoma, lymphoma, renal cancer and ovarian cancer have been evaluated in clinical trials; however, only limited clinical activity was achieved due to the lack of T cell expansion and long-term persistence in vivo. Currently, most clinical trials are using second- or third-generation CAR T cells, which lead to more positive clinical outcomes. Clinical evaluation of adoptive immunotherapy with second-generation GD2-specific CAR T cells showed that the long-term low-level presence of CAR-expressing T cells was associated with clinical benefits in 19 high-risk neuroblastoma patients, including CR in three patients [64]. CAR T-cell-based therapy is associated with the risk of developing SAEs because most targeted antigens are nonmutated self-antigens. Indeed, clinical results obtained from second-generation CAR T-cell-based therapy targeting carbonic anhydrase-9 (CAIX) induced liver toxicity in patients with renal carcinoma due to CAR T cell targeting of CAIX-positive bile duct epithelial cells [65]. Third-generation ERBB2-specific CAR T cells caused fatal respiratory distress syndrome in a colon cancer patient, which was associated with CAR T-cell-mediated cytokine release syndrome in response to ERBB2 expression in the lung epithelium [66].
The most promising results from CAR-transduced T-cell therapy have been with CD19-based targeting of B cell malignancies. The first patient with a B cell malignancy (lymphoma) successfully treated using an anti-CD19 CAR was reported by Kochenderfer et al. at the National Cancer Institute [67]. This patient still has an ongoing response 4 years later. Subsequently, six of eight patients with B cell lymphoma or chronic lymphocytic leukemia (CLL) treated with anti-CD19 CAR-transduced T cells achieved an objective clinical response, including one patient with a complete response [3,67]. Furthermore, several recent trials have also demonstrated positive clinical outcomes with anti-CD19 CAR T cell therapy in patients with B cell malignancies. A group at the University of Pennsylvania demonstrated that CD19 CAR T cells were able to eradicate large tumor burdens and mediate complete and ongoing CRs with long-term persistence of functional CAR T cells beyond 3 years in two of three CLL patients and one of two acute lymphoblastic leukemia (ALL) patients [69–71]. Another group at Memorial Sloan-Kettering Cancer Center reported that five relapsed B cell ALL patients with persistent morphological disease or minimal residual disease positive (MRD+) status treated with anti-CD19 CAR T cells achieved MRD− status, and four of five patients went on to receive allogeneic hematopoietic stem cell transplantation [72], subsequently they treated an additional 11 patients with relapsed or refractory ALL, further confirming that anti-CD19 CAR T cells induced CRs in the vast majority of patients [73], thus leading to the overall complete response in 88% of 16 patients. It should be noted that the former group used a second-generation CAR with 4-1BB and CD3ζ costimulatory domains, whereas the latter group used a second-generation CAR with CD28 and CD3ζ costimulatory domains. Although, both complete tumor responses and B cell aplasia have been observed in patients treated with T cells transduced with CARs containing either CD28 or 4-1BB, it is unclear whether both CAR configurations have the same anticancer efficacy in the clinical setting. Therefore, to identify the optimal CAR configuration, a direct comparison of T cells transduced with CARs containing different costimulatory endodomains should be performed in future clinical trials. In addition to B cell aplasia caused by infusion of anti-CD19 CAR T cells, the other associated toxicity is described as a cytokine release syndrome including high-grade fevers, hypotension, hypoxia as well as neurologic disturbances, which may require supportive treatment [68,71,72].
