

Abstract
Social subjugation has widespread consequences affecting behavior and underlying neural systems. We hypothesized that individual differences in stress responsiveness were associated with differential expression of neurotrophin associated genes within the hippocampus and amygdala. To do this we examined the brains of hamsters placed in resident/intruder interactions, modified by the opportunity to escape from aggression. In the amygdala, aggressive social interaction stimulated increased BDNF receptor TrKB mRNA levels regardless of the ability to escape the aggressor. In contrast, the availability of escape limited the elevation of GluR1 AMPA subunit mRNA. In the hippocampal CA1, the glucocorticoid stress hormone, cortisol, was negatively correlated with BDNF and TrKB gene expression, but showed a positive correlation with BDNF expression in the DG. Latency to escape the aggressor was also negatively correlated with CA1 BDNF expression. In contrast, the relationship between amygdalar TrKB and GluR1 was positive with respect to escape latency. These results suggest that an interplay of stress and neurotrophic systems influences learned escape behavior. Animals which escape faster seem to have a more robust neurotrophic profile in the hippocampus, with the opposite of this pattern in the amygdala. We propose that changes in the equilibrium of hippocampal and amygdalar learning result in differing behavioral stress coping choices.
Keywords: Social stress, Aggression, Learned escape, BDNF, TrKB, GluR1
1. Introduction
Stress shapes individual experience, and individuals respond to the stress of social aggression differently, therefore potential neural and behavioral consequences that are derived from social stress vary by individual [1,2]. Hamsters exhibit a marked territorial aggression that is influenced by social status, stress and/or defeat [3–5]. Exposing hamsters to social aggression alters their behavioral character in future social interactions. Specifically, if animals are defeated when they are juveniles they are more likely to attack non-threatening conspecifics later in life [5,6]. Hamsters that were not allowed to escape defeat by a conspecific show significantly increased social avoidance of adults thereafter [7,8]. Even limited social defeat experience in male adult hamsters influences subsequent social and aggressive behavior. It does this in such a way as to produce a conditioned defeat drastically inhibiting aggressive behavior [4,9]. Acquisition and expression of this conditioned defeat requires the basolateral amygdala and glutamatergic potentiation [10–12].
Neural plasticity describes events associated with synaptic remodeling and learning. As such, this kind of synaptic flexibility plays an important role in the development and expression of behavior associated with adaptive coping in response to social stress [13,14]. Conditioned defeat depends on a neurocircuitry that includes the amygdala, hippocampus, and prefrontal cortex modulated by glutamatergic (including NMDA), serotonergic, GABAergic, and corticotrophin releasing factor (CRF) systems [15–19]. Glutamate (via AMPA and/or NMDA receptors) and CRF stimulate CREB transcription factor and neurotrophic activity [20–26]. The behaviors associated with conditioned defeat are enhanced by overexpression of CREB in the basolateral amygdala (BLA) [27]. Neurotrophins such as brain-derived neurotrophic factor (BDNF) and its receptor tropomyosin related kinase B (TrKB) as well as the AMPA receptor subunit GluR1 contribute to molecular mechanisms of neural plasticity such as long-term potentiation, synaptic remodeling, and changes in learning [23,28–30]. What is more, BDNF and TrKB in the hippocampus and amygdala appear to be important for social defeat conditioning [31,32]. Neuroplastic changes in social and fear learning are especially prevalent in the hippocampus and amygdala [29,33–36]. Expression of the gene for BDNF is required for motivational aspects of social interaction, such that local knockdown in the nucleus accumbens eliminates the effects of repeated aggression, including social aversion [37]. Additionally, there appears to be a protective role for BDNF in early development. Knocking down the neurotrophin in the hippocampus of juveniles causes elevations in glucocorticoids [38]. Stress, and accompanying corticosteroids, typically inhibits hippocampal BDNF [39,40], as is the case in animals that display social avoidance [41]. A mutation of the human BDNF gene (Val66Met; G196A) is correlated with a higher susceptibility to stress-induced affective disorders in addition to heightened anticipatory stress responses [42,43].
The integrated relationships between social aggression, neuroendocrine stress responses, neuroplasticity, and the learning of adaptive coping strategies led to the development of a new conceptual model to help understand the decision-making process that occurs under stressful conditions [14]. This stress–choice model compares two adaptive behavioral responses to social aggression, submission, and escape, and allows examination of the neural and behavioral events that lead to each one. As neuroplastic changes in hippocampus promote improved spatial and operant learning [44,45] and changes in amygdala enhance fear learning [33,46], our hypothesis was that neural plasticity in the hippocampus and amygdala is important for producing adaptive social behavior. We also postulate that gene expression in the hippocampus and amygdala is counterbalanced, and the relative activity of those regions produce different kinds of behavioral adaptation. More specifically, we hypothesize that animals that actively make use of escape opportunities from aggressive interactions will have more BDNF expression in the hippocampus and less in the amygdala. On the other hand, animals that remain in the presence of aggressively dominant territorial opponents (do not escape) will exhibit the opposite pattern. As the BDNF protein stimulates numerous mechanisms that produce neural plasticity, we extend these hypotheses to the molecular elements involved in BDNF’s mechanism of action. Therefore, we hypothesize that expression of the BDNF receptor TrKB, its downstream 2nd messenger ERK, and the AMPA receptor subunit GluR1 will be modulated in a similar fashion to BDNF given that these genes are associated with learning and act downstream of BDNF [47,48].
2. Methods
2.1. Animals
Male golden hamsters used for behavioral experiments were singly housed in Plexiglas cages on a reversed light–day cycle (14L:10D lights off at 9:00 a.m.) with food and water provided ad libitum. Hamsters (both residents and intruders) were bred in the laboratory (from a stock originating from Harlan Sprague–Dawley; Indianapolis, IN) and weaned on postnatal day 25. Test animals were 42 days old and approximately 100 g when experiments began. Postnatal day 42 is a critical point in the development of the hamster where social behaviors and stress responsiveness start to resemble that of an adult [49]. All 10 residents used were retired breeders older than 12 weeks, previously tested and used for aggression, and weighed approximately 140 g. All animal experiments were executed in a manner that minimized suffering and the number of animals used, in accordance with National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23), and approved by the Institutional Animal Care and Use Committee of the University of Texas at Austin. Animals were kept at the Animal Resource Center, an AALAC-accredited facility.
2.2. Experimental design
Testing was accomplished using two dividers that separated hamster cages into three compartments (Fig. 1). A removable divider served to separate the two animals prior to the start of the interaction. The second divider, which was not removed, had a hole large enough only for the smaller test animal to utilize. Ensuring that the hole was small enough for the test animal to utilize while excluding the resident was another reason to begin with 42 day old animals. On the day before training test animals were habituated to the apparatus in a clean novel cage for 10 min with the escape hole blocked.
Fig. 1.
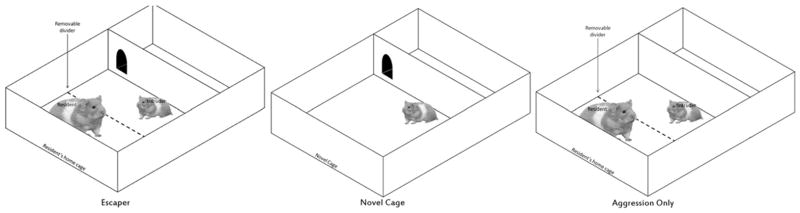
A schematic of the apparatus for testing social aggression and escape. These behavioral procedures took place in the home cage (20×33×13 cm) of a large aggressive male, fitted with dividers. One divider separated the test animal from the resident, prior to the social interaction (dotted line). The other divider provided an escape route large enough for only the test animal to pass through.
