

Abstract
This protocol describes a primary neuronal model of formation of α-synuclein (α-syn) aggregates that recapitulate features of Lewy Bodies and Lewy Neurites found in Parkinson’s disease brains and other synucleinopathies. This model allows investigation of aggregate formation, their impact on neuron function, and development of therapeutics. Addition of pre-formed fibrils (PFFs) synthesized from recombinant α-syn to neurons seeds recruitment of endogenous α-syn into aggregates characterized by detergent-insolubility, and hyperphosphorylation. Aggregate formation follows a lag phase of 2–3 days, followed by formation in axons by days 4–7, spread to somatodendritic compartments by days 7–10, and neuron death around 14 days post-PFF. Here, we provide methods and highlight critical steps for PFF formation, addition to cultured hippocampal neurons, and confirmation of aggregate formation. Neurons derived from various brain regions from non-transgenic and genetically-engineered mice and rats can be used, allowing interrogation of the impact of specific genes on aggregate formation.
Introduction
Aggregates of α-syn called Lewy Bodies and Lewy neurites are a defining feature of synucleinopathies, including Parkinson’s disease, Dementia with Lewy Bodies, and the Lewy Body variant of Alzheimer’s disease. Many questions including how these aggregates form and their contribution to the etiology of PD remain to be answered. For example, early formation of these aggregates and sequestration of normal α-syn from the presynaptic terminal may contribute to neuronal dysfunction well before neurodegeneration, contributing to the prodromal phase of the disease. In addition, it is unknown whether these aggregates sequester numerous soluble toxic species providing neuroprotection, at least in the short term1–3, or represent a toxic and transmissible species, with their formation and spread directly responsible for the neurodegenerative phenotypes. Understanding how these aggregates form and their impact on neuronal function could contribute to the development of therapeutic targets to prevent the progression of these devastating neurodegenerative diseases4–7.
Modelling aggregation of α-syn
A major obstacle to elucidating the role of α-syn pathology has been the lack of model systems to study the acute effects of α-syn aggregation occurring in real-time, especially in individual neurons. First, although aggregated α-syn, as detected by either amyloid dye staining (e.g. Thioflavin S) or resistance to protease digestion, has been reported in a variety of cell lines which overexpress α-syn, few show the characteristic amyloid fibril ultrastructure seen in human Lewy pathology. In addition, transgenic mice that overexpress disease-associated mutant α-syn produce pathologic aggregates with these features, but only in mice several months of age, and aggregate formation coincides with the rapid demise of the animal, making it impossible to understand the early effects of α-syn aggregation8–10. Finally, primary neuronal cultures from these mice also do not form aggregates that recapitulate features of those found in diseased brains. By contrast, the primary neuron model described herein, combines most of the features of inclusions found in diseased brains as well as key biochemical/molecular markers of Lewy Neurites and Lewy Bodies in a genetically unmodified neuronal culture system. The defining features of these aggregates, both in diseased brains and in our model system, are that they are: insoluble in detergent, hyperphosphorylated, ubiquitinated, and filamentous ultrastructure when examined by electron microscopy11–14.
Seeded formation of aggregates from α-syn endogenously expressed in primary neurons
The development of our model was based upon previous studies that utilized cell lines overexpressing mutant forms of disease-associated proteins to demonstrate that exogenous amyloid fibrils can seed transition of soluble proteins into inclusions15–18. In this model, small seeds of pre-formed α-syn fibrils (PFFs) generated from recombinant α-syn are added directly to primary neurons14. Small amounts of these PFFs are endocytosed by the neuron, without the addition of other factors to assist entry into the neuron. These seeds of PFFs induce recruitment of endogenously expressed α-syn into abnormal, phosphorylated, insoluble, ubiquitinated aggregates. Formation of these aggregates from endogenous α-syn in primary neurons derived from wild type, non-transgenic mice follows an initial lag phase of 2–4 days. By 4–7 days small, punctate insoluble, phosphorylated aggregates from in presynaptic terminals and axons. By 7–10 days post-PFF, the aggregates grow and become more elongated and serpentine in appearance, resembling Lewy Neurites. They also can be found in approximately 30% of neuronal soma and dendrites where they appear skein-like, but over time form condensed accumulations that resemble Lewy Bodies. Neuron death is negligible prior to 14 days after adding the PFFs. This permits the careful examination of α-syn aggregates from their initial early formation, to spread throughout the neuron, and ultimately neuron death, as well as how neuronal function may be perturbed at each of these stages. For example, calcium imaging experiments reveal major defects in neuronal synchronization at early time points when only small aggregates are present in axons14. In addition, one of the most predominant phenotypes is the increased expression of canonical autophagy markers19. However, although mature α-syn aggregates associate with components of autophagy machinery, they cannot be efficiently degraded by this pathway. Furthermore, mitochondrial oxidant stress contributes to PD pathogeneses and can be measured in dopaminergic neurons from the substantia nigra pars compacta by expression of mitochondrially targeted redox-sensitive green fluorescent protein (TH-mito-roGFP) expressed under control of a tyrosine hydroxylase promoter20. The use of live cell imaging of dopamine primary neurons derived from mice expressing TH-mito-roGFP, demonstrates that the presence of α-syn aggregates significantly increases mitochondrial oxidant stress, particularly in the soma and dendrites. Interestingly, the mitochondrial oxidant stress likely derives from increased lysosomal oxidase activity.
A critical aspect of this model is that none of the phenotypes described above occur when PFFs are added to primary neurons from α-syn knockout mice. For example,, the PFFs themselves are not phosphorylated, and therefore when they are added to α-syn knockout neurons, there is no p-α-syn visible by immunofluorescence or immunoblot14. Furthermore, addition of fibrils to neurons from α-syn knockout mice does not cause cell death, or changes in neuronal synchronous firing, excitation or connectivity. Thus, the pathological phenotypes are caused by “seeded” corruption of endogenous α-syn likely through both a loss of normal α-syn function and gains of toxic functions from the accumulation of the Lewy Neurite and Lewy Body-like inclusions, rather than exposure to the synthetic fibrils themselves.
Overall, this model provides researchers with the opportunity to understand how the presence of neuronal α-syn aggregates contributes to disease pathogenesis at cellular and subcellular level (Figure 1) with well-defined temporal and spatial resolution.
Figure 1.

Timeline of known events in PFF model of α-syn aggregation.
Applications of the Protocol
There are many other advantages afforded by this model in addition to those mentioned above. First, α-syn aggregation occurs in normal wild type neurons expressing endogenous α-syn, i.e. it does not require overexpression of wildtype or mutant α-syn14. Thus, researchers can potentially understand the impact of any gene of interest on α-syn aggregate formation utilizing knockout mice or transgenic mice. Second, α-syn aggregation occurs in primary neurons derived from hippocampus, cortex, midbrain and other brain regions and thus the impact of these aggregates on diverse neuronal populations and neuronal subtypes could be examined14, 20, 21. In addition, it provides spatial resolution to allow high resolution microscopy and/or live cell imaging, and thus future studies can elucidate of the cell biological and physiologic impact of α-syn aggregate formation. For example, transfection of fluorescent probes of localization and function of distinct organelles can be accomplished using various published protocols22–24. Furthermore, this method can potentially be used in conjunction with established methods in which neurons are compartmentalized (e.g. in microfluidic or Campenot chambers) to understand how α-syn aggregates may spread from neuron to neuron, which has recently been shown to contribute to disease progression25,26. Finally, another future potential application is the screening of putative therapeutics to prevent the formation of these aggregates.
