

Abstract
Background
Grape is one of the most valuable fruit crops and can serve for both fresh consumption and wine production. Grape cultivars have been selected and evolved to produce high-quality fruits during their domestication over thousands of years. However, current widely planted grape cultivars suffer extensive loss to many diseases while most wild species show resistance to various pathogens. Therefore, a comprehensive evaluation of wild grapes would contribute to the improvement of disease resistance in grape breeding programs.
Results
We performed deep transcriptome sequencing of three Chinese wild grapes using the Illumina strand-specific RNA-Seq technology. High quality transcriptomes were assembled de novo and more than 93% transcripts were shared with the reference PN40024 genome. Over 1,600 distinct transcripts, which were absent or highly divergent from sequences in the reference PN40024 genome, were identified in each of the three wild grapes, among which more than 1,000 were potential protein-coding genes. Gene Ontology (GO) and pathway annotations of these distinct genes showed those involved in defense responses and plant secondary metabolisms were highly enriched. More than 87,000 single nucleotide polymorphisms (SNPs) and 2,000 small insertions or deletions (indels) were identified between each genotype and PN40024, and approximately 20% of the SNPs caused nonsynonymous mutations. Finally, we discovered 100 to 200 highly confident cis-natural antisense transcript (cis-NAT) pairs in each genotype. These transcripts were significantly enriched with genes involved in secondary metabolisms and plant responses to abiotic stresses.
Conclusion
The three de novo assembled transcriptomes provide a comprehensive sequence resource for molecular genetic research in grape. The newly discovered genes from wild Vitis, as well as SNPs and small indels we identified, may facilitate future studies on the molecular mechanisms related to valuable traits possessed by these wild Vitis and contribute to the grape breeding programs. Furthermore, we identified hundreds of cis-NAT pairs which showed their potential regulatory roles in secondary metabolism and abiotic stress responses.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1442-3) contains supplementary material, which is available to authorized users.
Keywords: Grape, Chinese wild Vitis, De novo transcriptome, Disease related genes, Cis-NATs
Background
Grapes are among the most valuable fruit crops, grown on about 7 million ha with an annual production of approximately 67 million tonnes worldwide [1]. There are over 60 species of Vitis around the world [2] and Vitis vinifera is the most widely planted grapevine. However, most cultivars of V. vinifera are highly susceptible to various economically important diseases such as powdery mildew (PM) [3], downy mildew (DM) [4] and anthracnose [5]. Enhancing resistance to these diseases is the focus area in current grape breeding programs. Wild species may possess valuable genetic variations (e.g. new alleles, SNPs and indels) in disease resistance genes, particularly, in nucleotide-binding site leucine-rich repeat (NBS-LRR) proteins. It has been reported that the species-specific genes in wild and semi-wild watermelon were highly enriched with genes involved in disease-related processes [6]. In addition, the wild eggplant carries nearly 200 extra disease resistant genes compared to the cultivated eggplant [7]. Grape, unlike other domesticated crops, has retained high genetic diversity from wide progenitors [8]; however, most European cultivars are susceptible to many fungal diseases. Moreover, only limited disease resistant loci, such as Run1 [9], Ren1 [10], Ren2 [11] and Ren3 [12] that confer resistance to PM, and Rpv loci including Rpv1 [13], Rpv2 [14] and Rpv3 [15] that confer resistance to DM, have been identified, mainly from wild grapevine species. Currently, the widely planted grape cultivars are very sensitive to diverse pathogens. Thus, genetic engineering of disease resistance in grape has become an increasing need and this can be facilitated by the use of wild Vitis resources.