All training days (1–6) as well as the test day (7) were run in a resident’s home cage with the divider apparatus for the Escaper and Aggression Only groups. All trials for the novel cage group took place in a clean cage with access to an escape hole each day. On training days, the test animal was placed on the opposite side of the removable divider from the resident. After animals were in place a tone was sounded for 10 s, followed by 15 s of silence. After this, the divider separating the two animals was removed allowing the animals to interact for 15 min. The duration of the interactions varied for animals that had the possibility of escape, due to differences in individual escape latency. In these cases, duration of interaction was defined as the period from lifting of the divider to the moment that the animal exited, using the escape hole.
The tone served as a conditioned stimulus (CS) on training days, while aggression from the larger animal was the unconditioned stimulus (US). If the test animal utilized the hole during the experiment and made it into the escape chamber, a cover was placed over the hole for the remainder of the allotted 15 min. In addition to the animals that had an opportunity to use the hole (Aggression with Escape Possible = Escapers, N = 13), there were two other control groups. Animals in the same interaction chamber, except with no access to an escape hole (Aggression Only, N = 5), interacted with an aggressive male for 15 min after the dividers were removed. The same aggressive residents were used to interact with Escapers and Aggression Only animals. The second control group had access to the hole, but without a larger opponent and in a neutral cage (Novel Cage, N = 6). All groups underwent training for 6 days. On the seventh day, animals were tested by presenting the tone as in previous days, in the cage of a new, large aggressor, but without an attacker. Novel Cage controls received the same treatment as the first 6 days.
2.3. Behavior
Behavioral observations were manually and digitally recorded. Aggressive interactions between test animals and their aggressive opponents were conducted in the aggressive animal’s home cage, by removing the divider between them. Animal interactions were scored (Noldus Observer, Leesburg, VA) for self-directed contact time with their opponent, attacks made, latency to escape, and the duration that the resident was in close proximity to the escape hole. Attacks were defined as a successful bite by the resident on the test animal. For comparisons of behavior, the duration of bouts is variable among Escapers because each animal had a different latency to escape. In contrast, the bout duration remains consistent for Aggression Only animals (15 min). As such, for comparison the data from the two groups must be normalized (Fig. 2C). While resident male hamsters attack and bite their opponents, they typically leave no visible marks on the skin. These attacks are no doubt painful and stressful to the intruder. Even so, injury per se, is not necessarily the primary factor instigating behavioral responses to social stress, as it is in other rodent models [50].
Fig. 2.

(A) Latency to escape rapidly decreased and remained significantly less than day 1 for the duration of training and on test day. (B) The presence of an escape hole significantly increased the amount of time it took for the test animal to make contact with the resident. (C) The number of attacks received did not differ between groups. While Aggression Only animals experienced elevated attacks on day 1 (*), the attacks on Escapers were elevated for the first 2 days (+). (D) Training significantly (*, compared to all other days, α, compared to days 3–6, +, compared to day 6) decreased the proportion of time the test animal spent in contact with the aggressor for both the groups that had access to an escape hole (black dots) and those which did not (gray triangles). Hamsters that had access to an escape hole (Escapers) spent a significantly (#) larger portion of the interaction in contact with the aggressor, compared to animals that did not (Aggression Only). α, *, +, # P<0.05.
2.4. Cortisol analysis
Twenty minutes after behavioral testing on day seven, animals were decapitated with trunk blood and brains collected and frozen at −80 °C. We chose this time point so that the full effect of the interaction could be realized in terms of glucocorticoid release [51]. Plasma cortisol concentrations were quantified in duplicate using a cortisol enzyme-linked immunosorbent assay kit (Assay Designs, Ann Arbor, MI). Cross-reactivity to other endogenous steroids was low, but did include corticosterone at 27.68%, 11-deoxycortisol at 4.0%, and progesterone at 3.64%; intra-assay variance was within 6–10%. Absorbance readings were taken on a plate reader set to read a wavelength of 405 nm. Sample cortisol concentrations were calculated using a four point logistic curve fitted to the absorbance readings from the assay standards. Apriori hypotheses were generated for cortisol by gene expression interactions; such that we expected cortisol to be negatively correlated with BDNF expression in the CA1 region of the hippocampus [52]. This hypothesis was limited to Escapers given that only this group was engaged in a hippocampal learning task that might stimulate BDNF expression. Conversely, because Aggression Only, or Novel Cage was not considered learning situations, we expected no correlation between cortisol and BDNF. Similarly, since BDNF expression could lead to TrKB activity and expression, we hypothesized that cortisol would be negatively correlated with TrKB expression. As DG is often functionally distinct from CA1 [53], we posited a separate hypothesis, but also hypothesized that cortisol would be negatively correlated with BDNF expression [52].
2.5. Brain microdissection
Frozen brains were sliced coronally (300 μm), and neuroanatomy confirmed using a hamster brain atlas [54]. The entire amygdala (to ensure enough tissue for qPCR) and hippocampal regions, including Ammon’s horn region 1 (CA1) and dentate gyrus (DG), were microdissected with the blunt tip of a 23 gage needle on a freezing block and immediately injected into lysis buffer (RNeasy Micro kit; QIAGEN, Valencia, CA) before being homogenized with a pestle.
2.6. Quantitative polymerase chain reaction (qPCR)
Total RNA was extracted from microdissected samples using RNeasy Micro kit (QIAGEN, Valencia, CA) and quantified using Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). Fifty nanograms of purified RNA was further used for complementary DNA (cDNA) synthesis in 20 μl reactions using the High Capacity cDNA archive kit (Applied Biosystems, Carlsbad, CA) and purified using QIAquick PCR Purification Kit (QIAGEN, Valencia, CA). For all qPCR reactions 2 μl of total cDNA product was used in 25 μl reactions. Step One Plus Real-Time PCR System (Applied Biosystems, Carlsbad, CA) was used to perform all qPCR reactions using Taq-man Assay On Demand primer/probe sets (Applied Biosystems) for Glyceraldehyde 3-phosphate dehydrogenase (GAPDH; mm03302249q1), BDNF (mm01334047 m1), TrKB (mm00435422m1), ERK (mm00662375g1), and GluR1 (mm00433753m1). Each sample was run in duplicate and normalized to the expression of housekeeping gene, GAPDH. The TaqMan qPCR was performed at 50 °C for 2 min and 95 °C for 10 min, followed by 45 cycles at 95 °C for 15 s and 60 °C for 1 min. The animals in each group were considered biological replicates, and changes in gene expression were either represented individually (regressions) or averaged (group means). The qPCR reactions for each animal were repeated twice and results from individual reactions were averaged. Changes in gene expression quantified by qPCR were analyzed using the 2−ΔΔCT method [55], comparing all samples to the control average that was exposed to the behavioral apparatus without a larger opponent and in a novel cage. Values for qPCR data were expressed as mean fold change±standard error of the mean (SEM).
2.7. Data analysis
Behavioral and gene expression results were compared across groups (Escapers, Aggression Only, Novel Cage) by one-way ANOVA while comparisons across days were compared with one-way repeated measures analyses (using SPSS software). Significant effects between groups for one-way analyses were examined with Student–Newman–Keuls post hoc analyses (to minimize type I error) and Duncan’s Multiple Range Test (to minimize type II error). Comparisons of latency to contact (Escaper vs Aggression Only) and BDNF expression in fast and slow escapers were made using t-tests. Correlative effects from multiple factors were examined using regression analysis. Bonferroni corrections for these correlations within each apriori hypothesis yields an α level of 0.025 (see Section 2.4, comparisons of Aggression Only and Novel Cage groups). Comparisons of groups or times in the repeated measures analyses used Bonferroni post hoc tests (to minimize type I error) and Duncan’s Multiple Range Test (to minimize type II error).