Limitations of the protocol
In cultures from non-transgenic mice, there is a 2–4 day lag phase before insoluble, phosphorylated aggregates appear. Thus, any compounds tested during this lag phase using this model must not be detrimental to neuron health when used over this time course. Although, it may be possible to reduce the concentrations of test compounds such that they remain effective but do not lead to neuron death. Furthermore, this lag phase can be reduced by overexpression of α-syn.
Experimental design
Primary neuron culture
While we provide some information here regarding culturing of primary neurons, these methods have been presented in detail elsewhere and we reference these protocols for full details23, 27, 28. Other labs have reported strong PFF-seeding using their own culturing methods20, 21. Recommended plating densities are provided for culturing neurons as we have found that the efficiency of aggregate formation depends on synaptic densities; the higher the density of the cultures, the more aggregates are produced. The procedure describes culture of hippocampal neurons from 1 pregnant C57BL/6 mouse with approximately 6–8 E16–E17 embryos. We generally perform experiments using hippocampal neurons from non-transgenic mice because: α-syn aggregates are found in the hippocampus in diseased brains and correlate with cognitive impairments29–31 ; the morphological and physiological properties of these neurons in culture have been well defined32; and we can obtain enough neurons for both imaging and biochemical studies. Seeded α-syn inclusions also develop in primary neurons from cortex and tyrosine hydroxylase-positive neurons cultured midbrain14, 20. However, the presence of inclusions may depend on the subtype of neuron, for example, GAD-positive inhibitory neurons do not form inclusions because of low to no expression of α-syn in these neurons21.
PFFs
Storage of the PFFs is another crucial factor in the effectiveness of the protocol. Once generated, we recommend dividing the PFFs into aliquots and storing them at −80°C because we and others33 have observed that storage at 4°C results in loss of activity. Sonication with a probe tip sonicator is another critical step in this protocol to generate small seeds (Video 1, Figure 2). We recommend adding PFFs at DIV 5–10 because α-syn is only expressed at mature synapses34. PFFs can also be added at later time points and pathology will proceed more quickly because higher levels of α-syn are expressed at presynaptic terminals. However, because neurons in culture typically only last approximately 21–31 days in culture in general, we do not recommend adding PFFs at too late a time point because the neurons (including control neurons) may begin to die before the aggregates spread throughout the neuron, thus adding a confounding factor. Also, we provide our recommended concentrations of PFFs to add to neurons. However, we suggest testing a range of concentrations to determine which concentration is best for each researcher’s particular assay.
Figure 2.

Electron micrographs of PFFs before (A) and after (B) sonication. Scale bars= 500 nm42.
Choice of controls
Here, we describe using PBS as a control. Other controls may include treatment of the neurons with α-syn monomer which does not produce α-syn aggregates. However, given the propensity for α-syn to aggregate, it is important to centrifuge the monomer after thawing (benchtop ultracentrifuge, 100,000 × g, 1hr) to remove any small fibrils that may have formed, and use only the resulting supernatant. As mentioned above, cultures from α-syn knockout mice can be used as a control to confirm that phenotypes result from aggregates formed from endogenously expressed α-syn and not from addition of PFFs.
MATERIALS
Reagents
Recombinant α-syn which can be purified as previously published35–39 (the purification is also summarized in Box1). We recommend purification of α-syn because it can be prepared without a tag that may interfere with fibrillization (commercial sources of α-syn often contain a poly-His tag), high protein concentrations (up to 30 mg/mL) can be obtained, and the final buffer can be controlled. In addition, we have successfully used both recombinant human and mouse α-syn14. The advantage of human α-syn is the availability of human specific α-syn antibodies that allow the exogenously added fibrils to be specifically identified.
CAUTION: Although the safety of handling recombinant α-syn has not been assessed, we recommend using personal protective equipment and biohazard safety level 2.
Phosphate buffered saline (PBS; see REAGENT SETUP)
Laemmli buffer (see REAGENT SETUP)
4–20% polyacrylamide gels (Bio-Rad cat no. 456-1093; www.bio-rad.com)
Coomassie stain (see REAGENT SETUP)
Coomassie destain (see REAGENT SETUP)
-
Pregnant mice or rats carrying embryonic day E16 to E18 embryos. We have successfully used cultures from C57Bl/6 mice, CD1 mice, C57Bl6/Sv129, C57B/C3H mice and Sprague Dawley rats.
CRITICAL: The procedure describes culture from 1 pregnant C57BL/6 mouse with approx.. 6–8 E16–E17 embryos.
CAUTION: All experiments must be conducted in accordance with institutional and governmental guidelines and regulations.
Borate buffer: (SEE REAGENT SETUP)
Coverslips (12 mm, round; Carolina Biological Supply, cat no. 63-3029, www.carolina.com)
Poly-d-lysine (PDL) coated coverslips in culture dish for immunofluorescence: (SEE REAGENT SETUP)
Poly-d-lysine coated culture dishes for immunoblot (SEE REAGENT SETUP)
Hibernate E (Brainbits, HE, http://www.brainbitsllc.com)
Plating media: (SEE REAGENT SETUP)
Neuronal Media: (SEE REAGENT SETUP)
HBSS solution (SEE REAGENT SETUP)
DNase: Dissolve DNase to 10 U/mL (Sigma-Aldrich, DN25) in HBSS (above), aliquot and store at −20°C.
Papain solution (SEE REAGENT SETUP)
PBS, sterile filtered
5 mg/mL α-syn PFFS (SEE PROTOCOL)
Thaw at room temperature (20–25°C) immediately before use.
10% Tx-100 in PBS (vol/vol); store at 4°C (Sigma, T8787, www.sigmaaldrich.com)
4% Paraformaldehyde/4% sucrose/PBS (SEE REAGENT SETUP) (Electron Microscopy Sciences, cat no. 15713; http://www.emsdiasum.com/microscopy)
4% Paraformaldehyde/4% sucrose/1% Triton X-100/PBS (SEE REAGENT SETUP)
10% Bovine serum albumin dissolved in PBS (wt/vol) (BSA; Sigma, cat no. A790, www sigmaaldrich.com); filter sterilize; store at 4°C
Permeabilization Buffer: (SEE REAGENT SETUP)
Blocking buffer: SEE REAGENT SETUP)
Antibodies specific for α-syn phosphorylated at Ser129. These are sold by many companies. We have found the following work well: Covance, cat no. MMS-5091; store.crpinc.com; MJFR1, Epitomics, cat no. 138501; www.epitomics.com). CRITICAL: for both immunofluorescence and immunoblotting perform a dilution series to determine minimal concentration antibody needed for signal in PFF-treated neurons and minimal signal in PBS-treated neurons. Depending on the antibody, the dilutions can range from 1:1000 up to 1:10,000.
Antibodies specific for total α-syn (Syn1, BD biosciences, cat no. 610787; www.bdbiosciences.com or Cell Signaling Technologies 4179S).
Antibodies specific for human α-syn to distinguish exogenous PFFs: LB509 (Life Technologies cat no. 18-0214, Syn204 (Abcam, www.abcam.com, cat no. ab3309).
