

Abstract
Rubinstein-Taybi syndrome (RTS) is a rare developmental disorder comprising of mental retardation, unusual facial appearance, broad thumbs, and big toes. It is frequently associated with molecular lesions in the cAMP response element binding protein. Many cutaneous abnormalities are associated with RTS. Multiple spontaneous keloids are some of them. We hereby report a case of this rare syndrome associated with keloids without any preceding trauma.
Keywords: Rubinstein–Taybi syndrome, multiple spontaneous keloids, broad thumbs and halluces
What was known?
Rubinstein–Taybi syndrome is a rare congenital disorder characterized by broad thumb and toes, comical face, growth and mental retardation. It is associated with certain cutaneous features like keloids, capillary hemangioma, thick and highly arched eyebrows and long eyelashes.
Introduction
Rubinstein–Taybi syndrome (RTS) is a congenital disorder. It is characterized by mental retardation and physical abnormalities such as broad thumbs and halluces, short stature, and a peculiar facial expression—“comical face”, which is characterized by a beaked nose, down-slanting palpebral fissures, and hypoplastic maxilla.[1] It is caused by a microdeletion in the 16p13.3 gene encoding for transcriptional coactivator, cAMP response element binding protein (CREBP).[2] In addition, major dermatological manifestations associated with the disorder include hirsutism, capillary hemangiomas, and keloids.[3] Herein, we report a case of this rare syndrome associated with spontaneous keloids. To the best of our knowledge, a report of this nature is being reported for the first time in the Indian dermatology literature.
Case Report
A 45-year-old female, presented with 2 years history of itchy, hyperpigmented progressive raised skin lesions over shoulders, arms, legs, and back. She gave a history of a spinal deformity since childhood which has resulted in hunching. A thorough review of the family history was not contributory (Pedigree chart; Chart 1).
Chart 1.
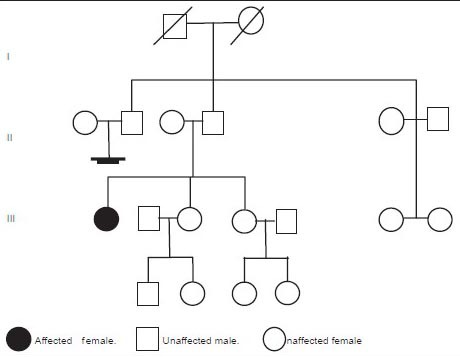
Pedigree chart showing the affected patient
On examination, multiple (more than 10), linear, hyperpigmented, nontender, firm, ill-defined nodules with regular margins were noted over the back, shoulders [Figure 1], arms [Figure 2], thighs, and legs (calf) [Figure 3].
Figure 1.
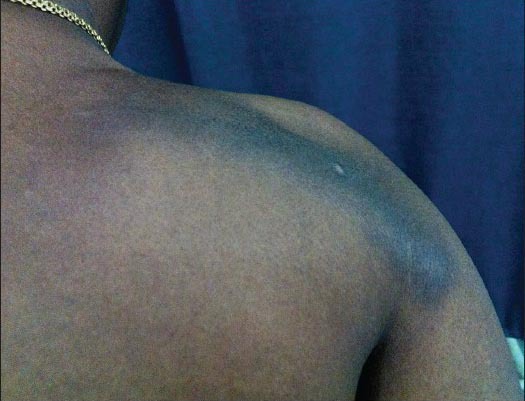
Linear keloids over the right shoulder
Figure 2.
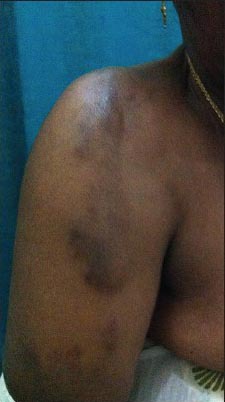
Keloids over the right arm
Figure 3.
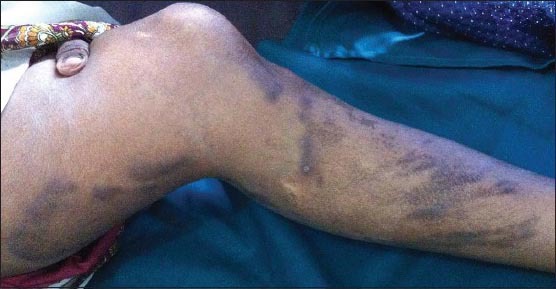
Multiple linear keloids over the left thigh and legs
Further examination revealed dysmorphic facial features such as down-slanting palpebral fissures, broad nasal bridge, deviated nasal septum, multiple atrophic facial scars [Figure 4], beaked nose with the nasal septum extending well below the alae, low set ears, mild micrognathism [Figure 5], grimacing smile, and high arched palate. Other features noted in the patient were broad halluces and radial deviation of the great toe [Figure 6], broad thumbs [Figure 7], short stature, and kyphoscoliosis [Figure 8]. Nervous system examination revealed hypotonia with mild mental retardation. Other systemic examinations were normal.