China, as one of the major centers of the origin of Vitis, has more than 35 native Vitis species [16]. Chinese wild Vitis are naturally distributed throughout the country and many of them can survive in regions of high humidity and moisture [2], under which the occurrence of fungal diseases is increased [17]. A number of disease resistant Chinese wild Vitis have been identified and characterized. The pioneering work from Wang et al. [18] and Wan et al. [19] identified a large number of Chinese wild grapes that displayed strong resistance to PM. Wan et al. [19] also found about half of the Chinese wild Vitis were resistant to DM and around one third of them have both PM and DM resistances. In addition, Wang et al. [20] and Li et al. [21] found that all the investigated Chinese wild Vitis exhibited much less susceptibility to anthracnose compared to the two V. vinifera cultivars (Cabernet Sauvignon and Chardonnay). Moreover, Chinese wild Vitis showed other specific characteristics, such as high photosynthetic efficiency in V. quiqangualaris Rehd [22] and high content of resveratrol (a phytoalexin that is beneficial to human health) in V. quiqangualaris Danfeng-2 [23]. However, the underlying genetic variations contributing to these phenotypic differences have not yet been explored.
Grapevine is the first fruit crop that had its genome sequenced. The high quality genome has been served as the reference for many genetic studies. However, recent deep sequencing experiments have shown that relying on a single reference genome may underestimate the variability among different genotypes [24]. For example, reconstructing the transcriptomes of different grape genotypes has revealed substantial heterogeneity in transcripts that associated with their phenotypes [24,25]. In this study, we aim to characterize Chinese wild Vitis transcriptomes to explore the potential genetic diversities such as SNPs and indels. To this end, two Chinese wild V. pseudoreticulata accessions “Baihe-13-1” (BH) and “Hunan-1” (HN), and one V. quinquangularis accession “Shang-24” (S) were selected for deep transcriptome sequencing. The selected accessions possess valuable resistances to various fungal pathogens including PM (BH and S) [18], DM (BH and HN) [19] and anthracnose (BH, HN and S) [20]. We de novo assembled the transcriptomes and conducted comparative analysis of SNPs and small indels between wild accessions and the reference genome, PN40024. Distinct genes, which represent genes that are absent or highly divergent from sequences in the reference PN40024 genome, were then identified from each accession. Using transcript information from strand-specific RNA-Seq libraries, we have identified cis-natural antisense transcript (cis-NAT) pairs, which were known to participate in a broad range of regulatory events.
Results
Transcriptome sequencing, de novo assembly, and comparison with the reference genome
In order to more broadly capture disease related genes, we infected young leaves of the three Chinese wild grapes with PM. Based on the proposed PM infection cycle [26,27], as well as our previous studies [28-30], we collected leaves at 0, 6, 12, 24, 48, 72, 96 and 120 hours post inoculation (hpi). We then prepared eight independent strand-specific RNA-Seq libraries for each accession. In total, 24 libraries were constructed and sequenced on the Illumina HiSeq 2000 platform. After removing adaptors, low quality sequences, and ribosomal RNA (rRNA) reads (see method), we obtained a total of 53,890,427, 68,593,803 and 70,718,358 high quality cleaned reads for BH, HN and S, respectively (Table 1). The transcriptomes of the three wild Chinese Vitis were constructed de novo separately. The final assembled transcript sets of BH, HN and S contained 34,914, 38,528 and 38,204 contigs, respectively, with N50 lengths of 814 bp, 876 bp and 980 bp and average lengths of 602 bp, 630 bp and 679 bp (Table 1). The GC-contents of these three transcript sets were similar (~44.5%; Table 1), but slightly higher than that of the PN40024 transcripts (42.0%). The assembled transcriptome sequences of the three Chinese wild Vitis can be downloaded and blasted at http://bioinfo.bti.cornell.edu/wild_vitis.
Table 1.