Fluorophore conjugated secondary antibodies: Goat anti-mouse Alexa 594 and Goat anti-rabbit Alexa 488 (highly preabsorbed, Life Technologies, cat no. A11032 and cat no. A11034; www.lifetechnologies.com)
Slides
Prolong Gold mounting media (Life technologies, cat no. P36930, www.lifetechnologies.com)
BCA Protein Assay kit (Thermo Scientific, cat no. 23225; http://www.thermoscientific.com/en/products/protein-biology.html)
4–20% polyacrylamide gels (can be purchased from Biorad; 456–1093; www.bio-rad.com)
Tris buffered Saline (TBS; 50 mM Tris, 150 mM NaCl, pH7.4)
10% SDS solution in distilled water (wt/vol): (Sigma dust-free pellets,75746, www.sigmaaldrich.com)
10% Tx-100 in TBS (vol/vol)
Protease Inhibitors (See REAGENT SETUP)
Phosphatase inhibitors (See REAGENT SETUP)
Ice cold 1% Tx-100 in TBS with protease and phosphatase inhibitors (SEE REAGENT SETUP) (make day of use)
Room temperature 2% SDS in TBS with protease and phosphatase inhibitors (SEE REAGENT SETUP) (make day of use)
Pierce BCA protein assay reagents (Thermoscientific 23227; www.piercenet.com)
SDS page running buffer (SEE REAGENT SETUP)
Transfer buffer (SEE REAGENT SETUP)
0.2 µm nitrocellulose membrane (Bio-Rad, cat no. 162-0112) CRITICAL: this pore size is important because of the small size of α-syn.
TBS/5% milk/0.2% Tween-20 (vol/vol) (Sigma, cat no. P9416, www.sigmaaldrich.com)
Goat anti-mouse HRP conjugated secondary antibody (Jackson Immunoresearch, cat no. 115-035-062 and cat no. 111-035-144; www.jacksonimmuno.com)
Enhanced chemiluminescence (ECL) substrate (Thermoscientific, cat no. PI-32109, www.fishersci.com)
Developer (Fuji Systems, Tokyo, Japan)
Box 1: Purification of α-syn.
The methods for purifying α-syn have been published in detail in several publications35–39. Here, we describe our protocol for purifying human full length α-syn (NM_000345) or mouse full length α-syn (NM_001042451). The final goal is to obtain a high concentration of untagged α-synuclein in a final buffer composed of Tris and NaCl.
Expression in e.coli
-
1)
Clone α-Syn into the ampicillin resistant bacterial expression vector, pRK172. Expression of α-syn from this plasmid does not require induction by IPTG and so you will likely have to modify your protocol if using another vector.
-
2)
Tranform plasmids into BL21(DE3)RIL competent E coli (Life Technologies, cat. No. 230245). Transfer a single colony to 4 mL of SOC medium and incubate 1–2 hrs at 37°C with shaking to create a starter culture.
-
3)
Add 0.5 mL of starter culture to each of 2 baffled 4L flasks with 500 mL of Terrific Broth (TB: 12g/L bacto-tryptone, 24g/L yeast extract 4% vol/vol glycerol; 17 mM KH2PO4 and 72mM K2HPO4) with ampicillin. Incubate overnight at 37°C with shaking.
Cell lysis and preclearing
-
4)
Spin bacteria for 10 minutes at 6000 ×g.
-
5)
Resuspend pellet in high salt buffer (750 mM NaCl, 10 mM Tris, pH 7.6, 1 mM EDTA) with protease inhibitors and 1 mM PMSF (50 mL for 1L of culture).
-
6)
Sonicate with at least a 0.25 inch probe tip at 60% power, for a total time of 5 min (30sec pulse on, 30 sec pulse off).
-
7)
Boil for 15 min to precipitate unwanted proteins. Cool on ice.
-
8)
Spin at 6000 ×g for 20 minutes.
-
9)
Dialyze supernatant with 10 mM Tris, pH 7.6, 50 mM NaCl, 1mM EDTA.
Purification
-
10)
Concentrate protein with 3.5K molecular weight cut-off Amicon Ultra Centrifuge filter devices (Millipore, UFC901008).
-
11)
Filter protein through a 0.22 µm syringe filter and load onto a Superdex 200 column (GE Healthcare Life Sciences).
-
12)
Collect fractions (about 30 fractions at 1 mL each). Combine 10 µL of each fraction with 10 µL of 2X laemmli buffer and analyze fractions by polyacrylamide gel electrophoresis with 4–20% gradient gels, followed by coomassie staining/destaining.
-
13)
Choose pure fractions with bands corresponding to α-syn which runs slightly above the 15 kDa marker, collect, and dialyze in 4L of 10 mM Tris, pH 7.6, 25 mM NaCl, 1mM EDTA.
-
14)
Apply protein to a Hi Trap Q HP anion exchange column (GE Healthcare Life Sciences) and run a linear gradient ranging from 25 mM NaCl to 1M NaCl. α-syn is eluted at approximately 300 mM NaCl. Again, collect fractions (about 50 fractions at 2 mL each). Combine 10 µL of each fraction with 10 µL of 2X laemmli buffer and analyze fractions by polyacrylamide gel electrophoresis with 4–20% gradient gels, followed by coomassie staining/destaining.
-
15)
Dialyze with 10 mM Tris, pH 7.6 and 50 mM NaCl.
-
16)
Concentrate fractions to approximately 30 mg/mL, aliquot and store at −80°C. Yield should be approximately 30 mg per 1 L culture.
Equipment
Pipets: 10 µL, 20 mL, 200 µL, 1000 µL
Sterile filtered pipet tips
Sterile serological pipets
Pipettor
−80 degree freezer
Benchtop centrifuge for 1.5 mL tubes (up to 15,000 × g)
Sterile (autoclaved) 1.5 mL microcentrifuge tubes
Standard lab incubator set at 37°C
Thermomixer with block for 1.5 mL tubes (Eppendorf, cat no. 5384 000.012; www.eppendorf.com) placed inside standard lab incubator
Benchtop ultracentrifuge up to at least 100,000 × g (Beckman Coulter cat no. A47882; www.beckmancoulter.com)
Beckman Coulter TLA-55 Fixed angle rotor (Beckman Coulter cat no. 366725; www.beckmancoulter.com)
Polyallomer tubes, snap-on cap, 1.5 mL for TLA-55 rotor (Beckman Coulter cat no. 357448; www.beckmancoulter.com)
Protein electrophoresis system (Biorad cat no. 165-8005; www.bio-rad.com)
Trans-blot electrophoretic transfer cell (Biorad cat no. 170-3930; www.bio-rad.com) or similar item
Fume hood
BSL-2 culture hoods
Tissue culture CO2 incubator designated for primary cultures
-
Sonic Dismembrator System with 0.16 inch microtip and 0.25 inch tip (Fisher Scientific cat no. FB120110, www.fishersci.com)
CRITICAL: a sonicator with a microtip such as this one cannot be substituted with a water bath sonicator. This protocol is dependent on the sonication step.
Cell scraper
95°C heat block
37°C water bath
Epifluorescent widefield microscope with filter sets for DAPI, FITC, TRITC
Reagent setup
Borate Buffer: 50 mM boric acid (Sigma, B6768) in ddH2O; adjust pH to 8.5 and filter sterilize. Can be stored indefinitely at room temperature.
PBS: 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM K2HPO4, pH to 7.4 with HCl, filter sterilize. Can be stored indefinitely at room temperature.