Figure 4.
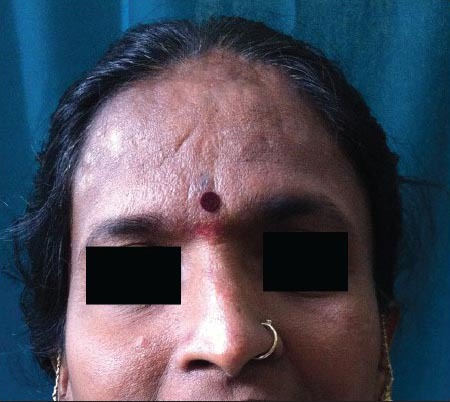
Down-slanting palpebral fissures, beaked nose, broad nasal bridge, multiple atrophic facial scars
Figure 5.
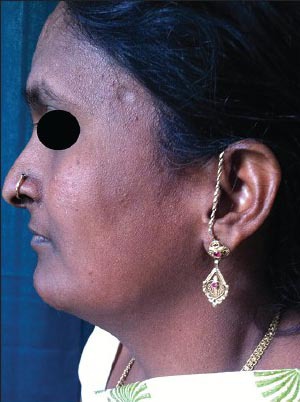
Low set ears, columella below the level of alae, and mild micrognathism
Figure 6.
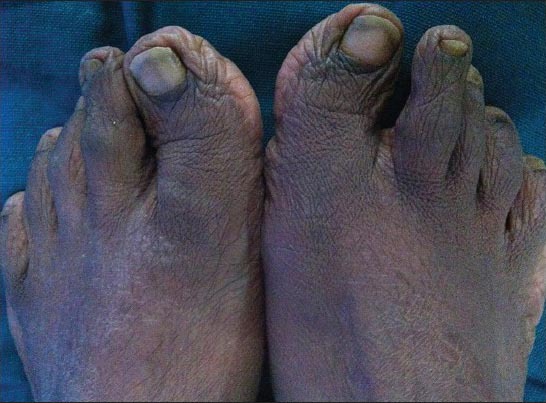
Broad toes with hallux valgus
Figure 7.
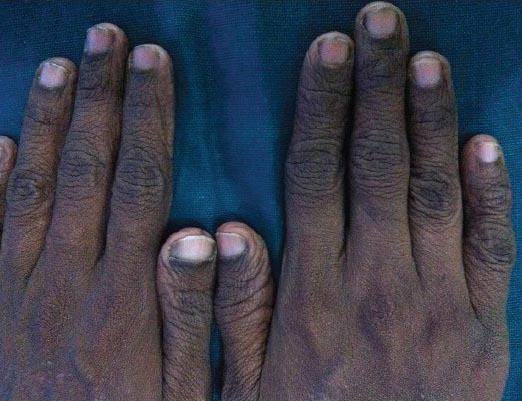
Broad thumbs
Figure 8.
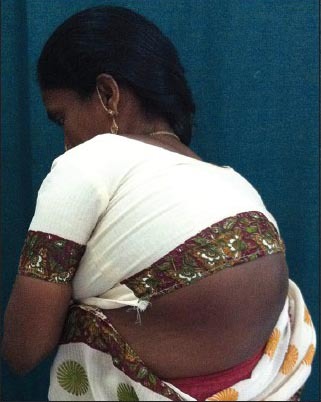
A spinal deformity (kyphoscoliosis)
Histopathology of the nodular lesion showed normal epidermis with increased dermal collagen and periadnexal inflammatory cell infiltrate. The collagen bundles were arranged haphazardly, which was consistent with the histological features of keloid [Figures 9 and 10].
Figure 9.