Despite the great success to date with anti-CD19 CAR T cells in the treatment of patients with B cell malignancies, clinical trials targeting solid cancers have achieved limited efficacy and observed ‘on-target, off-tumor responses’ with serious consequences [65,66]. The failure of CAR-transduced T cells to treat solid cancers may be due to several reasons including lack of ideal cancer antigens, short-term persistence of CAR T cells, and inefficient trafficking of sufficient numbers of CAR T cells to tumor sites. Furthermore, an immunosuppressive tumor environment also inhibits the functions of CAR T cells at tumor sites [98,99]. In addition, another potential hurdle for the successful use of CAR T cells is emergence of cancer cells that no longer express the target due to clonal evolution of cancer cells [71]. Strategies to overcome these barriers should be taken into consideration to construct CAR T cells capable of treating solid cancers as well as hematopoietic malignancies in future clinical trials. Since it is challenging to identify ideal targets allowing for specifically targeting cancer cells while avoiding damage to normal cells that express the same target, two novel approaches have been described to resolve this challenge [100]: one is to improve tumor targeting based on combinatorial antigen recognition [101], and the other is to use receptors that provide antigen-specific inhibition to divert T cells from the normal cells [102]. Another solution is to use suicide genes in the T-cell therapy to abort a threatening undesirable response [103–105], although this may impair subsequent therapeutical effects of the infused T cells.
Conclusion & future perspective
Although ACT-based cancer immunotherapy has exhibited encouraging results in clinical trials, this success has only been observed in a few types of cancer, predominantly in hematological cancers. Therefore, development of ACT-based immunotherapeutic approaches for the efficient and safe treatment of solid cancers is a major task in the near future. Identification of ideal cancer antigens, in vivo persistency and survival of adoptively transferred T cells, cancer biomarkers for ACT-based therapy, and combinational immunotherapy will also be major focuses of future research in ACT-based cancer immunotherapy.
Identification of ideal cancer antigens
Currently, most clinical trials of ACT–based immunotherapy have used self-antigens that are overexpressed in malignant cells compared with normal cells. Thus, infused antigen-specific T cells can target not only antigen-positive cancer cells in vivo but also normal tissues expressing the shared cancer antigen. The biggest challenge to overcome remains the identification of antigens with a strictly cancer cell-restricted expression, such as NY-ESO-1, to achieve selective tumor targeting while sparing normal tissues.
In vivo persistence & survival of adoptively transferred T cells
Results from clinical trials have suggested that cancer regression is positively associated with the long-term survival and persistence of adoptively transferred T cells in vivo. Strategies to enhance in vivo persistence and survival of transferred T cells can lead to improved anticancer efficacy. A recent report suggests that exposure to IL-21 during in vitro culture may improve the persistence and expansion efficiency of T cells in vivo [88]. Naive CD8+ T cells or human stem cell-like memory T cells may be the superior subset for use in adoptive immunotherapy, because they have a higher replicative potential and longer survival in vivo [106,107]. The proliferation and survival of adoptively transferred T cells may also be enhanced by shRNA-mediated silencing of genes that inhibit T cell function in the tumor microenvironment [108], thus leading to enhanced antitumor immunity. Another alternative is to generate rejuvenated antigen-specific T cells that have a high proliferative capacity by reprogramming to pluripotency and redifferentiation [109,110].
Cancer biomarkers for ACT-based therapy
Cancer biomarkers can support and guide clinical cancer treatment and development of the candidate products [111]. A recent study showed that expression of PD-L1 on tumor cells and/or other immune cells is positively correlated with anti-PD-1 monoclonal antibody-mediated therapy. Thus, identification of biological correlates of response, either TILs or tumor characteristics, will help not only to select the most efficacious cell types for infusion but also patients who are most likely to respond to ACT-based therapy.