Summary of transcriptome sequences and assemblies of the three Chinese wild Vitis
| Statistics | BH | HN | S |
|---|---|---|---|
| Sequences | |||
| Total raw reads | 63,210,010 | 80,577,978 | 81,282,272 |
| High-quality cleaned reads | 53,890,427 | 68,593,803 | 70,718,358 |
| High-quality cleaned nucleotides | 2,911,785,932 | 3,703,865,895 | 3,864,462,091 |
| Assemblies | |||
| Number of assembled transcripts | 34,914 | 38,528 | 38,204 |
| Number of assembled bases | 21,030,007 | 24,289,873 | 25,954,126 |
| Contig N50 | 814 | 876 | 980 |
| Average contig length | 602 | 630 | 679 |
| GC content (%) | 44.7 | 44.5 | 44.4 |
BH, V. pseudoreticulata accession “Baihe-13-1”; HN, V. pseudoreticulata accession “Hunan-1”; S, V. quinquangularis accession “Shang-24”.
To discover the variations between Chinese wild Vitis and PN40024, a genotype derived from Pinot Noir, all three transcriptomes were aligned to the PN40024 genome. There were 94% (32,892), 94% (36,151) and 93% (35,589) of the total transcripts can be uniquely aligned to the reference genome with ≥ 97% sequence identity for BH, HN and S, respectively (Figure 1). Only 1% of them were mapped to multiple locations of the PN40024 genome and most of them were mapped to two locations. The majority of transcripts (83%, 82% and 80% of the total transcripts from BH, HN and S, respectively) were mapped to the annotated gene regions (version 12× V0) with overlap fraction ≥ 90% and had the same strand with the corresponding PN40024 transcripts; whereas around 2% of the transcripts were aligned to gene regions in antisense orientations. Notably, a small proportion (5-7%) of the transcripts were aligned entirely to the intergenic regions. Among all the assembled transcripts, 1,758 (5%, BH), 2,083 (5%, HN) and 2,331 (6%, S) could not be aligned to the PN40024 genome. We considered these transcripts as candidates for distinct genes.
Figure 1.
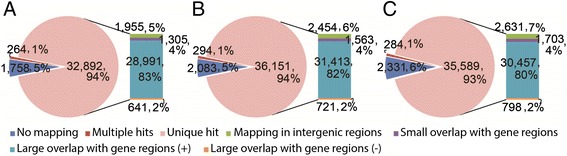
Mapping of de novo assembled transcripts of the three Chinese wild Vitis, BH (A), HN (B), and S (C), to the reference PN40024 genome. No mapping: Contigs not mapped; Multiple hits: Contigs mapped to multiple genomic locations; Unique hit: Contigs mapped to unique genomic locations; Mapping in intergenic regions: Contigs mapped to intergenic regions; Small overlap with gene regions: Contigs mapped to gene regions with low overlapping (<90% of contig length); Large overlap with gene regions (+): Contigs mapped to gene regions in sense directions with high overlapping (≥90% of contig length) ; Large overlap with gene regions (−): Contigs mapped to gene regions in antisense directions with high overlapping (≥90% of contig length). BH, V. pseudoreticulata accession “Baihe-13-1”; HN, V. pseudoreticulata accession “Hunan-1”; S, V. quinquangularis accession “Shang-24”.
Functional annotation of wild Chinese Vitis transcripts
All the transcripts from the three Chinese wild Vitis were annotated by comparing their sequences against the TrEMBL and Swiss-Prot protein databases [31]. In all three genotypes, around 88.5% of the transcripts had hits in TrEMBL, and around 53.5% had hits in Swiss-Prot (Table 2). The high percentage of transcripts that hit to known proteins, combined with the high mapping rate to the reference genome, indicated the high quality of the de novo assembled transcripts.
Table 2.
Annotations of assembled transcriptomes of the three Chinese wild Vitis
| Database | BH | HN | S |
|---|---|---|---|
| TrEMBL | 31,022 (88.85%) | 34,122 (88.56%) | 33,809 (88.50%) |
| Swiss-Prot | 18,704 (53.57%) | 20,491 (53.18%) | 20,703 (54.19%) |
| GO term | 26,936 (77.15%) | 29,491 (76.54%) | 29,195 (76.42%) |
| MetaCyc pathway | 3,114 (8.92%) | 3,343 (8.68%) | 3,208 (8.40%) |
BH, V. pseudoreticulata accession “Baihe-13-1”; HN, V. pseudoreticulata accession “Hunan-1”; S, V. quinquangularis accession “Shang-24”.