Laemmli buffer: 4X: 252 mM Tris-HCL (pH6.8), 8% SDS (vol/vol), 40% glycerol (vol/vol), 0.002% wt/vol bromophenol blue. Can be stored indefinitely at room temperature. Add fresh 0.4% 2-mercaptoethanol before using.
Laemmli buffer: 2X: Dilute 4X laemmli buffer 1:1 with water.
Coomassie stain: 0.2% Coomassie Brilliant Blue R250 (wt/vol), 50% methanol (vol/vol), dissolve dye, then add 10% acetic acid (vol/vol), and bring to final volume with water. Can be stored indefinitely at room temperature.
Coomassie destain: 50% methanol (vol/vol), 10% acetic acid (vol/vol), and bring to final volume with water. Can be stored indefinitely at room temperature.
Plating media: Neurobasal media (Invitrogen, 21103-049), 1X B27 from a 50X stock (Invitrogen, 17504-044), 1X Glutamax from a 100X stock (Invitrogen, 35050-061), 50 U ml−1 penicillin and 50 µg ml−1 streptomycin (1:200 dilution from a 100X stock, Invitrogen, 15140-122), 10% fetal bovine serum. Filter sterilize and store at 4°C for no longer than 3 weeks.
Neuronal Media: Neurobasal media (Invitrogen, 21103-049), 1X B27 from a 50X stock (Invitrogen, 17504-044), 1X Glutamax from a 100X stock (Invitrogen, 35050-061), 50 U ml−1 penicillin and 50 µg ml−1 streptomycin (1:200 dilution from a 100X stock, Invitrogen, 15140-122). Filter sterilize and store at 4°C for no longer than 3 weeks.
HBSS: To HBSS (Invitrogen, 14170-161), add 6 mL of 1M Hepes pH 7.4 (Invitrogen, 15630-106), 3 mL of 50 U ml−1 penicillin and 50 µg ml−1 streptomycin (Invitrogen, 15140-122), 6 mL of 100 mM Pyruvic acid (Invitrogen, 11360-070). Bring to 500 mL with remainder of HBSS, add100 mL of water (final volume 600 mL). Filter sterilize and store at 4°C for no longer than 3 weeks.
Papain solution: Make fresh before starting dissection. For tissue from 6–8 pups, make 10 mL. Add papain (Worthington Biochemical Corporation, LS 3126, http://www.worthington-biochem.com) to HBSS (above) to final concentration of 20 U/mL, 2 mg L-Cysteine (Sigma-Aldrich, C-7352, 22 µL of 500 mM EDTA, pH 8.0. Incubate with shaking at room temperature to allow papain to dissolve.
-
Poly-d-lysine (PDL) coated coverslips in culture dish for immunofluorescence.
Dissolve PDL (Sigma, cat no. P0899, www.sigmaaldrich.com) at 2mg/mL in Borate buffer. Filter sterilize. Make 2 mL aliquots in polystyrene tubes and freeze at −20 °C. Can be stored indefinitely.
-
Poly-d-lysine coated culture dishes for immunoblot
Place coverslips in 95%EtOH.Put single coverslips in individual wells of 24 well plate and rinse 2X with 0.5 mL sterile water. Alternatively, multiple coverslips can be placed in a 6 well or 6cm dish. Coat coverslips with 0.1mg/mL poly-d-lys for 1.5 h (or overnight) at 37°C (ensure that coverslips are completely covered with solution). Rinse 2X with water followed by neuronal plating media. Store in 37°C tissue culture incubator in plating media for no longer than one week. Prior to use, add sterile water in between wells to maintain humidity and prevent media evaporation.-
―Coat wells of 12 well, 6 well or 6 cm dishes as above for coverslips.
-
―
- 4% Paraformaldehyde/4% sucrose/PBS
-
―For 50 mL in PBS, pipet 10 mL of 20% paraformaldehyde (Electron Microscopy Sciences, 15713; http://www.emsdiasum.com/microscopy) into about 40 mL PBS, add 2g sucrose and mix until it dissolves. Bring volume to 50 mL with PBS. Best if used at room temperature, although others report optimal results when warmed at 37°C. Can be stored at 4°C, protected from light, for one week.
-
―
- 4% Paraformaldehyde/4% sucrose/1% Tx-100/PBS
-
―make 4% Paraformaldehyde/4% sucrose as above but add 1 mL of 10%Tx-100/PBS and then bring to final volume using PBS. Can be stored at 4°C, protected from light, for one week.
-
―
Permeabilization Buffer: 3%BSA, 0.1% Tx-100 in PBS: For 10 mL: 3 mL of 10% BSA/PBS, 100 µL of 10% Tx-100/PBS bring to 10 mL with PBS. Make fresh.
Blocking Buffer: 3% BSA in PBS: for 10 mL: 3 mL of 10% BSA/PBS, bring to 10 mL with PBS. Make fresh.
Dilute primary and secondary antibodies in blocking buffer-dilute day of use
Tris buffered Saline (TBS; 50 mM Tris, 150 mM NaCl, pH7.4). Can be stored indefinitely at room temperature.
Protease inhibitors: (1000X stock: 1 mg/mL pepstatin, 1 mg/mL Leupeptin, 1 mg/mL TPCK, 1 mg/mL TLCK, 1 mg/mL trypsin inhibitor, 0.1M EDTA; all from Sigma); make aliquots and store at −20°C.
Phosphatase inhibitors (100X stock: 200 mM Imidazole, 100 mM NaF, 100 mM NaOrthovanadate), make aliquots and store at −20°C.
Ice cold 1% Tx-100 in TBS with protease and phosphatase inhibitors (make day of use): For 10 mL, 1mL of 10% Tx-100 in TBS, bring to 10 mL with TBS, dilute protease inhibitors 1:1000, dilute phosphatase inhibitors 1:100, vortex, keep on iceRoom temperature 2% SDS in TBS with protease and phosphatase inhibitors, for 1 mL, 200 µL of 10% SDA, 800 µL of TBS, dilute protease inhibitors 1:1000, dilute phosphatase inhibitors 1:100, vortex, keep at room temperature to prevent precipitation of SDS, make day of use
SDS page running buffer: for 1X concentration 25 mM Tris, 192 mM glycine, 0.1% SDS, bring to final volume with water. Can be stored indefinitely at room temperature.
Transfer buffer: 25 mM Tris, 192 mM glycine, 10% methanol (vol/vol), bring to final volume with water. Can be stored in a bottle indefinitely at 4°C.
PROCEDURE
CAUTION: Appropriate personal protective equipment should be used at all times. WEAR GLOVES, LAB COAT, FACE MASK (such as: VWR, 414004-670), AND PROTECTIVE GOGGLES and PERFORM SONICATION IN A CLASS II BIOSAFETY CABINET TO AVOID INHALING OR GETTING AEROSOLIZED PFFS IN EYES (Video 1). Although studies have not performed on the safety of handling of PFFs, we recommend referring to the prion literature for the utmost safety precautions40, 41. Decontaminate PFFs or any spills with a final concentration of 1 N NaOH or 20,000 ppm bleach (two parts 5.25% household bleach to three parts water) for 1 hour. Make NaOH or bleach solutions fresh daily. Use disposable, absorbent pads whenever possible.