Combinational immunotherapy
Although emerging techniques allow for the generation of high affinity antigen-specific T cells capable of targeting cancer cells in vitro, immune suppression and negative regulation at tumor sites can impede the induction of effective anticancer immune responses in vivo. Suppressive cytokines/agents such as IL-10, transforming growth factor-b, and indoleamine 2,3-dioxygenase and cell populations such as MDSCs and Treg cells present in the tumor microenvironment induce immunosuppression, thereby blocking T cell function [45,46] and leading to poor antitumor efficacy of immunotherapy. Therefore, combining ACT therapy with other strategies that block negative regulators such as PD-1, reverse Treg cell/MDSC-mediated immunosuppression, or inhibit tumor cell proliferative and metabolic signaling pathways may induce potentially profound immune antitumor responses. In addition, our group has recently identified several coinhibitory molecules including NLRC5, NLRX1 and NLRP4 [112–114], which can negatively regulate NF-κB and type I interferon signaling pathways. Inhibition of these negative regulators can enhance DC functions and reverse immune suppression. Thus, combinational therapies of ACT with blockade of NLRC5, NLRX1 or NLRP4 signaling for cancer treatment merit further investigation.
In summary, ACT-based cancer immunotherapy has achieved encouraging results in clinical trials and has curative potential in some cancer patients. Further research is required to develop safe and efficient ACT-based immunotherapies with broad efficacy against a wide range of cancer types. We believe that the increasing interest and investments from pharmaceutical industries will accelerate research and development of ACT-based cancer immunotherapy to benefit patients with various types of cancers.
Executive summary.
Cancer antigen-specific T cells & activation
The success of cancer immunotherapy relies largely on the identification of suitable cancer antigens for the generation of effective cancer vaccines and antigen-specific T cells.
The key points in cancer immunotherapy are ‘one activation and two inhibitions’ where cancer-reactive T-cell activation by cancer antigen represents the one activation and blockade of coinhibitory molecules on T cells and reversal of Treg cell-mediated immunosuppression represent the two inhibitions.
Adoptive cell transfer-based cancer immunotherapy
Tumor-infiltrating lymphocyte based adoptive cell transfer (ACT) has resulted in objective response rates of 49–72% in patients with metastatic melanoma refractory to standard therapies.
Cancer antigen-induced specific T cells have been used for adoptive therapy to treat metastatic melanoma with little or no side effects.
Antigen-specific T-cell receptor-transduced T cells against several tumor antigens have been tested in clinical trials and have shown great promise in treating various types of cancers.
Chimeric antigen receptor transduced T cells have shown great success to treat patients with B cell malignancies, while clinical trials targeting solid cancers have achieved limited efficacy.
Conclusion
ACT-based cancer immunotherapy has achieved encouraging results in clinical trials and has curative potential in some cancer patients.
Further research is required to develop safe and efficient ACT-based immunotherapies with broad efficacy against a wide range of cancer types.
-
Future directions of cancer immunotherapy may include:
Identification of ideal cancer antigens.
Enhancing in vivo persistency and survival of adoptively transferred T cells.
Identification of cancer biomarkers for ACT-based therapy.
Combinational immunotherapy.
Acknowledgments
The authors would like to thank A-M Cimo for editing the manuscript and S Ye for assistance in figure preparation.
Footnotes
For reprint orders, please contact: reprints@futuremedicine.com
Financial & competing interests disclosure
This work was in part supported by grants from the National Cancer Institute, NIH and Cancer Prevention and Research Institute of Texas (CPRIT), and Houston Methodist Research Institute. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
• of interest
•• of considerable interest