The transcripts were further annotated by assigning them human-readable functional terms extracted from the functional descriptions of their homologous proteins using AHRD [32]. Approximately 80-81% of the transcripts from the three species could be assigned with functional terms. The Gene Ontology (GO) terms for each transcript were then extracted according to the annotations in Swiss-Prot and TrEMBL (Table 2). In total, about 77% transcripts from each accession could be assigned with GO terms from at least one of the three GO categories, biological process, cellular component and molecular function. The top five subcategories in biological process were “cellular process”, “biosynthetic process”, “response to stress”, “cellular component organization” and “nucleobase-containing compound metabolic process” (Additional file 1). Pathway analysis [33] revealed approximately 3,000 genes from each accession that involved in 505–513 biochemical pathways.
Distinct genes identified in the three wild Chinese Vitis
The assembled transcripts included redundant sequences, mainly due to alternative splicing. We found that 4.80% (BH), 6.81% (HN) and 8.94% (S) transcripts could be clustered with other transcripts (sequence identity ≥ 97% and overlap length ≥ 100 bp). Only one representative transcript from each cluster was used in the downstream functional enrichment/annotation analysis, to avoid the repetitive counting of the same genes. Finally, we obtained a total of 1,650-2,000 distinct unique transcripts in each of the three accessions. The coding potential of these transcripts was then assessed by Coding Potential Calculator (CPC) [34]. Consequently, we got 1,058, 1,296 and 1,315 distinct genes with high coding potential from BH, HN and S, respectively, as well as 596, 609, and 732 potential non-coding transcripts (Figure 2A). The list of these transcripts is provided in Additional file 2. We then performed the GO term enrichment analyses (corrected P-value ≤ 0.05) on these three distinct protein-coding gene datasets. A total of 19 GO terms under the category of biological process were enriched in distinct genes in all three accessions (Table 3 and Figure 2B), and the most representative GO term was response to stimulus (GO:0050896), which included nearly half of the distinct genes. Among the child terms of GO:0050896, the most significantly enriched one was defense response (GO:0006952; Table 3). We also identified 15, 18 and 30 enriched child GO terms associated with resistance-related biological processes in BH, HN and S, respectively (Additional file 3).
Figure 2.
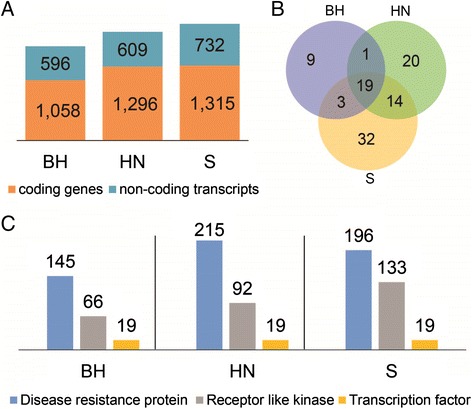
Distinct genes in the three Chinese wild Vitis , BH, HN and S. (A) Number of distinct protein-coding genes and non-coding transcripts from de novo transcriptome assemblies of the three Chinese Vitis leaf tissues, which were collected at 0, 6, 12, 24, 48, 72, 96, and 120 hours post inoculation with PM, respectively. (B) Venn diagram of GO terms enriched in the distinct protein-coding genes. (C) Number of distinct genes encoding disease resistance proteins, receptor like kinases and transcription factors. BH, V. pseudoreticulata accession “Baihe-13-1”; HN, V. pseudoreticulata accession “Hunan-1”; S, V. quinquangularis accession “Shang-24”.
Table 3.
