

Abstract
Conradi-Hünermann-Happle syndrome (CDPX2, OMIM 302960) is an inherited X-linked dominant variant of chondrodysplasia punctata which primarily affects the skin, bones, and eyes. CDPX2 patients display skin defects, including ichthyotic lesions, follicular atrophoderma, cicatricial alopecia, and less frequently ichthyosiform erythroderma, cataracts, and skeletal abnormalities consisting of short stature, asymmetric shortening of the limbs, epiphyseal stippling, and craniofacial defects. CDPX2 results from mutations in emopamil binding protein (EBP) gene. The aim of our study is to identify EBP mutation in a unique case of Conradi-Hünermann-Happle syndrome with rare psoriasiform lesions.
Keywords: Conradi-Hünermann-Happle syndrome, emopamil binding protein mutation, plaque-type psoriasis, X-linked dominant chondrodysplasia punctata
What was known?
Case reports of Conradi-Hunermann-Happle syndrome describing psoriasiform alterations in histopathological analysis of skin lesions are very rare. Also, a case of pustular psoriasis with Conradi-Hunermann-Happle syndrome had been reported.
Introduction
X-linked dominant chondrodysplasia punctata (CDPX2, OMIM 302960) is an inheritable systemic disease and is characterized by skeletal, ophthalmologic, and cutaneous manifestations.[1,2] In the late seventies, Rudolph Happle reported a female and brilliantly recognized a group of published patients with chondrodysplasia punctata whose mosaic phenotype and sex rate corresponded with an X-linked dominant pattern of inheritance.[3] In 1999, the gene emopamil binding protein (EBP) was identified and associated with CDPX2, so called Conradi-Hünermann-Happle syndrome.[4,5] Case reports and a few case series have been published on CDPX2. The EBP gene is located on the short arm of the X chromosome (Xp11.22-p11.23) and spans 7 kb of genomic DNA, containing five exons coding a mature transcript of 1 kb which is ubiquitously expressed. This protein consists of 230 amino acids and four transmembrane domains, it has a molecular weight of 27 kDa and it is implicated in cholesterol biosynthesis.[2]
Cutaneous lesions in CDPX2 usually start as erythematous scaly plaques following the lines of Blaschko that fade over time, and some patients may be born with congenital erythroderma. Rarely, ichthyosis may persist during the adulthood.[6] Retention hyperkeratos is a prominent finding in histopathological examination of the ichthyotic skin lesions. Follicular atrophoderma and scaring alopecia are also common in this disease.
While CDPX2 may exhibit variable phenotypes, psoriasiform skin lesions are exceptional and histopathologic investigations of psoriasiform skin lesions show epidermal hyperproliferation in addition to ichthyotic retention hyperkeratosis.[2,6,7] In this report, a case of a 12-year-old girl diagnosed with X-linked dominant chondrodysplasia punctata with a psoriasiform phenotype is reported with genetical confirmation. We discuss here a rare clinical presentation which helps in better depict this rare genodermatosis and on the lack of correlation between phenotype and genotype in this disease.
Case Report
A 12-year-old girl, who had been born with congenital erythroderma, had a 4-month history of linear and whorled inflammatory cutaneous lesions on her trunk, leg, and arms. Extensively distributed thickly adherent scaling had been developed at the age of one, and continued since then. The patient had been born at term following a normal pregnancy and delivery and family history was unremarkable.
She displayed growth retardation, bilateral conductive hearing, and unilateral congenital cataract in her right eye. She exhibited short stature with asymmetric shortening of her limbs, being her right arm shorter than her left. Dysmorphic facial features with frontal bossing, flat nasal bridge, and a depressed tip of the nose were observed. Dermatological examination revealed brown streaky hyperkeratotic plaques with thickly adherent scaling located along Blaschko's lines. Also, erythematous scaling and guttate lesions were observed on her trunk, arms, and legs. There were patchy areas of scarring alopecia on frontal and temporal regions of the scalp and lateral regions of eyebrows. She had fragile and coarse lusterless hair [Figure 1]. Blood biochemical and serological tests and whole blood counts were in normal ranges. Dorsal and lumbosacral magnetic resonance imaging showed scoliosis deformity, height losses of the T8 vertebrae left half and L3, L4 vertebral the right halves (tethered cord).