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Wagner PD, Srivastava S. New paradigms in translational science research in cancer biomarkers. Transl Res. 2012;159(4):343–353. doi: 10.1016/j.trsl.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kochenderfer JN, Rosenberg SA. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat Rev Clin Oncol. 2013;10(5):267–276. doi: 10.1038/nrclinonc.2013.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12(4):269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharma P, Wagner K, Wolchok JD, Allison JP. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps. Nat Rev Cancer. 2011;11(11):805–812. doi: 10.1038/nrc3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480–489. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 8.Hodi FS, O’Day SJ, Mcdermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwartzentruber DJ, Lawson DH, Richards JM, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364(22):2119–2127. doi: 10.1056/NEJMoa1012863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ruella M, Kalos M. Adoptive immunotherapy for cancer. Immunol Rev. 2014;257(1):14–38. doi: 10.1111/imr.12136. [DOI] [PubMed] [Google Scholar]
- 11.Jensen MC, Riddell SR. Design and implementation of adoptive therapy with chimeric antigen receptor-modified T cells. Immunol Rev. 2014;257(1):127–144. doi: 10.1111/imr.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12•.Hinrichs CS, Rosenberg SA. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol Rev. 2014;257(1):56–71. doi: 10.1111/imr.12132. Overview of adoptive T-cell-based immunotherapy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev. 2014;257(1):107–126. doi: 10.1111/imr.12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheadle EJ, Gornall H, Baldan V, Hanson V, Hawkins RE, Gilham DE. CAR T cells: driving the road from the laboratory to the clinic. Immunol Rev. 2014;257(1):91–106. doi: 10.1111/imr.12126. [DOI] [PubMed] [Google Scholar]
- 15•.Rosenberg SA, Packard BS, Aebersold PM, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. 1988;319(25):1676–1680. doi: 10.1056/NEJM198812223192527. The first tumor-infiltrating lymphocyte based clinical trial in melanoma patients. [DOI] [PubMed] [Google Scholar]
- 16.Rosenberg SA, Yang JC, Sherry RM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17(13):4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science. 2013;342(6165):1432–1433. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- 18.Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–271. doi: 10.1146/annurev-immunol-031210-101324. [DOI] [PubMed] [Google Scholar]
- 19.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331(6024):1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 20.Rosenberg SA. Cell transfer immunotherapy for metastatic solid cancer – what clinicians need to know. Nat Rev Clin Oncol. 2011;8(10):577–585. doi: 10.1038/nrclinonc.2011.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang HY, Wang RF. Enhancing cancer immunotherapy by intracellular delivery of cell-penetrating peptides and stimulation of pattern-recognition receptor signaling. Adv Immunol. 2012;114:151–176. doi: 10.1016/B978-0-12-396548-6.00006-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Den Eynde B, Lethe B, Van Pel A, De Plaen E, Boon T. The gene coding for a major tumor rejection antigen of tumor P815 is identical to the normal gene of syngeneic DBA/2 mice. J Exp Med. 1991;173(6):1373–1384. doi: 10.1084/jem.173.6.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vigneron N, Stroobant V, Van Den Eynde BJ, Van Der Bruggen P. Database of T cell-defined human tumor antigens: the 2013 update. Cancer Immun. 2013;13:15. [PMC free article] [PubMed] [Google Scholar]
- 24.Cancer Immunity: Peptide Database. www.cancerimmunity.org/peptide.
- 25.Matsueda S, Wang M, Weng J, et al. Identification of prostate-specific G-protein coupled receptor as a tumor antigen recognized by CD8(+) T cells for cancer immunotherapy. PLoS ONE. 2012;7(9):e45756. doi: 10.1371/journal.pone.0045756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Voo KS, Zeng G, Mu JB, Zhou J, Su XZ, Wang RF. CD4+ T-cell response to mitochondrial cytochrome B in human melanoma. Cancer Res. 2006;66(11):5919–5926. doi: 10.1158/0008-5472.CAN-05-4574. [DOI] [PubMed] [Google Scholar]