3. Results
3.1. Behavior
All hamsters (except Novel Cage controls) rapidly investigated each other following the removal of the opaque divider. The number of attacks by resident males toward the test animals did not vary by group (Escapers: 1.44±0.24/day; Aggression Only: 1.83±0.33/day; two-way repeated measures ANOVA, group effect: F1,61 = 2.8, P >0.118; t86 = 0.60, P >0.55). Although new aggressors were used for each day for each test animal, there was a significant decrease in aggressive interaction after the first day (two-way repeated measures ANOVA, time effect: F5,61 = 9.0, P < 0.001; no interaction effect F5,61 = 1.8, P >0.113) in the Aggression Only group (repeated measures ANOVA, F5,20 = 7.4, P<0.001), and after the second day in Escapers (repeated measures ANOVA, F5,41 = 5.3, P<0.001; Fig. 2D). Aggression by the resident on the test animal diminished over time; therefore, we examined avoidance of the resident. The latency to contacting the aggressive male in his home cage was significantly faster in those with no access to an escape hole (Escapers: 3.83±0.37 s; Aggression Only: 2.4±0.51 s; t15 = 2.18, P < 0.046; Fig. 2B). When an escape hole was provided, it was used. Specifically, of the 64 recorded trials that presented an animal with a chance to escape its aggressor (Aggression with escape possible = Escapers), in only 3 trials did any test animal (one animal twice, and one animal once) not escape; the data from these animals were not included in any analyses. In addition, there were six trials for which no social interaction ensued; no data were recorded from these trials. The amount of time that the resident spent in close proximity to the escape hole did not significantly affect any of the variables measured.
Test hamsters showed a significant reduction in latency to escape from their aggressors between days 1 and 2 (repeated measures ANOVA; F6,66 = 6.43, P < 0.001; Fig. 2A). This reduction in latency to escape was retained for all subsequent training days, as well as the test day. Animals exposed only to a hole in a novel cage, explored and passed through the hole rapidly, in approximately 60 s, suggesting that locating and using the hole did not significantly contribute to the latency to escape during social stress. During the 6 days of training while the animals reduced escape time, test animals also adjusted social contact with their aggressor, which was dramatically influenced by the availability of a hole for escaping social interaction (two-way repeated measures ANOVA: F1,61 = 9.5, P < 0.008) and affected by the day of training (two-way repeated measures ANOVA: F5,61 = 2.58, P<0.035; Fig. 2D). Specifically, hamsters that had the ability to escape aggression significantly decreased the proportion of time they spent in contact with the larger aggressor as training progressed (repeated measures ANOVA; F5,41 = 3.47, P < 0.011; Fig. 2D). There was also a significant reduction in test animal contact with the aggressor when no escape hole was present (Aggression Only, Repeated measures ANOVA; F5,20 = 2.77, P<0.047; Fig. 2D). However, in Escapers the latency to initiate contact was greater on day 1 (Fig. 2B), but the amount of self-initiated contact was also significantly greater on experimental day 1 (t14 = 2.43, P<0.029) and day 2 (t13 = 2.88, P < 0.013), but did not differ afterward, compared to hamsters with no possibility of escape.
3.2. Plasma cortisol
There were no significant effects of treatment on plasma cortisol across the three groups (Novel Cage 52.66±9.53 ng/ml, Aggression Only 30.1±2.93 ng/ml, Escapers 39.53±3.63 ng/ml; F2,21 = 3.05, P<0.069; data not shown), collected on test day when none of the animals were exposed to the stress of an aggressor.
3.3. Gene expression
3.3.1. Hippocampus — CA1 and DG
In the CA1 region of the hippocampus there were no significant differences in BDNF (F2,15 = 0.32, P >0.729), TrKB (F2,17 = 1.27, P >0.307), ERK (F2,18 = 0.37, P >0.696) or GluR1 (F2,18 = 0.31, P >0.740) when comparing across groups of animals (Escapers: BDNF = 1.39±0.3 fold expression, TrKB = 1.1±0.1, ERK = 1.06±0.1, GluR1 = 1.07±0.11; Aggression Only: BDNF = 1.42±0.55, TrKB = 0.78±0.13, ERK = 0.91±0.14, GluR1 = 1.26±0.21; Novel Cage: BDNF = 1.11±0.19, TrKB = 1.08±0.19, ERK = 1.03±0.11, GluR1 = 1.08±0.21 fold expression). There were also no significant effects on gene expression in the DG of the hippocampus for BDNF (F2,17 = 2.55, P>0.108), TRKB (F2,17 = 1.143, P>0.342), ERK (F2,17 = 1.143, P>0.342) and GluR1 (F2,17 = 2.309, P>0.13) across groups (Escapers: BDNF = 0.58±0.11 fold expression, TrKB = 0.8±0.08, ERK = 0.75±0.08, GluR1 = 0.69±0.11; Aggression Only: BDNF = 0.61±0.22, TrKB = 0.78±0.33, ERK = 0.72±0.26, GluR1 = 0.69±0.15; Novel Cage: BDNF = 1.09±0.22, TrKB = 1.04±0.16, ERK = 1.02±0.1, GluR1 = 1.33±0.51 fold expression).
Analyzing the relationship between plasma cortisol and gene expression in the CA1 produced significant results, but only in animals that were allowed to escape (Table 1). There was a significant negative regression (inverse second order polynomial; F2,7 = 10.50, P<0.016, r2 = 0.81; Table 1) between cortisol and BDNF mRNA expression in Escapers (Fig. 3A). Similarly in the CA1 of escaping animals, mRNA expression of the BDNF receptor, TrKB, was negatively correlated with plasma cortisol (linear regression; F1,8 = 7.41, P < 0.026, r2 = 0.48; Fig. 3B). There were no significant correlations in the CA1 region for BDNF or TrKB mRNA expression against plasma cortisol concentrations when there was no possibility of escape (Aggression Only; linear regression of BDNF mRNA expression×cortisol; F1,3 = 1.78, P >0.315, r2 = 0.47; linear regression of TrKB mRNA expression×cortisol; F1,3 = 0.11, P>0.76, r2 = 0.05) or Novel Cage treatment (linear regression of BDNF mRNA×cortisol; F1,5 = 0.0002, P >0.99, r2 = 0.000004; linear regression of TrKB mRNA× cortisol; F1,3 = 0.38, P >0.57, r2 = 0.11).
Table 1.
Regression analyses for gene expression, cortisol, and escape latency.
| Comparison | Brain region | Group | Regression correlation
|
Correlation direction | ||
|---|---|---|---|---|---|---|
| F | P | r2 | ||||
| Cortisol×BDNF | CA1 | Escapers | 10.496 | 0.016 | 0.810 | − |
| Cortisol×BDNF | CA1 | Aggression Only | 1.775 | 0.314 | 0.470 | ns |
| Cortisol×BDNF | CA1 | Novel Cage | <0.001 | 0.991 | <0.001 | ns |
| Cortisol×TRKB | CA1 | Escapers | 7.407 | 0.026 | 0.481 | − |
| Cortisol×TRKB | CA1 | Aggression Only | 0.112 | 0.770 | 0.053 | ns |
| Cortisol×TRKB | CA1 | Novel Cage | 0.385 | 0.579 | 0.114 | ns |
| Cortisol×BDNF | DG | Escapers | 5.486 | 0.041 | 0.354 | + |
| Cortisol×BDNF | DG | Aggression Only | 0.179 | 0.713 | 0.082 | ns |
| Cortisol×BDNF | DG | Novel Cage | 18.129 | 0.050 | 0.900 | + |
| Escape latency×BDNF | CA1 | Escapers | 30.371 | 0.0007 | 0.910 | − |
| Escape latency×TRKB | Amygdala | Escapers | 5.184 | 0.049 | 0.366 | + |
| Escape latency×GluR1 | Amygdala | Escapers | 14.407 | 0.004 | 0.590 | + |
Fig. 3.

Hamsters that had access to an escape hole (Escapers) had a significant correlation in the CA1 between cortisol and both (A) BDNF and (B) TrKB mRNA expressions. (C) Cortisol and BDNF were also significantly correlated in the DG.