Preparation of PFFs
Timing: 7 days
-
1
Rapidly thaw purified α-syn monomer in 1.5 mL microcentrifuge tube in 37°C water bath until small ice cube visible. Place on ice until completely thawed
-
2
Centrifuge at 100,000 × g at 4°C for 60 minutes to pellet any aggregated material. Remove the supernatant to use to generate PFFs
-
3
Dilute supernatant from step 2 in PBS in new, sterile 1.5 mL microcentrifuge tube to final volume of 500 µL and final concentration of 5mg/mL.
-
4
Place in Thermomixer that is set inside a standard lab 37°C incubator (this helps prevent condensation which could affect fibril concentration)
-
5
Shake for 7 days at 1000 RPM. Should appear turbid i.e. slightly cloudy after the 7 days.
-
6
Aliquot (10–20 µL) into sterile microcentrifuge tubes and store at −80.
CRITICAL: Store at −80 and not higher temperatures. Do not keep PFFs on ice or at 4°C as this causes dissociation and reduces activity.
PAUSEPOINT: We have stored our PFFs at −80 for up to one year.
Sedimentation assay for confirmation of pelletable, active PFFs
-
7
Pipet 20 µL of PFFs into polyallomer tube for ultracentrifuge.
-
8
Centrifuge at 100,000 × g for 60 minutes
-
9
Carefully remove supernatant (20 µL) and transfer to a new tube. Add 20 µL of 2X Laemmli buffer.
-
10
Suspend pellet in 20 µL of PBS, centrifuge again at 100,000 × g for 30 minutes. Discard supernatant.
-
11
Suspend pellet in 20 µL PBS, add 20 µL 2X Laemmli buffer to this fraction.
-
12
Load 15 µL of supernatant from step 9 and pellet from step 11 onto a 4–20% acrylamide gel with a 15 well comb and perform SDS-PAGE. Coomassie stain and destain. There should be approximately equivalent amounts of α-syn (typically runs above 15 kDa marker) in the supernatant and pellet (Figure 3), or more in the pellet. If there is more α-syn in the supernatant, than the pellet, the PFFs will be less efficient. If this is the case, check the pH of the PBS, as we found this to be critical in PFF formation.
Figure 3. Typical results seen at step 12 following the sedimentation assay of PFFs.

PFFs were prepared as described in steps 1–12 and the supernatant and pellet resolved by SDS-PAGE and stained with Coomassie Blue. This batch of PFFs had approximately equivalent amounts of α-syn in the supernatant and pellet was efficient at seeding aggregates from endogenous α-syn in primary neurons.
Culturing neurons
Timing: About 8 hours
-
13
Calculate amount of plating media and neuronal media needed (20 mL for rinses, 0.5 mL for each well of 24 well dish, 2 mL for each well of 6 well dish). Pipet plating media and neuronal media into separate T75 sterile tissue culture flask, label flasks clearly, and place in tissue culture C02 incubator for 2–24 hours. This step will equilibrate the pH of the media, it should be slightly orange/pink in color.
-
14
Dissect hippocampi as previously demonstrated using cold Hibernate E27, 28.
-
15
Place dissected tissue in 15 mL Falcon tube with 10 mL of Hibernate E on ice.
PAUSEPOINT Tissue is stable on ice for up to 8 hours. We have kept tissue on ice for 24 hours, and successfully obtained viable neurons, but more cell death was observed.
-
16
Rinse 2X with HBSS. Remove final HBSS (can leave around 1 mL).
-
17
Filter sterilize Papain solution using 0.20 µm syringe filter (Fisher Scientific, SLLG025SS) and add 10 mL to tissue. Incubate in tissue culture incubator for 30–60 min.
-
18
Place tube with tissue in culture hood, allow tissue to sink to bottom of tube. Carefully remove papain solution (tissue is very sticky).
-
19
Add 50 µL DNase solution to 10 mL plating solution. Add this to tissue. The tissue will now easily sink to bottom of tube. Rinse one more time with plating media.
-
20
Rinse 2X with HBSS. Remove final HBSS so that around 1 mL remains.
-
21
Triturate tissue 10X.
-
22
Add 5 mL HBSS and filter through 40 mm nylon mesh cell strainer (Fisher Scientific, 22363547). Perform cell count
-
23
Plate neurons as follows and place in tissue culture incubator
For immunofluorescence with coverslips:Culture Vessel Number of neurons plated 24 well 100,000 cells/well 6 well 0.5 ×10^6 cells/well 6 cm 1×10^6 cells/well For biochemistry/immunoblotting:Culture Vessel Number of neurons plated 6 well 1 ×10^6 cells/well 6 cm 2 ×10^6 cells/dish -
24
Between 2 and 18 hours after plating, completely change plating media to neuronal media.. We use slightly more media (1 mL for 24 well, 2.5 mL for 6 well) so that it can be collected later as “conditioned media”.
-
25
Continue to culture neurons. If culturing for over a week, remove 50% of the media after 7 days, and add the equivalent volume of fresh neuronal media (pre-equilibrated as described above). We have found the neurons are healthier with fewer media changes and can last for up to 31 days after plating.
Adding PFFs to neurons
Timing: about 60 min
CRITICAL Perform 5–10 days after plating neurons
-
26
When neurons are 5–10 days in vitro (DIV 5–10) add PFFs. When PFFs are added at later time points, aggregate formation occurs more quickly14.
-
27
Warm neuronal media in a 37 °C water bath.
-
28
Thaw aliquot of 5 mg/mL PFFs at room temperature immediately before use.
CRITICAL STEP Perform all remaining steps at room temperature. Placing fibrils on ice causes their dissociation into monomer33.
-
29
Add PFFs to sterile PBS to final concentration of 0.1 mg/mL e.g. 10 L in 490 µL PBS or 20 mL in 980 µL PBS. The minimal volume that can be used is 200 µL.
-
30
Sonicate with 60 pulses at 10% power (total of 30 sec, 0.5 sec on, 0.5 sec off)
CRITICAL STEP: Sonication with a micro probe tip sonicator is crucial (Video 1, Figure 2).
-
31
Dilute sonicated PFFs or an equivalent volume of PBS as a control, into pre-warmed neuronal media as follows:
For Immunofluorescence/Imaging:Culture Vessel Volume media µL PFFs (or PBS) to add to media 24 well 0.5 mL/well 5 (1:100 dilution) 6 well 2 mL/well 20 (1:100 dilution) 6 cm 4 mL/dish 40 (1:100 dilution) For Biochemistry/Immunoblotting:Culture Vessel Volume media µL PFFs (or PBS) to add to media 6 well 2 mL/well 40 (1:25 dilution) 6 cm 4 mL/dish 80 (1:25 dilution) -
32
Aspirate approximately 80% of media from the well of the neurons from step 26 and add the appropriate volume of media and PFFs (determined by tables in previous step). Only aspirate 3–6 wells at a time.
-
33
Incubate neurons for a further 7–23 days. Change ~50% of media once a week. The fresh media does not need to contain more fibrils.
Confirmation of PFF transduction and seeding
Takes 2–3 days. CRITICAL Perform 7–23 days after transduction
PFF transduction and seeding can be confirmed by immunofluorescence (option A) or sequential extraction and immunoblotting (option B). Abnormal α-syn derived from endogenous α-syn can be detected via immunofluorescence with a p-α-syn specific antibody. These phosphorylated inclusions will not be visible when PFFs are added to α-syn knockout neurons (Figure 4). Alternatively, the neurons can be fixed with 4%paraformaldehyde/4%sucrose/1% Tx-100 and stained with an antibody to total α-syn. The normal, “synaptic” α-syn will be extracted and will not be visible, but the PFF-induced α-syn inclusions will not be extracted and will be visible by immunofluorescence (Figure 5).To distinguish exogenously added human PFFs from inclusions formed from endogenous α-syn, neurons can be co-stained using an antibody that is specific for human α-syn (LB509 or Syn204; both raised in mouse) and an antibody for p-α-syn (MJFR1, raised in rabbit) (Figure 6).