- 27.Wang M, Yin B, Matsueda S, et al. Identification of special AT-rich sequence binding protein 1 as a novel tumor antigen recognized by CD8(+) T cells: implication for cancer immunotherapy. PLoS ONE. 2013;8(2):e56730. doi: 10.1371/journal.pone.0056730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang RF. Enhancing antitumor immune responses: intracellular peptide delivery and identification of MHC class II-restricted tumor antigens. Immunol Rev. 2002;188:65–80. doi: 10.1034/j.1600-065x.2002.18807.x. [DOI] [PubMed] [Google Scholar]
- 29.Wang RF, Appella E, Kawakami Y, Kang X, Rosenberg SA. Identification of TRP-2 as a human tumor antigen recognized by cytotoxic T lymphocytes. J Exp Med. 1996;184(6):2207–2216. doi: 10.1084/jem.184.6.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang RF, Johnston SL, Southwood S, Sette A, Rosenberg SA. Recognition of an antigenic peptide derived from tyrosinase-related protein-2 by CTL in the context of HLA-A31 and -A33. J Immunol. 1998;160(2):890–897. [PubMed] [Google Scholar]
- 31.Wang RF, Johnston SL, Zeng G, Topalian SL, Schwartzentruber DJ, Rosenberg SA. A breast and melanoma-shared tumor antigen: T cell responses to antigenic peptides translated from different open reading frames. J Immunol. 1998;161(7):3598–3606. [PubMed] [Google Scholar]
- 32.Wang RF, Parkhurst MR, Kawakami Y, Robbins PF, Rosenberg SA. Utilization of an alternative open reading frame of a normal gene in generating a novel human cancer antigen. J Exp Med. 1996;183(3):1131–1140. doi: 10.1084/jem.183.3.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang RF, Robbins PF, Kawakami Y, Kang XQ, Rosenberg SA. Identification of a gene encoding a melanoma tumor antigen recognized by HLA-A31-restricted tumor-infiltrating lymphocytes. J Exp Med. 1995;181(2):799–804. doi: 10.1084/jem.181.2.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang RF, Rosenberg SA. Human tumor antigens for cancer vaccine development. Immunol Rev. 1999;170:85–100. doi: 10.1111/j.1600-065x.1999.tb01331.x. [DOI] [PubMed] [Google Scholar]
- 35.Peng W, Wang HY, Miyahara Y, Peng G, Wang RF. Tumor-associated galectin-3 modulates the function of tumor-reactive T cells. Cancer Res. 2008;68(17):7228–7236. doi: 10.1158/0008-5472.CAN-08-1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang RF. The role of MHC class II-restricted tumor antigens and CD4+ T cells in antitumor immunity. Trends Immunol. 2001;22(5):269–276. doi: 10.1016/s1471-4906(01)01896-8. [DOI] [PubMed] [Google Scholar]
- 37.Wang RF, Wang X, Atwood AC, Topalian SL, Rosenberg SA. Cloning genes encoding MHC class II-restricted antigens: mutated CDC27 as a tumor antigen. Science. 1999;284(5418):1351–1354. doi: 10.1126/science.284.5418.1351. [DOI] [PubMed] [Google Scholar]
- 38.Wang RF, Wang X, Rosenberg SA. Identification of a novel major histocompatibility complex class II-restricted tumor antigen resulting from a chromosomal rearrangement recognized by CD4(+) T cells. J Exp Med. 1999;189(10):1659–1668. doi: 10.1084/jem.189.10.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12(4):265–277. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13(4):227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Royal RE, Levy C, Turner K, et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. 2010;33(8):828–833. doi: 10.1097/CJI.0b013e3181eec14c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shevach EM. Application of IL-2 therapy to target T regulatory cell function. Trends Immunol. 2012;33(12):626–632. doi: 10.1016/j.it.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang HY, Wang RF. Regulatory T cells and cancer. Curr Opin Immunol. 2007;19(2):217–223. doi: 10.1016/j.coi.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 46.Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. 2010;10(7):490–500. doi: 10.1038/nri2785. [DOI] [PubMed] [Google Scholar]
- 47.Peng G, Wang HY, Peng W, Kiniwa Y, Seo KH, Wang RF. Tumor-infiltrating gammadelta T cells suppress T and dendritic cell function via mechanisms controlled by a unique toll-like receptor signaling pathway. Immunity. 2007;27(2):334–348. doi: 10.1016/j.immuni.2007.05.020. [DOI] [PubMed] [Google Scholar]
- 48•.Peng G, Guo Z, Kiniwa Y, et al. Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science. 2005;309(5739):1380–1384. doi: 10.1126/science.1113401. The first report demonstrating that suppressive function of Treg cells can be reversed by Toll-like receptor 8. [DOI] [PubMed] [Google Scholar]