There were also significant relationships between cortisol and gene expression in the dentate gyrus of animals that were allowed to escape aggression (Escapers). Interestingly, while the BDNF/cortisol relationship among the animals that could escape was negative in the CA1, the correlation in the DG was positive (linear regression; F1,10 = 5.49, P<0.041, r2 = 0.35; Fig. 3C). Furthermore, there was also a positive correlation in the DG of animals that were exposed to a novel cage (linear regression; F1,2 = 18.13, P < 0.05, r2 = 0.90). Animals that did not have an opportunity to escape their aggressor (Aggression Only) did not have any significant BDNF×cortisol effects (linear regression; F1,2 = 0.18, P >0.71, r2 = 0.08).
There was a significant negative relationship between escape time and BDNF expression in test animals that had an escape route available. The amount of time that it took for test hamsters to use the escape hole on test day following the tone, was negatively correlated with BDNF mRNA expression (F1,8 = 27.12, P < 0.002, r2 = 0.90; Fig. 4A; Table 1). Hamsters that escaped faster than 110 s (groups split based on significantly different time intervals between data points, t6 = 2.7, P < 0.035) had significantly greater BDNF expression than those escaping slower than 150 s (t7 = 7.0, P < 0.001; Fig. 4B).
Fig. 4.

Hamsters showed a (A) significant (*P<0.05) negative correlation between BDNF mRNA expression in the CA1 and the latency to escape on test day 7, and (B) significant differences in BDNF expression between fast (<110 s; N = 6) and slow (>150 s; N = 3) escapers.
3.3.2. Amygdala
There was a significant elevation of the BDNF receptor TrKB expression in the amygdala (F2,18 = 3.54, P < 0.05; Fig. 5A) for both groups in which there was a social interaction. However, BDNF mRNA expression was not significantly affected (F2,16 = 0.798, P >0.46). Specifically, TrKB mRNA expression was elevated in animals that were allowed to escape the aggressor (Escapers), and animals that were not (Aggression Only). Furthermore, a statistically significant elevation in AMPA subunit GluR1 mRNA expression was measured in the Aggression Only animals who were exposed to the social interaction, but not allowed to escape (F2,20 = 8.450, P < 0.002; Fig. 5C), when compared with hamsters that were exposed to social aggression with the possibility of escape (Escapers), and those exposed to a Novel Cage only.
Fig. 5.

A social interaction was enough to induce a significant (*P<0.05) increase in amygdalar TRKB mRNA expression relative to the Novel Cage only group (A). Furthermore, there was a significant positive correlation between TRKB mRNA expression and latency to escape on test day 7 (B). Only the presence of aggression with no opportunity to escape (Aggression Only) induced a significant increase in amygdalar AMPA subunit GluR1 mRNA expression relative to Novel Cage controls (C). There was a significant positive correlation between GluR1 mRNA expression and latency to escape on test day 7 (D).
Escape behavior had a significant positive relationship with gene expression in animals that were allowed to escape an aggressor. However, there were no gene by cortisol correlations in the amygdala, nor did BDNF expression significantly correlate with escape times on test day (F1,9 = 1.21, P >0.30; data not shown). Positive correlations exist for both TrkB and GluR1 between latency to escape and gene expression (linear regression TrKB mRNA expression; F1,9 = 5.184, P < 0.048; Fig. 5B; linear regression GluR1 mRNA expression; F1,10 = 14.41, P<0.004; Fig. 5D; Table 1).
4. Discussion
Hamsters exhibit a vast improvement in escape latency between the first and second times they used an available escape hole (Fig. 2A). These escape latencies remained fast on all subsequent training days; and also on the test day when no larger aggressive male was present (Fig. 2A). Similarly, McCann et al., demonstrated that hamsters escape over the cage wall much faster after the first resident/intruder trial [8]. Trout exposed to the stress–choice model also show an improvement in latency to escape time, but unlike hamsters do so gradually over the course of 7 days [14]. A critical aspect of the stress–choice model is that escape allows for control in a socially stressful situation. Adaptively, it seems reasonable that an animal that escapes faster will suffer fewer negative consequences from intense social stressors. As results from other species have demonstrated in the stress–choice paradigm, hamsters learned to escape more quickly as their training proceeded (Fig. 2A). What is more, social stress-induced latency to escape was delayed compared with the approximate time animals in the novel cage first explored and passed through the available hole. The delay suggests that stress-induced escape latency has more to do with social interaction than it does with simply finding and using the hole [14]. Furthermore, the ability to escape changed the degree to which test animals were prepared to interact with their aggressive opponent (Fig. 2B, D). It’s important to note that all hamsters in the Escape group passed through the hole on test day (day 7) even though there was no aggressor present. The lack of an aggressive stimulus on test day, when only the tone conditioned stimulus was present suggests that the escape behavior was learned. Also, escape occurred with a similar short latency following the tone on the last day of training when there was an aggressor, strengthening the case for a learning event.
While aggressive social interaction drives the motivation for escape, the availability of escape also influences the social relationships that play out during an interaction (Fig. 2B, D). Surprisingly, the latency to first contact initiated by the smaller test animal to the larger aggressor was increased by the availability of an escape route, perhaps due to the additional novel item in the cage (Fig. 2B). Furthermore, while it would seem natural that animals with the ability to escape would experience fewer attacks, in actuality there were no significant differences between the number of attacks between the two groups. In addition, the number of attacks in both groups rapidly decline after the first day (Fig. 2C). The reason for this decline may be obvious for the animals that were able to leave the interaction, thereby subsequently limiting aggression. However, the same reason cannot explain the decline in attacks in animals that could not escape. The answer is likely to lie in the observation that the Aggression Only group spent a significantly smaller proportion of time in contact with the aggressor for the first 2 days, effectively decreasing aggression by avoiding the resident (Fig. 2D). This would suggest that both groups are using different, yet equally effective strategies to reduce aggression. Whereas the escaping group can afford to use a more proactive social strategy, because they can actively leave the interaction and thus limit aggression, animals that cannot escape use a more passive/reactive strategy, minimizing the amount of time they are in contact with the aggressor while also reducing the number of attacks they receive [56]. It is important to note that the escapers are also using avoidance behavior as they spend a significantly smaller proportion of their time in contact with the aggressor after day 2. Therefore, two simultaneous learning events were necessary to produce the behavioral results: 1) learning how to use the escape hole, while concomitantly 2) learning to avoid the new aggressive opponent each day.
Aggressive social interaction, modified by the possibility of escape, also appears to influence the expression of genes related to learning and synaptic remodeling types of neuroplasticity. Repeated social defeat fosters amygdalar plasticity and fear learning [57]. However, we found that the ability to escape produced a unique pattern of gene expression that was not seen with defeat alone. In the amygdala, a brain region known to be important in fear learning, social defeat has been demonstrated to stimulate BDNF mRNA expression in the basolateral and medial amygdalae of losing male hamsters, and in the dentate gyrus of winners [31]. Specific TrKB receptor drugs influence defeat and submission in hamsters [31,32]. In our small test animals, aggressive social interaction from a larger male stimulated increased amygdalar BDNF receptor TrKB mRNA levels (Fig. 5A). This increase in TrKB mRNA was measured in animals with (Escapers) and without the possibility of escape (Aggression Only); suggesting that it was the aggressive social interaction that stimulated the TrKB gene expression. In addition, the availability of escape specifically impacts neuroplastic gene expression (Fig. 5C). Hamsters that experience aggression without the possibility of escape (Aggression Only) exhibit significantly elevated GluR1 AMPA subunit mRNA in amygdala. By contrast, although they also experience limited aggression, Escapers demonstrate repressed GluR1 expression compared to the Aggression Only group (Fig. 5C). Hamsters subjected to social aggression without the possibility of escape (Aggression Only) exhibited increased expression of both TrKB and GluR1 mRNA. Therefore, faster escapers expressed both TRKB and GluR1 at lower levels in amygdala (Fig. 5B, D). These receptors are influenced by BDNF and both are necessary for fear learning [58–60]. It is important to note, that TrKB and GluR1 mRNA were measured on the test day, when no aggressor was present. It is possible that the results reflect gene expression changes that are due to the reprieve from social stress on test day. However, if this was the case, the effect would be reflected similarly in both gene expression experiments (i.e. similar results for TrKB and GluR1 mRNA). The results suggest an increase in neuroplastic gene expression in amygdala associated with fearful, stressful stimuli, and relieved in part by the possibility of escape.