GO terms enriched in the distinct gene sets of all three Chinese wild Vitis
| GO ID | GO term | GO term level | BH | HN | S | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| C* | B | Corrected P-value | C | B | Corrected P-value | C | B | Corrected P-value | |||
| GO:0050896 | response to stimulus | 2 | 47% | 35% | 3.73E-13 | 50% | 35% | 8.09E-26 | 51% | 35% | 1.08E-27 |
| GO:0006950 | response to stress | 3 | 38% | 24% | 1.65E-22 | 42% | 24% | 2.41E-45 | 41% | 24% | 9.38E-39 |
| GO:0006952 | defense response | 4 | 26% | 11% | 4.26E-37 | 31% | 12% | 2.01E-78 | 31% | 12% | 2.34E-75 |
| GO:0098542 | defense response to other organism | 4 | 11% | 7% | 2.01E-02 | 13% | 7% | 2.54E-09 | 14% | 7% | 3.21E-14 |
| GO:0009617 | response to bacterium | 4 | 9% | 5% | 1.11E-03 | 10% | 5% | 5.25E-07 | 11% | 6% | 8.50E-11 |
| GO:0045087 | innate immune response | 4 | 8% | 4% | 5.56E-04 | 8% | 4% | 4.34E-07 | 9% | 4% | 6.53E-11 |
| GO:0006955 | immune response | 3 | 8% | 4% | 3.59E-03 | 8% | 5% | 7.25E-07 | 9% | 5% | 1.87E-10 |
| GO:0042742 | defense response to bacterium | 6 | 8% | 4% | 5.15E-03 | 9% | 5% | 5.20E-09 | 10% | 5% | 4.03E-12 |
| GO:0009814 | defense response, incompatible interaction | 5 | 4% | 2% | 9.50E-03 | 4% | 2% | 2.86E-04 | 4% | 2% | 7.07E-05 |
| GO:0009816 | defense response to bacterium, incompatible interaction | 6 | 3% | 1% | 5.15E-04 | 3% | 1% | 3.91E-05 | 3% | 1% | 1.36E-05 |
| GO:0009626 | plant-type hypersensitive response | 5 | 2% | 1% | 4.62E-02 | 3% | 1% | 8.40E-07 | 3% | 1% | 3.87E-06 |
| GO:0098581 | detection of external biotic stimulus | 5 | 1% | 0% | 2.98E-02 | 1% | 0% | 1.03E-02 | 2% | 0% | 4.12E-11 |
| GO:0009804 | coumarin metabolic process | 6 | 2% | 0% | 1.31E-03 | 2% | 0% | 1.84E-08 | 1% | 0% | 2.91E-03 |
| GO:0009805 | coumarin biosynthetic process | 6 | 2% | 0% | 1.31E-03 | 2% | 0% | 1.84E-08 | 1% | 0% | 2.91E-03 |
| GO:0015074 | DNA integration | 6 | 3% | 0% | 5.21E-23 | 2% | 0% | 5.65E-10 | 3% | 0% | 3.69E-15 |
| GO:0015916 | fatty-acyl-CoA transport | 7 | 1% | 0% | 7.87E-08 | 0% | 0% | 3.88E-05 | 0% | 0% | 1.12E-03 |
| GO:1901337 | thioester transport | 4 | 1% | 0% | 7.87E-08 | 0% | 0% | 3.88E-05 | 0% | 0% | 1.12E-03 |
| GO:0080001 | mucilage extrusion from seed coat | 5 | 1% | 0% | 1.08E-05 | 1% | 0% | 2.57E-03 | 1% | 0% | 4.89E-03 |
| GO:0009835 | fruit ripening | 5 | 1% | 0% | 4.40E-03 | 1% | 0% | 1.33E-05 | 1% | 0% | 1.52E-04 |
BH, V. pseudoreticulata accession “Baihe-13-1”; HN, V. pseudoreticulata accession “Hunan-1”; S, V. quinquangularis accession “Shang-24”.