- 49.Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364(26):2517–2526. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 50.Yee C. The use of endogenous T cells for adoptive transfer. Immunol Rev. 2014;257(1):250–263. doi: 10.1111/imr.12134. [DOI] [PubMed] [Google Scholar]
- 51.Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298(5594):850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yee C, Thompson JA, Byrd D, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci USA. 2002;99(25):16168–16173. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mackensen A, Meidenbauer N, Vogl S, Laumer M, Berger J, Andreesen R. Phase I study of adoptive T-cell therapy using antigen-specific CD8+ T cells for the treatment of patients with metastatic melanoma. J Clin Oncol. 2006;24(31):5060–5069. doi: 10.1200/JCO.2006.07.1100. [DOI] [PubMed] [Google Scholar]
- 54.Hunder NN, Wallen H, Cao J, et al. Treatment of metastatic melanoma with autologous CD4+ T cells agaInst. NY-ESO-1. N Engl J Med. 2008;358(25):2698–2703. doi: 10.1056/NEJMoa0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khammari A, Labarriere N, Vignard V, et al. Treatment of metastatic melanoma with autologous Melan-A/MART-1-specific cytotoxic T lymphocyte clones. J Invest Dermatol. 2009;129(12):2835–2842. doi: 10.1038/jid.2009.144. [DOI] [PubMed] [Google Scholar]
- 56.Chapuis AG, Thompson JA, Margolin KA, et al. Transferred melanoma-specific CD8+ T cells persist, mediate tumor regression, and acquire central memory phenotype. Proc Natl Acad Sci USA. 2012;109(12):4592–4597. doi: 10.1073/pnas.1113748109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57•.Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–129. doi: 10.1126/science.1129003. The first report of an antigen-specific T-cell receptor-transduced T-cell-based clinical trial in patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johnson LA, Morgan RA, Dudley ME, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114(3):535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Parkhurst MR, Yang JC, Langan RC, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19(3):620–626. doi: 10.1038/mt.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Robbins PF, Morgan RA, Feldman SA, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29(7):917–924. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morgan RA, Chinnasamy N, Abate-Daga D, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36(2):133–151. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cameron BJ, Gerry AB, Dukes J, et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med. 2013;5(197):197ra103. doi: 10.1126/scitranslmed.3006034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Linette GP, Stadtmauer EA, Maus MV, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122(6):863–871. doi: 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Louis CU, Savoldo B, Dotti G, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118(23):6050–6056. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lamers CH, Sleijfer S, Van Steenbergen S, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. 2013;21(4):904–912. doi: 10.1038/mt.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kochenderfer JN, Wilson WH, Janik JE, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116(20):4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119(12):2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69••.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–733. doi: 10.1056/NEJMoa1103849. Chimeric antigen receptor (CAR)-transduced T-cell-based clinical trial showing great success in treatment of chronic lymphoid leukemia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71••.Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–1518. doi: 10.1056/NEJMoa1215134. CAR-transduced T-cell-based clinical trial showing great success in treatment of acute lymphoid leukemia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72••.Brentjens RJ, Davila ML, Riviere I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5(177):177ra138. doi: 10.1126/scitranslmed.3005930. CAR-transduced T-cell-based clinical trial showing great success in treatment of acute lymphoblastic leukemia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra225. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Grimm EA, Mazumder A, Zhang HZ, Rosenberg SA. Lymphokine-activated killer cell phenomenon. Lysis of natural killer-resistant fresh solid tumor cells by interleukin 2-activated autologous human peripheral blood lymphocytes. J Exp Med. 1982;155(6):1823–1841. doi: 10.1084/jem.155.6.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schmidt-Wolf IG, Negrin RS, Kiem HP, Blume KG, Weissman IL. Use of a SCID mouse/human lymphoma model to evaluate cytokine-induced killer cells with potent antitumor cell activity. J Exp Med. 1991;174(1):139–149. doi: 10.1084/jem.174.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rosenberg SA, Lotze MT, Muul LM, et al. A progress report on the treatment of 157 patients with advanced cancer using lymphokine-activated killer cells and interleukin-2 or high-dose interleukin-2 alone. N Engl J Med. 1987;316(15):889–897. doi: 10.1056/NEJM198704093161501. [DOI] [PubMed] [Google Scholar]
- 77.Hontscha C, Borck Y, Zhou H, Messmer D, Schmidt-Wolf IG. Clinical trials on CIK cells: first report of the international registry on CIK cells (IRCC) J Cancer Res Clin Oncol. 2011;137(2):305–310. doi: 10.1007/s00432-010-0887-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rosenberg SA, Yannelli JR, Yang JC, et al. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J Natl Cancer Inst. 1994;86(15):1159–1166. doi: 10.1093/jnci/86.15.1159. [DOI] [PubMed] [Google Scholar]
- 79.Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. 1986;233(4770):1318–1321. doi: 10.1126/science.3489291. [DOI] [PubMed] [Google Scholar]
- 80•.Robbins PF, Lu YC, El-Gamil M, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. 2013;19(6):747–752. doi: 10.1038/nm.3161. Highlights mutated antigen-specific T cells present in tumor-infiltrating lymphocytes from melanoma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tran E, Turcotte S, Gros A, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344:641–645. doi: 10.1126/science.1251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dudley ME, Yang JC, Sherry R, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26(32):5233–5239. doi: 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Paulos CM, Wrzesinski C, Kaiser A, et al. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J Clin Invest. 2007;117(8):2197–2204. doi: 10.1172/JCI32205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee PP, Yee C, Savage PA, et al. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med. 1999;5(6):677–685. doi: 10.1038/9525. [DOI] [PubMed] [Google Scholar]
- 85.Pittet MJ, Valmori D, Dunbar PR, et al. High frequencies of naive Melan-A/MART-1-specific CD8(+) T cells in a large proportion of human histocompatibility leukocyte antigen (HLA)-A2 individuals. J Exp Med. 1999;190(5):705–715. doi: 10.1084/jem.190.5.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sonpavde G, Wang M, Peterson LE, et al. HLA-restricted NY-ESO-1 peptide immunotherapy for metastatic castration resistant prostate cancer. Invest New Drugs. 2014;32(2):235–242. doi: 10.1007/s10637-013-9960-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yee C, Thompson JA, Roche P, et al. Melanocyte destruction after antigen-specific immunotherapy of melanoma: direct evidence of t cell-mediated vitiligo. J Exp Med. 2000;192(11):1637–1644. doi: 10.1084/jem.192.11.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang S, Ji Y, Gattinoni L, et al. Modulating the differentiation status of ex vivo-cultured anti-tumor T cells using cytokine cocktails. Cancer Immunol Immunother. 2013;62(4):727–736. doi: 10.1007/s00262-012-1378-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wolfl M, Kuball J, Ho WY, et al. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. Blood. 2007;110(1):201–210. doi: 10.1182/blood-2006-11-056168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ye Q, Song DG, Poussin M, et al. CD137 accurately identifies and enriches for naturally occurring tumor-reactive T cells in tumor. Clin Cancer Res. 2014;20(1):44–55. doi: 10.1158/1078-0432.CCR-13-0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Oates J, Jakobsen BK. ImmTACs: novel bi-specific agents for targeted cancer therapy. Oncoimmunology. 2013;2(2):e22891. doi: 10.4161/onci.22891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mccormack E, Adams KJ, Hassan NJ, et al. Bi-specific TCR-anti CD3 redirected T-cell targeting of NY-ESO-1- and LAGE-1-positive tumors. Cancer Immunol Immunother. 2013;62(4):773–785. doi: 10.1007/s00262-012-1384-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liddy N, Bossi G, Adams KJ, et al. Monoclonal TCR-redirected tumor cell killing. Nat Med. 2012;18(6):980–987. doi: 10.1038/nm.2764. [DOI] [PubMed] [Google Scholar]