Figure 4. Visualization of PFF-induced formation of α-syn aggregates using an antibody to p-α-syn.

A. Fourteen days following the addition of PFFs to nontransgenic primary neurons, p-α-syn inclusions, visualized by immunofluorescence using mAB81A, were abundant throughout neurites and soma (top panels)14. These inclusions were also insoluble as determined by fixation in paraformaldehyde/4% sucrose/1% Tx (top, right). p-α-Syn inclusions were not apparent in PBS or PFF treated (14 days post-treatment) in primary neurons from α-syn knockout mice (bottom panels). Scale bar = 50 µm. B. PFFs (top panels) or monomeric, non-fibrillar α-syn (bottom panels) were added to primary neurons. Immunofluorescent imaging was performed using antibodies MJFR1, which recognizes p-α-syn, and NeuN, to visualize neuronal soma. Inclusions positive for p-α-syn (green) were abundant in PFF-treated neurons, but not neurons treated with soluble, monomeric α-syn. Scale bar = 20 µm.
Figure 5. Immunofluorescence of α -syn aggregates.

A. Neurons were fixed in 4% paraformaldehyde/4% sucrose and immunofluorescence was performed using an antibody to total α-syn (Syn1) and an antibody which recognizes the presynaptic marker, VAMP2
In control PBS-treated neurons, α-syn colocalized extensively with VAMP2 at presynaptic terminals. Fourteen days following PFF treatment, α-syn no longer localized to the presynaptic terminal, but was found in longer serpentine aggregates in axons and skein-like filaments in the soma. Scale bar = 10 µm. B. Neurons were fixed in 4% paraformaldehyde/4% sucrose/1% Tx-100 and immunofluorescence was performed using an antibody to total α-syn and an antibody which recognizes the neuron specific marker, NeuN. In control neurons, α-syn was completely extractable but in PFF treated neurons (here, 10 days post), α-syn was insoluble and thus visible throughout the culture. Scale bar = 50 µm. C. 4% paraformaldehyde/4% sucrose/1% Tx-100 and co-stained using an antibody to total α-syn and NeuN. Scale bar = 10 µm. D. Seven days following PFF treatment, neurons were fixed with 4% paraformaldehyde/4% sucrose and co-stained with a p- α-syn (red) and tau (green) antibody to label axons.Scale bar = 10 µm.
Figure 6. Visualization of exogenously added PFFs and p-α-syn aggregates formed from endogenous α-syn.
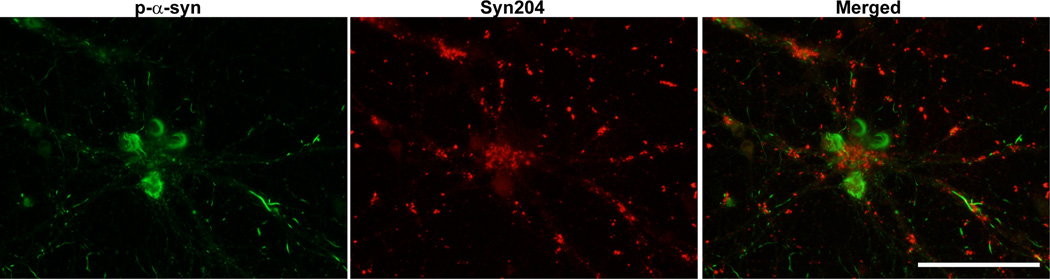
Neurons were fixed 14 days after treatment with PFFs and double immunofluorescence was performed using a rabbit antibody to p-α-syn generated in the Lee lab (green)14, and a mouse antibody, Syn204, that specifically recognizes the exogenously added human PFFs (red). There was minimal colocalization between the exogenous PFFs and the p-α-syn inclusions derived from endogenous α-syn. Scale bar = 50 µm.
- Immunofluorescence
- Aspirate media from coverslips (aspirate from only a few coverslips at a time)
- Add either 4%paraformaldehyde/4%sucrose (0.5mL for 24 well plate) or 4%paraformaldehyde/4%sucrose/1% Tx-100 (0.5mL for 24 well plate), called “Tx-100 extracted neurons”.
- Incubate room temperature for 15 minutes. CRITICAL STEP Unless otherwise noted, this and all subsequent steps should be performed at room temperature.
- Rinse 5X with 0.5 mL PBS/well
- Permeabilize and block neurons with 3%BSA/0.1% Tx-100 for 15 minutes.
- Rinse 3X with PBS
- Dilute antibody in blocking buffer. We prefer to perform double staining immunofluorescence using 2 antibodies such as LB509/p-α-syn (as described above) or an α-syn antibody with MAP2 or tau to help resolve the spatial relationship of inclusions within dendrites or axons, respectively, or combined with an antibody to any other marker of interest. Incubate neurons in primary antibody for 2 hours at room temperature or 4°C overnight.
- Rinse 5X with PBS
- Dilute goat anti-mouse Alexafluor conjugated secondary antibody and/or goat anti-rabbit Alexafluor conjugated in blocking buffer 1:500 (Life Technologies). (The choice of Alexafluor depends on the microscope filters or lasers, if using confocal microscopy). Incubate neurons in secondary antibody for 1 hour.
- Rinse 5X with PBS
- Mount coverslips onto glass slides with Prolong Gold mounting media. Visualize pathology with epifluorescent microscope.
- Sequential Extraction and Immunoblotting
- Rinse neurons 2X with PBS
-
Place dish on ice. Working one well at a time, completely aspirate PBS and add following volumes of ice cold 1%Tx-100/TBS with protease and phosphatase inhibitors:6 well: 250 µL per well6 cm dish: 500 µL
- Use cell scraper to thoroughly scrape all neurons from well
- Place in polyallomar tube for table top ultracentrifuge, keep on ice
- Sonicate 10X, 0.5 sec pulse, power at 10%
- Incubate on ice for 30 minutes.
- Centrifuge at 100,000 × g at 4°C for 30 minutes.
- Add 4X Laemmli to about 150–200 µL of Tx-100 supernatant (some is usually lost during steps iii-viii, we recommend measuring remaining supernatant before determining how much to remove for laemmli buffer). Save about 20 µL of supernatant for protein assay. Retain on ice or in −20°C freezer.
-
To the pellet, add the same volume of ice cold 1% Tx-100 with protease and phosphatase inhibitors6 well: 250 µL6 cm dish: 500 µL
- Sonicate 10X, 0.5 sec pulse, power 10%. Keep tip of probe toward bottom of tube to prevent frothing. Make sure that pellet is completely dispersed.
- Centrifuge at 100,000 × g at 4°C for 30 minutes.
- Discard supernatant.
- Add 2% SDS/TBS to pellet with protease and phosphatases inhibitors. To a 6 well plate add 125 µL. To a 6 cm dish add 250 µL.
- Sonicate 15X, 0.5 sec pulse, power 10%. Keep tip of probe toward bottom of tube. Make sure that pellet is completely dispersed.