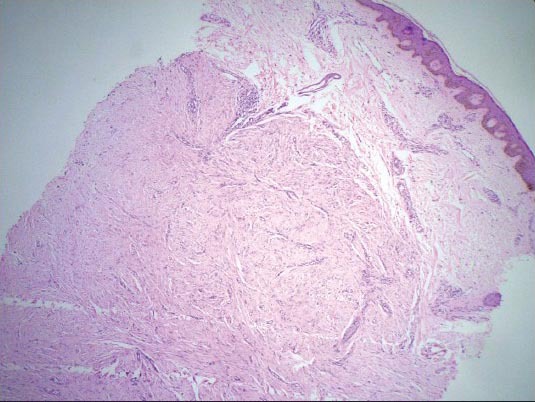
H and E staining showing skin with unremarkable epidermis and nodular proliferation in the dermis (×4)
Figure 10.
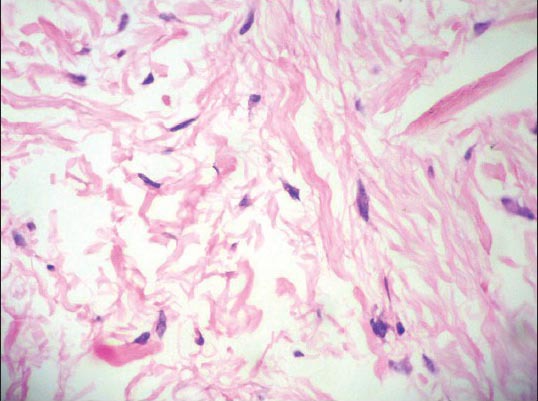
H and E staining showing wavy collagen bundles arranged haphazardly (×40)
On the basis of constellation of these clinicohistopathological features a diagnosis of RTS with multiple keloids was made. Chromosomal analysis was not performed in our patient because of lack of facilities in our setup.
Discussion
In 1963, Rubinstein and Taybi described a new syndrome characterized by broad thumbs and toes, facial abnormalities, and mental retardation.[4] The associated symptoms described earlier by Michail et al.[5] were precisely delineated later as a syndrome by Rubinstein and Taybi.[2] This syndrome usually occurs sporadically, although it can be inherited as an autosomal dominant disorder.[6] Birth prevalence is one in 100,000-125,000.[7] The syndrome has been linked to a variety of mutations at 16p13.3, the gene that codes for the cAMP response element binding protein (CREBP). CREBP functions in multiple signal transduction pathways and is thought to regulate the expression of many genes.[8]
Individuals with RTS have other distinctive features, which include: apparent hypertelorism, malpositioned ears with dysplastic helices, grimacing smile, high arched palate, broad, short terminal phalanges of the thumbs and halluces with or without angulation deformity, persistent digital fetal pads, postnatal growth retardation, and head circumference below the 50th percentile.[9]
Cutaneous anomalies in the RTS are numerous, which includes keloid formation, thick and highly arched eyebrows and unusually long eyelashes, brown spots in the lumbar region, capillary hemangiomas, transverse palmar creases, abnormal dermatoglyphics, spatulate nails, chronic paronychia of the fingernails and toenails, hypoplasia of the toenails, hirsutism, supernumerary nipples, keratosis pilaris, atopic eczema, and seborrheic dermatitis.[10]
There are also a number of other medical issues that occur commonly in individuals with RTS such as gastroesophageal reflux, feeding difficulties, constipation, hypotonia, congenital heart disease, renal anomalies, problems with anesthesia, ophthalmologic problems, orthopedic problems, developmental delay, and mental retardation.[9] There is an increased risk to develop tumors, mainly meningioma and other brain tumors and leukemia; however, life expectancy seems to be normal.[7]
Our case presented with many typical features of RTS like down-slanting palpebral fissures, broad nasal bridge, deviated nasal septum, multiple atrophic facial scars, beaked nose with the nasal septum extending well below the alae, low set ears, mild micrognathism, grimacing smile, and high arched palate. Other features noted in the patient were broad halluces and radial deviation of great toes and broad thumbs. The systemic associations were kyphoscoliosis, hypotonia, growth, and mental retardation. The most striking dermatologic feature in our patient was multiple spontaneous colossal keloids without any preceding trauma, an observation which has been only reported by Hendrix and Greer[11] and Siraganian et al.[12] In a study conducted by Siraganian et al., the incidence of keloids was only 4.8% and they were mainly concentrated on upper trunk, shoulder, and arms.[12] However, in our patient, keloids were also present on trunk and lower limbs which is an unusual site in RTS. Spontaneous multiple keloids are also reported in the Goeminne syndrome, which is characterized by torticollis, cryptorchidism, renal dysplasia, and multiple nevi.[8] In addition to RTS, keloids are also described in Pachydermoperiostosis, Ehlers–Danlos syndrome, Turner's syndrome, and Noonan's syndrome.[12]
The diagnosis is still essentially a clinical diagnosis and rests on recognition of the characteristic features. In a recent review article by Hennekam RCM, the author has put forth clinical criteria to aid the diagnosis of RTS. According to him, the major items to be looked for are the beaked nose with low hanging septum, grimacing smile, broad thumbs and big toes, and mental retardation. Dental inspection for the presence of talon cusps can be very useful, and the same holds to a lesser extent for the presence of larger keloids on the upper thorax and arms. Chromosomal analysis for mutation of CREBP gene is detected only in 55% of cases.[8] Currently, there is no known cure for RTS. Management is usually supportive and tailored to the problem of each patient.[3] The interesting features of this case report are that RTS being a rare syndrome. Further, it is associated with spontaneous keloids without preceding trauma and the presence of keloids at unusual sites like trunk and lower limbs.