- 94.Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci USA. 1989;86(24):10024–10028. doi: 10.1073/pnas.86.24.10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chmielewski M, Hombach AA, Abken H. Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev. 2014;257(1):83–90. doi: 10.1111/imr.12125. [DOI] [PubMed] [Google Scholar]
- 96.Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011;71(17):5697–5706. doi: 10.1158/0008-5472.CAN-11-0103. [DOI] [PubMed] [Google Scholar]
- 97.Pegram HJ, Lee JC, Hayman EG, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119(18):4133–4141. doi: 10.1182/blood-2011-12-400044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.John LB, Devaud C, Duong CP, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. 2013;19(20):5636–5646. doi: 10.1158/1078-0432.CCR-13-0458. [DOI] [PubMed] [Google Scholar]
- 99.Peng W, Liu C, Xu C, et al. PD-1 blockade enhances T-cell migration to tumors by elevating IFN-gamma inducible chemokines. Cancer Res. 2012;72(20):5209–5218. doi: 10.1158/0008-5472.CAN-12-1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fedorov VD, Sadelain M, Kloss CC. Novel approaches to enhance the specificity and safety of engineered T cells. Cancer J. 2014;20(2):160–165. doi: 10.1097/PPO.0000000000000040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31(1):71–75. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. 2013;5(215):215ra172. doi: 10.1126/scitranslmed.3006597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lupo-Stanghellini MT, Provasi E, Bondanza A, Ciceri F, Bordignon C, Bonini C. Clinical impact of suicide gene therapy in allogeneic hematopoietic stem cell transplantation. Hum Gene Ther. 2010;21(3):241–250. doi: 10.1089/hum.2010.014. [DOI] [PubMed] [Google Scholar]
- 104.Di Stasi A, Tey SK, Dotti G, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365(18):1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Morgan RA. Live and let die: a new suicide gene therapy moves to the clinic. Mol Ther. 2012;20(1):11–13. doi: 10.1038/mt.2011.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gattinoni L, Lugli E, Ji Y, et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17(10):1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hinrichs CS, Borman ZA, Gattinoni L, et al. Human effector CD8+ T cells derived from naive rather than memory subsets possess superior traits for adoptive immunotherapy. Blood. 2011;117(3):808–814. doi: 10.1182/blood-2010-05-286286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhou P, Shaffer DR, Alvarez Arias DA, et al. In vivo discovery of immunotherapy targets in the tumour microenvironment. Nature. 2014;506(7486):52–57. doi: 10.1038/nature12988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vizcardo R, Masuda K, Yamada D, et al. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8(+) T cells. Cell Stem Cell. 2013;12(1):31–36. doi: 10.1016/j.stem.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 110.Nishimura T, Kaneko S, Kawana-Tachikawa A, et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 2013;12(1):114–126. doi: 10.1016/j.stem.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 111.Kalos M. Biomarkers in T cell therapy clinical trials. J Transl Med. 2011;9:138–146. doi: 10.1186/1479-5876-9-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cui J, Li Y, Zhu L, et al. NLRP4 negatively regulates type I interferon signaling by targeting the kinase TBK1 for degradation via the ubiquitin ligase DTX4. Nat Immunol. 2012;13(4):387–395. doi: 10.1038/ni.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Xia X, Cui J, Wang HY, et al. NLRX1 negatively regulates TLR-induced NF-kappaB signaling by targeting TRAF6 and IKK. Immunity. 2011;34(6):843–853. doi: 10.1016/j.immuni.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cui J, Zhu L, Xia X, et al. NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell. 2010;141(3):483–496. doi: 10.1016/j.cell.2010.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]