Figure 1.
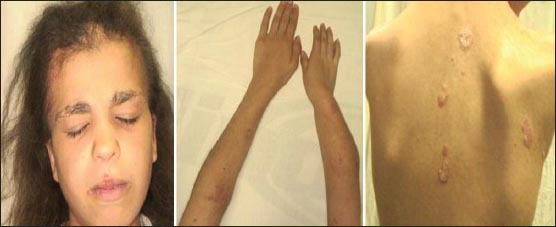
Clinical features of the patient with Conradi–Hünermann–Happle Syndrome (X-linked dominant chondrodysplasia punctata)
Genomic DNA of the patient was extracted from peripheral blood leukocytes via a conventional phenol-chloroform method. Exons 1-5 and the intronic splicing regions from the EBP gene were amplified by polymerase chain reaction (PCR) as previously described.[2] The reaction was conducted in a 2720 Thermal Cycler (Applied Biosystems, Foster City, CA, U.S.A.). Automatic sequencing was performed with the ABI PRISM 310 Genetic Analyzer (Applied Biosystems). We identified a previously reported EBP mutation, c. 440G >A (p.R147H) in the patient, but not in her parents [Figure 2]. According to the clinical and laboratory findings, the diagnosis of Conradi-Hünermann-Happle syndrome was established. A skin biopsy of one of the guttate, erythematous, and scaling plaques revealed retention hyperkeratosis, parakeratosis, acanthosis with regular elongation of rete ridges, capillary proliferation, and mononuclear cell infiltration in papillary dermis, being this consistent with a psoriasiform pattern.
Figure 2.
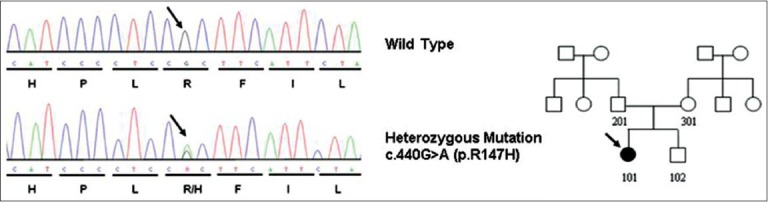
The pedigree and DNA sequencing showed a heterozygous mutation c. 440G > A resulting in p.R147H in EBP gene
Discussion
In Conradi-Hünermann-Happle syndrome, cutaneous findings are remarkable and characterized by hyperkeratotic lesions on an erythematous background following the Blaschko's lines. Characteristic ichthyosiform lesions are present at birth and fade spontaneously over time.[6,7]
There is limited number of cases with a psoriasiform phenotype in CDPX2.[6,7,8] Bruch et al. described a unique adult patient presenting both with ichthyotic and pustular psoriasiform lesions together.[6] However, mutational analysis was not performed in that report.[6] Later, in a large Spanish series, a patient with ichthyosis in a psoriasiform pattern was reported with the genetic analysis showed c. 187C > T (p.Arg63X) de novo mutation.[2]
We here present an exceptional case of CDPX2 with psoriasiform and ichthyotic skin lesions together in an adolescent, the first case from Tunisia. Histopathological features of guttate, erythematous, and scaling plaques followed a psoriasiform pattern.
In 1999, the gene EBP was identified and associated with the Conradi-Hünermann-Happle syndrome (Xp11.22-p11.23).[4,5] To date, about 70 different mutations in the EBP gene have been described related with this disease.[2] We found a c.(440G > A) missense de novo mutation in the EBP gene (p.Arg147His), placed in the second endoplasmic reticulum domain (ER2) of the protein. This is a recurrent mutation which has been recognized in several cases of CDPX2.[2] However, none of these cases displayed ichthyosis following a psoriasiform pattern. The unique case of CDPX2 with a similar phenotype to ours displayed a c.(187C > T), p.Arg63X nonsense mutation,[2] which had also been described in two patients with typical skin phenotype of CDPX2.[4,5] The c. 440G seems to be a nucleotide with particularly high mutational susceptibility within the EBP gene, a hot spot. However, the mechanisms underlying this phenomenon are not well understood. On the other hand, the clinical phenotype associated with this mutation has been variable, which subscribes the lack of correlation between genotype and phenotype in this disease.