*C: frequency of distinct genes belonging to the corresponding GO terms; B: frequency of all transcripts in each wild Vitis species belonging to the corresponding GO terms.
Plants produce three main secondary metabolites, terpenes, phenolics and nitrogen- and sulfur-containing compounds, which provide a major barrier against the attack of pathogens and herbivores [35,36]. Here, we found that several biological processes related to biosynthesis of phenolic compounds such as coumarin, were enriched in the distinct genes in all three accessions (Table 3). Biosynthesis of flavonoids, a class of phenolic compounds, were enriched in distinct genes of both BH and S. It is worth noting that callose deposition in cell wall and cell wall thickening biological processes and two other processes involved in the metabolism of anthocyanin-containing compounds were enriched in the distinct genes of the S accession. In addition, terpenoid compound metabolism and glucosinolate metabolic related processes were only enriched in the HN distinct genes (Additional file 3).
We found an important category of enriched biological processes that were related to the metabolism and action of plant hormones, a group of small molecules which function as versatile regulators of plant growth, development, reproduction and response to abiotic or biotic stresses [37,38]. Specifically, jasmonic acid related processes were enriched in distinct genes of BH and S, whereas ethylene biosynthetic and metabolic processes were overrepresented in distinct genes of both HN and S. Moreover, abscisic acid related process was only enriched in HN while processes related to salicylic acid and brassinosteroid were enriched in S distinct genes (Additional file 3).
We identified 145 (BH), 215 (HN) and 196 (S) distinct genes that were predicted to encode disease-related proteins (Figure 2C). Interestingly, a large number of them were NBS-LRR genes (81 in BH, 114 in HN and 107 in S), which play important roles in plant effector-triggered immunity (ETI) as they can directly or indirectly detect pathogen-associated proteins [39]. In addition, a large number of genes contained known protein domains related to disease resistance, such as NB-ARC [40], LRR, TIR and Dirigent [41]. Another group of distinct disease-related genes were those encoding receptor like protein kinases (66, 92 and 133 in BH, HN and S, respectively) (Figure 2C). In addition to the genes described above, we discovered several genes that are involved in plant-pathogen interaction, such as Mlo, phytoalexin-deficient 4 (PAD4), enhanced disease susceptibility 1 (EDS1), RPW8.2 and genes encoding lipoxygenases [42-45].
Transcription factors play a central role in mediating biological activity in plant cells. We discovered 19 transcription factors in each of the three Vitis genotypes. More than 70% of these transcription factors belonged to the ethylene-responsive factor family, which has been reported to be involved in the control of primary and secondary metabolism, growth and developmental programs, as well as responses to environmental stresses [46].
SNPs and small indels between wild Chinese Vitis and PN40024
Genomic variations, such as SNPs and small insertions and deletions (indels) are important driving force of genetic diversity. We identified SNPs and small indels through mapping the RNA-Seq reads from each accession to the reference PN40024 genome. We obtained a total of 110,450 SNPs and 2,354 small indels between BH and PN40024, 87,583 SNPs and 2,079 small indels between HN and PN40024, and 89,024 SNPs and 2,085 small indels between S and PN40024. Among these variations, 52% (BH), 48% (HN) and 49% (S) of SNPs, and 3.19% (BH), 3.56% (HN) and 4.22% (S) of small indels were located in the annotated coding regions (Figure 3A and 3B). In BH, we identified ~24,000 nonsynonymous substitutions, which potentially affect approximately 10,000 genes. In the other two accessions, we detected more than 18,000 nonsynonymous mutations that may alter the function of ~9,000 genes. The overall ratio of nonsynonymous to synonymous sites was ~0.7 in all three accessions. Interestingly, this ratio in NBS-LRR genes was substantially higher (1.3 in BH, 2.1 in HN and 3.0 in S). SNPs located at specific regions might have large effects on corresponding genes, such as gain or loss of start/stop codons, and disruption of splice site acceptors or donors [47,48]. We found 32, 24 and 24 genes in BH, HN and S, respectively, contained loss of start codon SNPs which would change the length of protein products (Figure 3C). About 40 genes with loss of stop codons and 70 genes with gain of stop codons were identified in each accession. Moreover, small indels mapped to coding regions (Figure 3B) led to the frame shifts of 74, 72 and 87 genes in BH, HN and S, respectively (Figure 3C). The identified SNPs and small indels were listed in Additional file 4.