Hence, the aim of this case report is to emphasize the importance of the cutaneous manifestations in RTS and to highlight the fact that in a patient reporting with spontaneous multiple keloids possibility of RTS should be kept in mind.
What is new?
Keloids occurring in patients of RTS are multiple, spontaneous, colossal and are at unusual sites.
Footnotes
Source of support: Nil
Conflict of Interest: Nil.
References
- 1.Rubinstein JH, Taybi H. Broad thumbs and toes and facial abnormalities. A possible mental retardation syndrome. Am J Dis Child. 1963;105:588–608. doi: 10.1001/archpedi.1963.02080040590010. [DOI] [PubMed] [Google Scholar]
- 2.Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RC, Masuno M, et al. Rubinstein–Taybisyndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–51. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- 3.Rubinstein JH. Broad thumb-hallux (Rubinstein-Taybi syndrome) 1957-1988. Am J Med Genet Suppl. 1990;6:3–16. doi: 10.1002/ajmg.1320370603. [DOI] [PubMed] [Google Scholar]
- 4.Cantani A, Gagliesi D. Rubinstein–Taybi syndrome. Review of 732 cases and analysis of the typical traits. EurRevMed PharmacolSci. 1998;2:81–7. [PubMed] [Google Scholar]
- 5.Michail J, Matsoukas J, Theodorou S. Arched, clubbed thumb in strong abduction–extension and other concomitant symptoms. Article in French. Rev Chir Orthop Reparatrice Appar Mot. 1957;43:142–6. [PubMed] [Google Scholar]
- 6.Shawky RM, Elsayed NS, Seifeldin NS. Facial dysmorphism, skeletal anomalies, congenitalglaucoma, dysplastic nails: Mild Rubinstein–Taybisyndrome. Egypt J Med Hum Genet. 2012;13:233–7. [Google Scholar]
- 7.Hennekam RC. Rubinstein–Taybi syndrome. Eur J Hum Genet. 2006;14:981–5. doi: 10.1038/sj.ejhg.5201594. [DOI] [PubMed] [Google Scholar]
- 8.Clark JA, Turner ML, Howard L, Stanescu H, Kleta R, Kopp JB. Description of familial keloids in five pedigrees: Evidence for autosomal dominant inheritance and phenotypic heterogeneity. BMC Dermatol. 2009;9:8. doi: 10.1186/1471-5945-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wiley S, Swayne S, Rubinstein JH, Lanphear NE, Stevens CA. Rubinstein–Taybisyndrome medical guidelines. Am J Med Genet A. 2003;119A:101–10. doi: 10.1002/ajmg.a.10009. [DOI] [PubMed] [Google Scholar]
- 10.Selmanowitz VJ, Stiller MJ. Rubinstein–Taybi syndrome. Cutaneous manifestations and colossal keloids. Arch Dermatol. 1981;117:504–6. doi: 10.1001/archderm.117.8.504. [DOI] [PubMed] [Google Scholar]
- 11.Hendrix JD, Jr, Greer KE. Rubinstein–Taybi syndrome with multiple flamboyant keloids. Cutis. 1996;57:346–8. [PubMed] [Google Scholar]
- 12.Siraganian PA, Rubinstein JH, Miller RW. Keloids and neoplasms in the Rubinstein–Taybi syndrome. Med Pediatr Oncol. 1989;17:485–91. doi: 10.1002/mpo.2950170526. [DOI] [PubMed] [Google Scholar]