Although the relationship between the various genotypes and phenotypes in Conradi-Hünermann-Happle syndrome has not been fully elucidated, detailed clinical and molecular analyses are helpful for providing data to be used in genetic counseling. Our case expands the clinical phenotypic variation in CDPX2 and further demonstrates the lack of correlation between genotype and phenotype in this disease. More case series would be desirable to better understand this complex genodermatosis. Unique additional clinical manifestations like plaque-type psoriasis may present with different mutations.
What is new?
Conradi-Hunermann-Happle may represent extensive plaque-type psoriasis in adolescent patients.
Acknowledgment
The authors want to acknowledge the patient's family members for their participation in the study.
Footnotes
Source of support: Nil
Conflict of Interest: Nil.
References
- 1.Yajima M, Muroga E, Nomura T, Arakawa A, Takahashi K, Matsubara K, et al. Case of Conradi-Hünermann-Happle syndrome with alopecia: Histological examination of affected follicles. J Dermatol. 2012;39:1059–60. doi: 10.1111/j.1346-8138.2012.01593.x. [DOI] [PubMed] [Google Scholar]
- 2.Cañueto J, Girós M, Ciria S, Pi-Castán G, Artigas M, García-Dorado J, et al. Clinical, molecular and biochemical characterization of nine Spanish families with Conradi-Hünermann-Happle syndrome: New insights into X-linked dominant chondrodysplasia punctata with a comprehensive review of the literature. Br J Dermatol. 2012;166:830–8. doi: 10.1111/j.1365-2133.2011.10756.x. [DOI] [PubMed] [Google Scholar]
- 3.Happle R. X-linked dominant chondrodysplasia punctata. Review of literature and report of a case. Hum Genet. 1979;53:65–73. doi: 10.1007/BF00289453. [DOI] [PubMed] [Google Scholar]
- 4.Braverman N, Lin P, Moebius FF, Obie C, Moser A, Glossmann H, et al. Mutations in the gene encoding 3 beta-hydroxysteroid-delta 8, delta 7-isomerase cause X-linked dominant Conradi-Hünermann syndrome. Nat Genet. 1999;22:291–4. doi: 10.1038/10357. [DOI] [PubMed] [Google Scholar]
- 5.Derry JM, Gormally E, Means GD, Zhao W, Meindl A, Kelley RI, et al. Mutations in a delta 8-delta 7 sterol isomerase in the tattered mouse and X-linked dominant chondrodysplasia punctata. Nat Genet. 1999;22:286–90. doi: 10.1038/10350. jderry@immunex.com . [DOI] [PubMed] [Google Scholar]
- 6.Bruch D, Megahed M, Majewski F, Ruzicka T. Ichthyotic and psoriasiform skin lesions along Blaschko's lines in a woman with X-linked dominant chondrodysplasia punctata. J Am Acad Dermatol. 1995;33:356–60. doi: 10.1016/0190-9622(95)91433-1. [DOI] [PubMed] [Google Scholar]
- 7.Hoang MP, Carder KR, Pandya AG, Bennett MJ. Ichthyosis and keratotic follicular plugs containing dystrophic calcification in newborns: Distinctive histopathologic features of x-linked dominant chondrodysplasia punctata (Conradi-Hünermann-Happle syndrome) Am J Dermatopathol. 2004;26:53–8. doi: 10.1097/00000372-200402000-00007. [DOI] [PubMed] [Google Scholar]
- 8.Kalter DC, Atherton DJ, Clayton PT. X-linked dominant Conradi-Hünermann syndrome presenting as congenital erythroderma. J Am Acad Dermatol. 1989;21:248–56. doi: 10.1016/s0190-9622(89)70169-9. [DOI] [PubMed] [Google Scholar]