- Remove supernatant and place into new microcentrifuge tube.
- Perform BCA/protein assay on Tx-100 supernatant and SDS extract. Typically a 1:5 dilution for the BCA assay is sufficient.
- Dilute 2% SDS extract from step Xv into Laemmli buffer to 2X volume for corresponding Tx-100 fraction (regardless of protein concentration of SDS fraction). For example, if you have 180 µL Tx-100 extract (from step viii) at 1 mg/mL and 90 µL of SDS extract, add 60 µL of 4X Laemmli to the Tx-100 extract and 30 µL of 4X Laemmli to the SDS extract. Load 12.5 µL of both the Tx-100 extract (10 µg) and SDS extract. We suggest 2X volume because it makes the insoluble α-syn species more abundant and thus easier to visualize and quantify by immunoblot.
- Load samples on 4–20% gel and run according to manufacturer’s directions. We use 85V constant voltage until the dye front runs off the gel. Be sure not to let the 10 kDa marker to run off the gel.
- Transfer following manufacturers instructions at 100 V for 75 minutes or overnight at 40V.
- Block membrane for 1 hour with TBS/5% milk
- Dilute primary antibodies in TBS/5% milk and incubate overnight at 4°C with shaking.
- Rinse 3X with TBS/T, 10 minutes each rinse.
- Incubate with HRP conjugated secondary antibodies for 1 hour at room temperature.
- Rinse 4X with TBS/T, 10 minutes each rinse.
- Develop with ECL
Anticipated results
Immunofluorescence
Significant α-syn pathology is detectable 4–10 days following addition of PFFs to primary hippocampal neurons from non-transgenic, wildtype mice using this protocol. When immunofluorescence is performed with an antibody to total α-syn, α-syn will localize to presynaptic puncta in control neurons, and to longer serpentine aggregates that do not colocalize with presynaptic markers in PFF-treated neurons (Figure 4,5). Note that some presynaptic a-syn that has not converted to aggregates will remain, especially at early time points after PFF addition. This is why we recommend also performing Tx-100 extraction. In neurons fixed with 4% paraformaldehyde/4% sucrose/1% Tx-100, there will be no staining for α-syn in the control neurons, but the aggregates will remain in Tx-100 extracted PFF-treated neurons. When immunofluorescence is performed using antibodies to p-α-syn, the control neurons will show light, diffuse staining. However the staining in the PFF-treated neurons will be substantially more intense and discrete puncta and longer serpentine and skein-like aggregates will be apparent.
Immunoblots
When probed using an antibody against total α-syn, PBS-treated control neurons will show a band slightly above the 15 kDa band corresponding to monomeric α-syn exclusively in the Tx-100 soluble fraction (Figure 7). Following 14 days treatment with PFFs, neuronal lysates will show reduced levels of α-syn in the Tx-100 soluble fraction relative to control neuronal lysates, and an appearance of α-syn in the SDS-soluble fraction. These bands typically appear as a series of bands within the SDS fraction, likely corresponding to α-syn oligomers. PBS-treated control neurons will not show p-α-syn immunoreactivity in the Tx-100 or the SDS fraction. PFF-treated neurons will show pathologic p-α-syn immunoreactivity predominately in the SDS soluble fraction that again will appear as a series of bands.
Figure 7. Typical immunoblot results seen following sequential extraction of neurons in 1% Tx-100 followed by SDS.
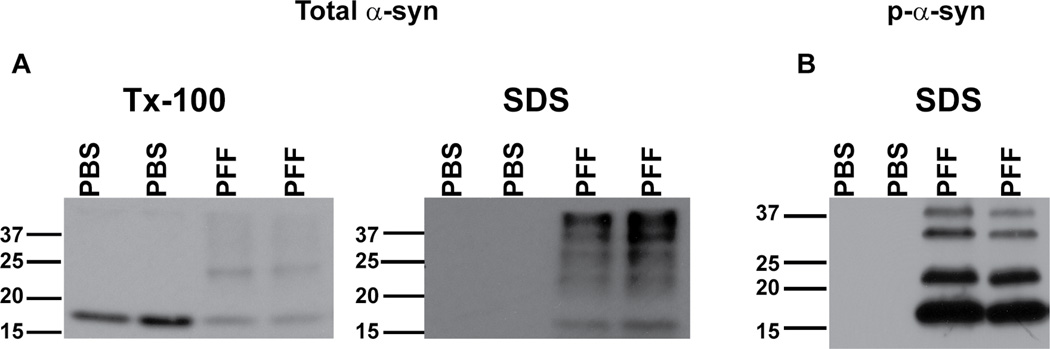
A. Immunoblotting was performed using Syn1, an antibody that recognizes total α-syn. In control neurons, α-syn was completely extractable in Tx-100. In PFF treated neurons, there was a decrease in Tx-100 soluble α-syn, and increase in SDS-soluble α-syn. B. Immunoblots of SDS extracts (Tx-100 insoluble) using mAB81a that recognizes p-α-syn. There was no p-α-syn in control neurons but a substantial amount of p-α-syn in PFF treated neurons.
Troubleshooting
Preparation of PFFs
Preparation of PFFs from recombinant, purified protein without tags is the most effective. Generating PFFs from α-syn purchased from various suppliers has not been successful in our hands, likely because many suppliers utilize a tag to assist in purification. The composition of the buffers used to purify α-syn and generate PFFs, including salt concentrations and pH, is also crucial.
Inclusion formation
The density of the neurons has been optimized for efficient inclusion formation. Using lower neuronal densities decreases the abundance of inclusions. Also, as mentioned throughout the protocol, sonication with a probe tip sonicator is critical for this protocol. Investigators who have tried bath sonicators for PFF sonication have not been as successful in generating abundant inclusions. Note that PFFs may lose activity over multiple freeze thaw cycles. Verify that PFFs were diluted and stored correctly prior to addition to neurons. In addition, our system has been optimized for primary hippocampal neurons from C57Bl/6 mice or CD1 mice. Some neuronal populations may be more or less sensitive to the effects of the PFFs and therefore it may be necessary to perform a time course and a concentration curve after PFF addition to determine the time point and concentration at which pathology is maximal.
Immunoblots
p-α-Syn can sometimes be difficult to detect by immunoblot. If this is difficult, we recommend loading as much protein as possible (≥10µg). Using BSA in the blocking buffer instead of milk may also be helpful. Using lysates from α-syn knockout mice is also recommended as a control to distinguish specific bands.
Supplementary Material
This video demonstrates sonication PFFs using a probe tip sonicator, a critical step for the success of this protocol.
Summary.
Lewy bodies and Lewy neurites are found in the brains of patients with Parkinson’s disease and other synucleinopathies. This protocol describes how to induce α-synuclein aggregates in a primary neuronal culture system, creating a cellular model of these synucleinopathies.
Acknowledgements
We would like to thank Andrew B. West for allowing us to create of video of him demonstrating the sonication of the fibrils. We also thank the reviewers whose careful attention to details and suggestions greatly improved this protocol. This study was supported by NIH grant, P50 NS053488 to V.M.-Y.L.
Footnotes
Author Contributions: L.V.D. carried out the experiments that formed the basis of the protocol. K.C.L. provided the electron microscopy images of the sonicated fibrils. V.M.-Y.L. supervised the project. L.V.D., K.L. and V.M.-Y.L. provided intellectual input that contributed to the development of the protocol. L.V.D. wrote the paper, and K.L. and V.M.-Y.L. provided valuable editorial input.