The expression of BDNF, TrKB, and GluR1 mRNAs was also significantly correlated with the latency to escape. It is important to note that animals were sampled on test day 7, and the largest improvement of escape times took place between days 1 and 2. The sharp change in behavioral performance likely reflects a learning event which would undoubtedly involve increased BDNF signaling. It is possible that more robust neurotrophic activity would have been seen at this time point. Still, the slopes of regression for amygdala contrast with those from hippocampus when comparing gene expression over the latency to escape (Figs. 4A; 5B, D). A negative curvilinear regression of BDNF in hippocampal CA1 against escape latency (Fig. 4A), is distinct from positive linear regressions of TrKB and GluR1 in the amygdala against escape latency (Fig. 5B, D). While gene expressions of BDNF in CA1, and GluR1 in amygdala were not stimulated by escape in these animals, nor was elevated TrKB expression in amygdala exclusively found in Escapers, significant relationships between hippocampal BDNF as well as amygdalar TrKB and GluR1 expression with latency to escape suggest that neural plasticity is involved nevertheless. Animals that took longer to escape in our model had lower levels of hippocampal CA1 BDNF mRNA expression (Fig. 4B), an event also seen when animals are exposed to chronic stress corresponding to depression and anxiety [61]. Given that this type of behavioral escape involves stress and fear, while at the same time requiring spatial, social, and operant learning, it seems reasonable that both amygdala and hippocampus are involved [14,62–65].
In male golden hamsters there is a negative relationship correlating glucocorticoid stress hormones with neurotrophin (BDNF) and receptor (TrKB) gene expression (Fig. 3A, B). While this was true for BDNF and TrKB gene expression in the CA1 (Fig. 3A, B), we observed the opposite effect on BDNF expression in the DG (Fig. 3C). Other work has demonstrated that stress and/or glucocorticoids decrease hippocampal BDNF mRNA and protein [39,40,52,66–71]. In regions outside the hippocampus, such as the amygdala, stress-induced alterations of BDNF levels are also found [61]. The changes we observed suggest that context is critical in determining the relationship between gene expression and glucocorticoid activity. Animals that were exposed to the stress while having a means to actively escape the stressor had a significant correlation between BDNF and TrkB mRNA and cortisol. Other regions of the brain that are also involved in regulation of the neuroendocrine stress response via the HPA axis produce BDNF that is influenced by glucocorticoids, sometimes positively [72], as was the case in dentate gyrus. The inhibitory effects of stress on BDNF in the hippocampus affect learning [73]. However, learning can also protect hippocampal BDNF mRNA expression from exogenous corticosterone [53]. This may help explain the confounding results from the subregions of the hippocampus. Namely, while higher cortisol levels were associated with lower levels of BDNF gene expression in the CA1 of our animals, the opposite pattern was seen in the dentate gyrus. Schaaf and colleagues have shown that the CA1 and DG react differently with respect to BDNF gene expression in response to glucocorticoids [53]. Specifically, while both CA1 and DG BDNF mRNA expressions are suppressed by exogenous corticosterone, only CA1 was suppressed by endogenous corticosterone levels (as revealed by adrenalectomy). Yet when corticosterone levels were elevated from a spatial learning task, the Morris Water Maze, neither the CA1 nor DG had different BDNF expression patterns relative to controls. However, we measured no significant group effects on cortisol levels, including a lack of influence from social learning or stress. As cortisol levels were measured after test day, with the express goal of examining the likelihood of classical conditioning [14], the influence of immediate social stress was not evident in plasma glucocorticoid concentrations. It should also be noted that hamsters express corticosterone in addition to cortisol. Kollack-Walker, Watson and Akil have shown that social aggression, in addition to other stressors, induces release of both forms of glucocorticoid in a highly correlated positive linear fashion [74]. As social aggression produces parallel responses in these two glucocorticoids, our measurements of cortisol alone should be reflected in corticosterone as well. These experiments suggest that hippocampal granular (DG) and pyramidal (CA) cells may be differentially sensitive to corticosteroids and neurotrophins.
The changes we measured in BDNF, TrKB and GluR1 gene expressions following escapable or non-escapable social defeat may reflect broader changes in the circuitry that produces conditioned defeat in hamsters. While defeat is expressed differently in juvenile and adult male hamsters [6], our experiment used young adult test animals, and thus may reflect a transitional state. During puberty three different periods mark the development of aggressive behavior, starting with attacks targeted toward face and cheeks during play fighting transitioning to adult fighting characterized by attacks focused on the belly and rear. It is important to note that repeated social defeat accelerates the onset of adult aggression style in juvenile animals [5,6]. An adult hamster experiencing conditioned defeat will assume submissive/defensive postures, even in the presence of a smaller nonaggressive intruder [9]. As in our stress–choice model, plasticity in the hippocampus and amygdala is critical for the expression of conditioned defeat behaviors [18]. Furthermore, the two models are similar in that the changes in plasticity are driven by the stress of social aggression. Social defeat stimulates amygdalar BDNF expression in losers, but increases hippocampal BDNF gene expression in winners [31]. While systemic TrKB receptor stimulation promotes resilience to social defeat [32], perhaps through hippocampal mechanisms, intra-BLA injection of a TrKB antagonist reduced acquisition of conditioned defeat [31]. Taken together, these results suggest opposing actions of BDNF in hippocampus and amygdala, similar to the contrasting regressions we measured for hippocampal BDNF and amygdalar TrKB and GluR1. While glucocorticoid receptors appear to play a negligible role in acquisition of conditioned defeat [15], cortisol was correlated with hippocampal neurotrophic gene expression in escaping animals in our study.
This work highlights neurotrophic changes that take place in the hippocampus and amygdala during a task that involves spatial, social, and operant learning. Opposing activity in these two brain regions makes these results potentially important for understanding the mechanisms that produced divergent stress coping behavior. The data show that differential BDNF, TrKB and GluR1 gene expressions in amygdala versus hippocampal CA1 contrast specifically with respect to social interaction and the possibility of escape, and thereby may influence the adaptive celerity of escape. We hypothesize that differential activation of emotional brain regions manage alternatively adaptive learning tasks. The hippocampus has been demonstrated to be necessary for elements of spatial, social, and operant learning [18,75–78], and the amygdala for social and fear learning [18,79–81]. Where these types of learning overlap, there must be interaction between these brain regions, and the behavioral output must be dynamically regulated between them. The gene expression changes in the amygdala may help explain the change in proportion of time spent interacting with the intruder due to classical fear learning, and changes in hippocampus may help explain individual differences in escape latency. Those hamsters with greater stimulation of hippocampal CA1 BDNF expression may have had an advantage for learning how to use the escape hole (Fig. 4). Of the Escapers, those more sensitive to stress, and therefore with higher cortisol levels, escaped more slowly; the combination of reduced BDNF and TrKB expression in the CA1 along with increased TrKB expression in the amygdala, potentially make learning to avoid the aggressor more difficult (Figs. 3A, B, 4A, 5C). Only in those animals that received aggression without the possibility of escape did both TrKB and the AMPA receptor subunit GluR1 expression significantly increase, suggesting the possibility that learning submissive behaviors under these conditions is necessary [14]. This hypothesis is supported by the evidence that both and TrKB and GluR1 expressions are positively correlated with increasing latency to escape (Fig. 5C, D).
In conclusion, we hypothesize a neurocircuitry that utilizes the specified functionalities of hippocampus and amygdala to produce a behavioral phenotype. The results also suggest a relationship between gene expression and escape behavior for male golden hamsters, where higher CA1 BDNF expression is associated with faster escape (Fig. 4). For slowly escaping hamsters, gene expression data suggest a putative imbalance in this proposed circuitry, favoring the amygdalar functions. Such negative stimuli would prompt amygdalar learning, which may limit explorations and motivated escape. Furthermore, our model shows that the exact context is critical in determining the response to a stressor. Devising a way to cope with such stressors would undoubtedly involve compensatory hippocampal learning and activity. This work raises the possibility that individual variation in behavioral stress coping strategies is mediated by this hippocampal/amygdalar balance.
Acknowledgments
From the 20th Anniversary meeting of the International Behavioral Neuroscience Society at Steamboat Springs, Colorado. This research was funded by NIH grant P20 RR15567.
References
- 1.Affleck G, Tennen H. Construing benefits from adversity: adaptational significance and dispositional underpinnings. J Pers. 1996;64(4):899–922. doi: 10.1111/j.1467-6494.1996.tb00948.x. [DOI] [PubMed] [Google Scholar]
- 2.Bartolomucci A, Palanza P, Sacerdote P, Panerai AE, Sgoifo A, Dantzer R, et al. Social factors and individual vulnerability to chronic stress exposure. Neurosci Bio-behav Rev. 2005;29(1):67–81. doi: 10.1016/j.neubiorev.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 3.Delville Y, Melloni RH, Jr, Ferris CF. Behavioral and neurobiological consequences of social subjugation during puberty in golden hamsters. J Neurosci. 1998;18(7):2667–72. doi: 10.1523/JNEUROSCI.18-07-02667.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huhman KL, Solomon MB, Janicki M, Harmon AC, Lin SM, Israel JE, et al. Conditioned defeat in male and female Syrian hamsters. Hormones and Behavior. 2003;44(3):293–9. doi: 10.1016/j.yhbeh.2003.05.001. [DOI] [PubMed] [Google Scholar]
- 5.Delville Y, David JT, Taravosh-Lahn K, Wommack JC. Stress and the development of agonistic behavior in golden hamsters. Hormones and Behavior. 2003;44(3):263–70. doi: 10.1016/s0018-506x(03)00130-2. [DOI] [PubMed] [Google Scholar]
- 6.Wommack JC, Taravosh-Lahn K, David JT, Delville Y. Repeated exposure to social stress alters the development of agonistic behavior in male golden hamsters. Horm Behav. 2003;43(1):229–36. doi: 10.1016/s0018-506x(02)00029-6. [DOI] [PubMed] [Google Scholar]
- 7.Bastida CC, Puga F, Delville Y. Risk assessment and avoidance in juvenile golden hamsters exposed to repeated stress. Horm Behav. 2009;55(1):158–62. doi: 10.1016/j.yhbeh.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 8.McCann KE, Huhman KL. The effect of escapable versus inescapable social defeat on conditioned defeat and social recognition in Syrian hamsters. Physiol Behav. 2011;105(2):493–7. doi: 10.1016/j.physbeh.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Potegal M, Huhman K, Moore T, Meyerhoff J. Conditioned defeat in the Syrian golden hamster (Mesocricetus auratus) Behav Neural Biol. 1993;60(2):93–102. doi: 10.1016/0163-1047(93)90159-f. [DOI] [PubMed] [Google Scholar]
- 10.Jasnow AM, Huhman KL. Activation of GABA A receptors in the amygdala blocks the acquisition and expression of conditioned defeat in Syrian hamsters. Brain Research. 2001;920(1–2):142–50. doi: 10.1016/s0006-8993(01)03054-2. [DOI] [PubMed] [Google Scholar]
- 11.Day DE, Cooper MA, Markham CM, Huhman KL. NR2B subunit of the NMDA receptor in the basolateral amygdala is necessary for the acquisition of conditioned defeat in Syrian hamsters. Behav Brain Res. 2011;217(1):55–9. doi: 10.1016/j.bbr.2010.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jasnow AM, Cooper MA, Huhman KL. N-methyl-D-aspartate receptors in the amygdala are necessary for the acquisition and expression of conditioned defeat. Neuroscience. 2004;123(3):625–34. doi: 10.1016/j.neuroscience.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 13.Djordjevic A, Adzic M, Djordjevic J, Radojcic MB. Chronic social isolation is related to both upregulation of plasticity genes and initiation of proapoptotic signaling in Wistar rat hippocampus. J Neural Transm. 2009;116(12):1579–89. doi: 10.1007/s00702-009-0286-x. [DOI] [PubMed] [Google Scholar]
- 14.Carpenter RE, Summers CH. Learning strategies during fear conditioning. Neurobiology of Learning and Memory. 2009;91:415–23. doi: 10.1016/j.nlm.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cooper MA, Huhman KL. Blocking corticotropin-releasing factor-2 receptors, but not corticotropin-releasing factor-1 receptors or glucocorticoid feedback, disrupts the development of conditioned defeat. Physiol Behav. 2010;101(4):527–32. doi: 10.1016/j.physbeh.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cooper MA, McIntyre KE, Huhman KL. Activation of 5-HT1A autoreceptors in the dorsal raphe nucleus reduces the behavioral consequences of social defeat. Psychoneuroendocrinology. 2008;33(9):1236–47. doi: 10.1016/j.psyneuen.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cooper MA, Huhman KL. Corticotropin-releasing factor receptors in the dorsal raphe nucleus modulate social behavior in Syrian hamsters. Psychopharmacology (Berl) 2007;194(3):297–307. doi: 10.1007/s00213-007-0849-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Markham CM, Taylor SL, Huhman KL. Role of amygdala and hippocampus in the neural circuit subserving conditioned defeat in Syrian hamsters. Learn Mem. 2010;17(2):109–16. doi: 10.1101/lm.1633710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Markham CM, Luckett CA, Huhman KL. The medial prefrontal cortex is both necessary and sufficient for the acquisition of conditioned defeat. Neuropharmacology. 2011 doi: 10.1016/j.neuropharm.2011.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cammarota M, Bevilaqua LR, Ardenghi P, Paratcha G, Levi de Stein M, Izquierdo I, et al. Learning-associated activation of nuclear MAPK, CREB and Elk-1, along with Fos production, in the rat hippocampus after a one-trial avoidance learning: abolition by NMDA receptor blockade. Brain Res Mol Brain Res. 2000;76(1):36–46. doi: 10.1016/s0169-328x(99)00329-0. [DOI] [PubMed] [Google Scholar]
- 21.Stern CM, Meitzen J, Mermelstein PG. Corticotropin-releasing factor and urocortin I activate CREB through functionally selective Gbetagamma signaling in hippocampal pyramidal neurons. Eur J Neurosci. 2011;34(5):671–81. doi: 10.1111/j.1460-9568.2011.07812.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hubbard DT, Nakashima BR, Lee I, Takahashi LK. Activation of basolateral amygdala corticotropin-releasing factor 1 receptors modulates the consolidation of contextual fear. Neuroscience. 2007;150(4):818–28. doi: 10.1016/j.neuroscience.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu K, Nakashima BR, Lee I, Takahashi LK. Brain-derived neurotrophic factor acutely enhances tyrosine phosphorylation of the AMPA receptor subunit GluR1 via NMDA receptor-dependent mechanisms. Brain Res Mol Brain Res. 2004;130(1–2):178–86. doi: 10.1016/j.molbrainres.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 24.Aguilar-Valles A, Sanchez E, de Gortari P, Balderas I, Ramirez-Amaya V, Bermudez-Rattoni F, et al. Analysis of the stress response in rats trained in the water-maze: differential expression of corticotropin-releasing hormone, CRH-R1, glucocorticoid receptors and brain-derived neurotrophic factor in limbic regions. Neuroendocrinology. 2005;82(5–6):306–19. doi: 10.1159/000093129. [DOI] [PubMed] [Google Scholar]