Competing Financial Interests: The authors report no competing financial interests
Contributor Information
Kelvin C. Luk, Email: kelvincl@mail.med.upenn.edu.
Virginia M.-Y. Lee, Email: vmylee@upenn.edu.
References
- 1.McNaught KS, Shashidharan P, Perl DP, Jenner P, Olanow CW. Aggresome-related biogenesis of Lewy bodies. Eur J Neurosci. 2002;16:2136–2148. doi: 10.1046/j.1460-9568.2002.02301.x. [DOI] [PubMed] [Google Scholar]
- 2.Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- 3.Kramer ML, Schulz-Schaeffer WJ. Presynaptic alpha-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J Neurosci. 2007;27:1405–1410. doi: 10.1523/JNEUROSCI.4564-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luk KC, Lee VM. Modeling Lewy pathology propagation in Parkinson's disease. Parkinsonism Relat Disord. 2014;20(Suppl 1):S85–S87. doi: 10.1016/S1353-8020(13)70022-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bendor JT, Logan TP, Edwards RH. The function of alpha-synuclein. Neuron. 2013;79:1044–1066. doi: 10.1016/j.neuron.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaufman SK, Diamond MI. Prion-like propagation of protein aggregation and related therapeutic strategies. Neurotherapeutics. 2013;10:371–382. doi: 10.1007/s13311-013-0196-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.George S, Rey NL, Reichenbach N, Steiner JA, Brundin P. alpha-Synuclein: the long distance runner. Brain Pathol. 2013;23:350–357. doi: 10.1111/bpa.12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Masliah E, et al. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 9.Giasson BI, et al. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron. 2002;34:521–533. doi: 10.1016/s0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- 10.Lee MK, et al. Human alpha-synuclein-harboring familial Parkinson's disease-linked Ala-53 -->Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc Natl Acad Sci U S A. 2002;99:8968–8973. doi: 10.1073/pnas.132197599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spillantini MG, et al. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson's disease and dementia with Lewy bodies. Neurosci Lett. 1998;251:205–208. doi: 10.1016/s0304-3940(98)00504-7. [DOI] [PubMed] [Google Scholar]
- 12.Baba M, et al. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am J Pathol. 1998;152:879–884. [PMC free article] [PubMed] [Google Scholar]
- 13.Fujiwara H, et al. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- 14.Volpicelli-Daley LA, et al. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Desplats P, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009;106:13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284:12845–12852. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luk KC, et al. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A. 2009;106:20051–20056. doi: 10.1073/pnas.0908005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ren PH, et al. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol. 2009;11:219–225. doi: 10.1038/ncb1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tanik SA, Schultheiss CE, Volpicelli-Daley LA, Brunden KR, Lee VM. Lewy body-like alpha-synuclein aggregates resist degradation and impair macroautophagy. J Biol Chem. 2013;288:15194–15210. doi: 10.1074/jbc.M113.457408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dryanovski DI, et al. Calcium entry and alpha-synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. J Neurosci. 2013;33:10154–10164. doi: 10.1523/JNEUROSCI.5311-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taguchi K, et al. Differential expression of alpha-synuclein in hippocampal neurons. PLoS One. 2014;9:e89327. doi: 10.1371/journal.pone.0089327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buerli T, et al. Efficient transfection of DNA or shRNA vectors into neurons using magnetofection. Nat Protoc. 2007;2:3090–3101. doi: 10.1038/nprot.2007.445. [DOI] [PubMed] [Google Scholar]
- 23.Zeitelhofer M, et al. High-efficiency transfection of mammalian neurons via nucleofection. Nat Protoc. 2007;2:1692–1704. doi: 10.1038/nprot.2007.226. [DOI] [PubMed] [Google Scholar]
- 24.Jiang M, Chen G. High Ca2+-phosphate transfection efficiency in low-density neuronal cultures. Nat Protoc. 2006;1:695–700. doi: 10.1038/nprot.2006.86. [DOI] [PubMed] [Google Scholar]
- 25.Campenot RB, Lund K, Mok SA. Production of compartmented cultures of rat sympathetic neurons. Nat Protoc. 2009;4:1869–1887. doi: 10.1038/nprot.2009.210. [DOI] [PubMed] [Google Scholar]
- 26.Luk KC, et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beaudoin GM, 3rd, et al. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat Protoc. 2012;7:1741–1754. doi: 10.1038/nprot.2012.099. [DOI] [PubMed] [Google Scholar]
- 28.Seibenhener ML, Wooten MW. Isolation and culture of hippocampal neurons from prenatal mice. J Vis Exp. 2012 doi: 10.3791/3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Irwin DJ, et al. Neuropathologic substrates of Parkinson disease dementia. Ann Neurol. 2012;72:587–598. doi: 10.1002/ana.23659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Churchyard A, Lees AJ. The relationship between dementia and direct involvement of the hippocampus and amygdala in Parkinson's disease. Neurology. 1997;49:1570–1576. doi: 10.1212/wnl.49.6.1570. [DOI] [PubMed] [Google Scholar]
- 31.Braak H, et al. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 32.Banker GA. Trophic interactions between astroglial cells and hippocampal neurons in culture. Science. 1980;209:809–810. doi: 10.1126/science.7403847. [DOI] [PubMed] [Google Scholar]
- 33.Bousset L, et al. Structural and functional characterization of two alpha-synuclein strains. Nat Commun. 2013;4:2575. doi: 10.1038/ncomms3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murphy DD, Rueter SM, Trojanowski JQ, Lee VM. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci. 2000;20:3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giasson BI, Murray IV, Trojanowski JQ, Lee VM. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J Biol Chem. 2001;276:2380–2386. doi: 10.1074/jbc.M008919200. [DOI] [PubMed] [Google Scholar]
- 36.Murray IV, et al. Role of alpha-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry. 2003;42:8530–8540. doi: 10.1021/bi027363r. [DOI] [PubMed] [Google Scholar]
- 37.Fauvet B, et al. alpha-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J Biol Chem. 2012;287:15345–15364. doi: 10.1074/jbc.M111.318949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Conway KA, Harper JD, Lansbury PT., Jr Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson's disease are typical amyloid. Biochemistry. 2000;39:2552–2563. doi: 10.1021/bi991447r. [DOI] [PubMed] [Google Scholar]
- 39.Kloepper KD, Woods WS, Winter KA, George JM, Rienstra CM. Preparation of alpha-synuclein fibrils for solid-state NMR: expression, purification, and incubation of wild-type and mutant forms. Protein Expr Purif. 2006;48:112–117. doi: 10.1016/j.pep.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 40.Bellon A, et al. Decontamination of prions in a plasma product manufacturing environment. Transfusion. 2013;54:1028–1036. doi: 10.1111/trf.12381. [DOI] [PubMed] [Google Scholar]
- 41.Murphy RG, et al. Alkaline hydrolysis of mouse-adapted scrapie for inactivation and disposal of prion-positive material. J Anim Sci. 2009;87:1787–1793. doi: 10.2527/jas.2008-1492. [DOI] [PubMed] [Google Scholar]
- 42.Luk KC, et al. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med. 2012;209:975–986. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This video demonstrates sonication PFFs using a probe tip sonicator, a critical step for the success of this protocol.