- 25.Ma YL, Chen KY, Wei CL, Lee EH, Chen KY, Wei CL, Lee EH, et al. Corticotropin-releasing factor enhances brain-derived neurotrophic factor gene expression to facilitate memory retention in rats. Chin J Physiol. 1999;42(2):73–81. [PubMed] [Google Scholar]
- 26.Li W, Keifer J. BDNF-induced synaptic delivery of AMPAR subunits is differentially dependent on NMDA receptors and requires ERK. Neurobiol Learn Mem. 2009;91(3):243–9. doi: 10.1016/j.nlm.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jasnow AM, Shi C, Israel JE, Davis M, Huhman KL. Memory of social defeat is facilitated by cAMP response element-binding protein overexpression in the amygdala. Behav Neurosci. 2005;119(4):1125–30. doi: 10.1037/0735-7044.119.4.1125. [DOI] [PubMed] [Google Scholar]
- 28.Diogenes MJ, Costenla AR, Lopes LV, Jeronimo-Santos A, Sousa VC, Fontinha BM, et al. Enhancement of LTP in aged rats is dependent on endogenous BDNF. Neuropsychopharmacology. 2011;36(9):1823–36. doi: 10.1038/npp.2011.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Broad KD, Mimmack ML, Keverne EB, Kendrick KM. Increased BDNF and trk-B mRNA expression in cortical and limbic regions following formation of a social recognition memory. Eur J Neurosci. 2002;16(11):2166–74. doi: 10.1046/j.1460-9568.2002.02311.x. [DOI] [PubMed] [Google Scholar]
- 30.Schjetnan AG, Escobar ML. In vivo BDNF modulation of hippocampal mossy fiber plasticity induced by high frequency stimulation. Hippocampus. 2010 doi: 10.1002/hipo.20866. [DOI] [PubMed] [Google Scholar]
- 31.Taylor SL, Stanek LM, Ressler KJ, Huhman KL. Differential brain-derived neurotrophic factor expression in limbic brain regions following social defeat of territorial aggression. Behav Neurosci. 2011;125(6):911–20. doi: 10.1037/a0026172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeffress EC, Norvelle A, Huhman KL. 7,8-Dihydroxyflavone, a TrKB receptor agonist, decreases submission following social defeat in Syrian hamsters. Soc Neurosci Abs. 2011;936(13) [Google Scholar]
- 33.Lonsdorf TB, Weike AI, Golkar A, Schalling M, Hamm AO, Ohman A. Amygdala-dependent fear conditioning in humans is modulated by the BDNFval66met polymorphism. Behav Neurosci. 2010;124(1):9–15. doi: 10.1037/a0018261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fanous S, Hammer RP, Jr, Nikulina EM. Short- and long-term effects of intermittent social defeat stress on brain-derived neurotrophic factor expression in mesocortico-limbic brain regions. Neuroscience. 2010;167(3):598–607. doi: 10.1016/j.neuroscience.2010.02.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Razzoli M, Domenici E, Carboni L, Rantamaki T, Lindholm J, Castren E, et al. A role for BDNF/TrkB signaling in behavioral and physiological consequences of social defeat stress. Genes Brain Behav. 2011;10(4):424–33. doi: 10.1111/j.1601-183X.2011.00681.x. [DOI] [PubMed] [Google Scholar]
- 36.Roth TL, Zoladz PR, Sweatt JD, Diamond DM. Epigenetic modification of hippocampal Bdnf DNA in adult rats in an animal model of post-traumatic stress disorder. J Psychiatr Res. 2011;45(7):919–26. doi: 10.1016/j.jpsychires.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311(5762):864–8. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- 38.Taliaz D, Loya A, Gersner R, Haramati S, Chen A, Zangen A. Resilience to chronic stress is mediated by hippocampal brain-derived neurotrophic factor. J Neurosci. 2011;31(12):4475–83. doi: 10.1523/JNEUROSCI.5725-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murakami S, Imbe H, Morikawa Y, Kubo C, Senba E. Chronic stress, as well as acute stress, reduces BDNF mRNA expression in the rat hippocampus but less robustly. Neurosci Res. 2005;53(2):129–39. doi: 10.1016/j.neures.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 40.Shi SS, Shao SH, Yuan BP, Pan F, Li ZL. Acute stress and chronic stress change brain-derived neurotrophic factor (BDNF) and tyrosine kinase-coupled receptor (TrkB) expression in both young and aged rat hippocampus. Yonsei Med J. 2010;51(5):661–71. doi: 10.3349/ymj.2010.51.5.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9(4):519–25. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- 42.Colzato LS, Van der Does AJ, Kouwenhoven C, Elzinga BM, Hommel B. BDNF Val(66)Met polymorphism is associated with higher anticipatory cortisol stress response, anxiety, and alcohol consumption in healthy adults. Psychoneuroendocrinology. 2011;36(10):1562–9. doi: 10.1016/j.psyneuen.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 43.Schenkel LC, Segal J, Becker JA, Manfro GG, Bianchin MM, Leistner-Segal S. The BDNF Val66Met polymorphism is an independent risk factor for high lethality in suicide attempts of depressed patients. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(6):940–4. doi: 10.1016/j.pnpbp.2010.04.023. [DOI] [PubMed] [Google Scholar]
- 44.Li H, Zhong X, Chau KF, Williams EC, Chang Q. Loss of activity-induced phosphorylation of MeCP2 enhances synaptogenesis, LTP and spatial memory. Nat Neurosci. 2011;14(8):1001–8. doi: 10.1038/nn.2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xie X, Ramirez DR, Lasseter HC, Fuchs RA. Effects of mGluR1 antagonism in the dorsal hippocampus on drug context-induced reinstatement of cocaine-seeking behavior in rats. Psychopharmacology (Berl) 2010;208(1):1–11. doi: 10.1007/s00213-009-1700-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Monfils MH, Cowansage KK, LeDoux JE. Brain-derived neurotrophic factor: linking fear learning to memory consolidation. Mol Pharmacol. 2007;72(2):235–7. doi: 10.1124/mol.107.038232. [DOI] [PubMed] [Google Scholar]
- 47.Slipczuk L, Bekinschtein P, Katche C, Cammarota M, Izquierdo I, Medina JH. BDNF activates mTOR to regulate GluR1 expression required for memory formation. PLoS One. 2009;4(6):e6007. doi: 10.1371/journal.pone.0006007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takei S, Morinobu S, Yamamoto S, Fuchikami M, Matsumoto T, Yamawaki S. Enhanced hippocampal BDNF/TrkB signaling in response to fear conditioning in an animal model of posttraumatic stress disorder. J Psychiatr Res. 2011;45(4):460–8. doi: 10.1016/j.jpsychires.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 49.Wommack JC, Delville Y. Stress, aggression, and puberty: neuroendocrine correlates of the development of agonistic behavior in golden hamsters. Brain Behav Evol. 2007;70(4):267–73. doi: 10.1159/000105490. [DOI] [PubMed] [Google Scholar]
- 50.Blanchard RJ, Wall PM, Blanchard DC. Problems in the study of rodent aggression. Horm Behav. 2003;44(3):161–70. doi: 10.1016/s0018-506x(03)00127-2. [DOI] [PubMed] [Google Scholar]
- 51.Keeney A, Jessop DS, Harbuz MS, Marsden CA, Hogg S, Blackburn-Munro RE. Differential effects of acute and chronic social defeat stress on hypothalamic–pituitary–adrenal axis function and hippocampal serotonin release in mice. J Neuroendocrinol. 2006;18(5):330–8. doi: 10.1111/j.1365-2826.2006.01422.x. [DOI] [PubMed] [Google Scholar]
- 52.Smith MA, Makino S, Kvetnansky R, Post RM. Stress and glucocorticoids affect the expression of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J Neurosci. 1995;15(3 Pt 1):1768–77. doi: 10.1523/JNEUROSCI.15-03-01768.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schaaf MJ, Sibug RM, Duurland R, Fluttert MF, Oitzl MS, De Kloet ER, et al. Corticosterone effects on BDNF mRNA expression in the rat hippocampus during Morris water maze training. Stress. 1999;3(2):173–83. doi: 10.3109/10253899909001121. [DOI] [PubMed] [Google Scholar]
- 54.Smith OA, Jr, Bodemer CN. A stereotaxic atlas of the brain of the golden hamster (Mesocricetus auratus) J Comp Neurol. 1963;120:53–63. doi: 10.1002/cne.901200106. [DOI] [PubMed] [Google Scholar]
- 55.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 56.Koolhaas JM, de Boer SF, Buwalda B, van Reenen K. Individual variation in coping with stress: a multidimensional approach of ultimate and proximate mechanisms. Brain Behav Evol. 2007;70(4):218–26. doi: 10.1159/000105485. [DOI] [PubMed] [Google Scholar]
- 57.Clark GA. Emotional learning. Fear and loathing in the amygdala. Curr Biol. 1995;5(3):246–8. doi: 10.1016/s0960-9822(95)00050-9. [DOI] [PubMed] [Google Scholar]
- 58.Rattiner LM, Davis M, French CT, Ressler KJ. Brain-derived neurotrophic factor and tyrosine kinase receptor B involvement in amygdala-dependent fear conditioning. J Neurosci. 2004;24(20):4796–806. doi: 10.1523/JNEUROSCI.5654-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Humeau Y, Reisel D, Johnson AW, Borchardt T, Jensen V, Gebhardt C, et al. A pathway-specific function for different AMPA receptor subunits in amygdala long-term potentiation and fear conditioning. J Neurosci. 2007;27(41):10947–56. doi: 10.1523/JNEUROSCI.2603-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Frank L, Wiegand SJ, Siuciak JA, Lindsay RM, Rudge JS. Effects of BDNF infusion on the regulation of TrkB protein and message in adult rat brain. Exp Neurol. 1997;145(1):62–70. doi: 10.1006/exnr.1997.6440. [DOI] [PubMed] [Google Scholar]
- 61.Yu H, Chen ZY. The role of BDNF in depression on the basis of its location in the neural circuitry. Acta Pharmacol Sin. 2011;32(1):3–11. doi: 10.1038/aps.2010.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fekete EM, Zhao Y, Li C, Sabino V, Vale WW, Zorrilla EP. Social defeat stress activates medial amygdala cells that express type 2 corticotropin-releasing factor receptor mRNA. Neuroscience. 2009;162(1):5–13. doi: 10.1016/j.neuroscience.2009.03.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wommack JC, Delville Y. Chronic social stress during puberty enhances tyrosine hydroxylase immunoreactivity within the limbic system in golden hamsters. Brain Research. 2002;933(2):139–43. doi: 10.1016/s0006-8993(02)02311-9. [DOI] [PubMed] [Google Scholar]
- 64.Wommack JC, Salinas A, Melloni RH, Jr, Delville Y. Behavioural and neuroendocrine adaptations to repeated stress during puberty in male golden hamsters. J Neuroendocrinol. 2004;16(9):767–75. doi: 10.1111/j.1365-2826.2004.01233.x. [DOI] [PubMed] [Google Scholar]
- 65.Markham CM, Huhman KL. Is the medial amygdala part of the neural circuit modulating conditioned defeat in Syrian hamsters? Learn Mem. 2008;15(1):6–12. doi: 10.1101/lm.768208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stranahan AM, Arumugam TV, Mattson MP. Lowering corticosterone levels reinstates hippocampal brain-derived neurotropic factor and Trkb expression without influencing deficits in hypothalamic brain-derived neurotropic factor expression in leptin receptor-deficient mice. Neuroendocrinology. 2011;93(1):58–64. doi: 10.1159/000322808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang Y, Gu F, Chen J, Dong W. Chronic antidepressant administration alleviates frontal and hippocampal BDNF deficits in CUMS rat. Brain Res. 2010;1366:141–8. doi: 10.1016/j.brainres.2010.09.095. [DOI] [PubMed] [Google Scholar]
- 68.Nitta A, Ohmiya M, Sometani A, Itoh M, Nomoto H, Furukawa Y, et al. Brain-derived neurotrophic factor prevents neuronal cell death induced by corticosterone. J Neurosci Res. 1999;57(2):227–35. doi: 10.1002/(SICI)1097-4547(19990715)57:2<227::AID-JNR8>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 69.Jacobsen JP, Mork A. Chronic corticosterone decreases brain-derived neurotrophic factor (BDNF) mRNA and protein in the hippocampus, but not in the frontal cortex, of the rat. Brain Res. 2006;1110(1):221–5. doi: 10.1016/j.brainres.2006.06.077. [DOI] [PubMed] [Google Scholar]
- 70.Prickaerts J, van den Hove DL, Fierens FL, Kia HK, Lenaerts I, Steckler T. Chronic corticosterone manipulations in mice affect brain cell proliferation rates, but only partly affect BDNF protein levels. Neurosci Lett. 2006;396(1):12–6. doi: 10.1016/j.neulet.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 71.Zhou J, Zhang F, Zhang Y. Corticosterone inhibits generation of long-term potentiation in rat hippocampal slice: involvement of brain-derived neurotrophic factor. Brain Res. 2000;885(2):182–91. doi: 10.1016/s0006-8993(00)02934-6. [DOI] [PubMed] [Google Scholar]
- 72.Smith MA, Makino S, Kim SY, Kvetnansky R. Stress increases brain-derived neurotropic factor messenger ribonucleic acid in the hypothalamus and pituitary. Endocrinology. 1995;136(9):3743–50. doi: 10.1210/endo.136.9.7649080. [DOI] [PubMed] [Google Scholar]
- 73.Song L, Che W, Min-Wei W, Murakami Y, Matsumoto K. Impairment of the spatial learning and memory induced by learned helplessness and chronic mild stress. Pharmacol Biochem Behav. 2006;83(2):186–93. doi: 10.1016/j.pbb.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 74.Kollack-Walker S, Watson SJ, Akil H. Social stress in hamsters: defeat activates specific neurocircuits within the brain. J Neurosci. 1997;17(22):8842–55. doi: 10.1523/JNEUROSCI.17-22-08842.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dupret D, Revest JM, Koehl M, Ichas F, De Giorgi F, Costet P, et al. Spatial relational memory requires hippocampal adult neurogenesis. PLoS One. 2008;3(4):e1959. doi: 10.1371/journal.pone.0001959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lai WS, Ramiro LL, Yu HA, Johnston RE. Recognition of familiar individuals in golden hamsters: a new method and functional neuroanatomy. J Neurosci. 2005;25(49):11239–47. doi: 10.1523/JNEUROSCI.2124-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rapanelli M, Frick LR, Zanutto BS. Differential gene expression in the rat hippocampus during learning of an operant conditioning task. Neuroscience. 2009;163(4):1031–8. doi: 10.1016/j.neuroscience.2009.07.037. [DOI] [PubMed] [Google Scholar]
- 78.Rapanelli M, Frick LR, Zanutto BS. Learning an operant conditioning task differentially induces gliogenesis in the medial prefrontal cortex and neurogenesis in the hippocampus. PLoS One. 2011;6(2):e14713. doi: 10.1371/journal.pone.0014713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hitchcock JM, Davis M. Fear-potentiated startle using an auditory conditioned stimulus: effect of lesions of the amygdala. Physiol Behav. 1987;39(3):403–8. doi: 10.1016/0031-9384(87)90242-3. [DOI] [PubMed] [Google Scholar]
- 80.LeDoux JE, Iwata J, Cicchetti P, Reis DJ. Different projections of the central amygdaloid nucleus mediate autonomic and behavioral correlates of conditioned fear. J Neurosci. 1988;8(7):2517–29. doi: 10.1523/JNEUROSCI.08-07-02517.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Petrulis A. Neural mechanisms of individual and sexual recognition in Syrian hamsters (Mesocricetus auratus) Behav Brain Res. 2009;200(2):260–7. doi: 10.1016/j.bbr.2008.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]