Figure 3.
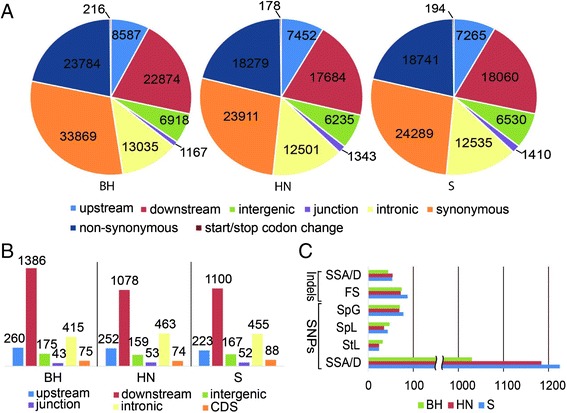
SNPs and small indels between the three Chinese wild Vitis , BH, HN and S, and PN40024. (A) Number of SNPs at different annotated regions of the reference PN40024 genome. (B) Number of small indels at different annotated regions of the reference PN40024 genome. (C) Number of genes affected by SNPs and small indels. SSA/D, splice site acceptor or splice site donor; FS, frame shift; SpG, stop codon gained; SpL, stop codon lost; StL, start codon lost. BH, V. pseudoreticulata accession “Baihe-13-1”; HN, V. pseudoreticulata accession “Hunan-1”; S, V. quinquangularis accession “Shang-24”.
Cis-NATs in wild Chinese Vitis
Natural antisense transcripts (NATs) are endogenous RNA sequences partially or entirely complemented to other transcripts and cis-NAT pairs are two transcripts from the same genomic locus but on opposite strands [49]. We explored cis-NATs in our strand specific RNA-Seq data and identified 112, 145 and 196 cis-NAT pairs in BH, HN and S, respectively (Additional file 5). The classification of cis-NAT pairs based on their overlapping patterns was shown in Table 4. In addition, 84 (75.0%; BH), 115 (79.3%; HN) and 156 (79.6%; S) cis-NAT pairs were designated as coding-noncoding pairs depending on their coding potential calculated by CPC, while 25 (22.3%), 22 (15.2%) and 33 (16.8%) were classified as coding-coding pairs. Cis-NATs were previously reported to be involved in modulating various abiotic and biotic stresses [50,51]. Through functional analysis of the cis-NAT pairs we identified in the three Chinese wild grapes, we also found those related to abiotic stress responses were significantly enriched. In addition, cis-NATs involved in secondary metabolite biosynthetic processes were highly enriched; specifically, among the top five enriched biological processes, four (GO:0009698, GO:0009813, GO:0009812 and GO:0009699), three (GO:0009813, GO:0009812 and GO:0044550) and two (GO:0009813 and GO:0009812) enriched biological processes in BH, HN and S, respectively, were associated with flavonoid metabolic processes (Additional file 6).
Table 4.
Structure analysis of cis -NAT pairs in the three Chinese wild Vitis
| Category | Number of pairs | ||
|---|---|---|---|
| BH | HN | S | |
| Tail to tail (3’ to 3’) | 31 | 43 | 58 |
| Head to head (5’ to 5’) | 28 | 47 | 63 |
| Contained | 53 | 55 | 75 |
| Total | 112 | 145 | 196 |
BH, V. pseudoreticulata accession “Baihe-13-1”; HN, V. pseudoreticulata accession “Hunan-1”; S, V. quinquangularis accession “Shang-24”.
Discussion
In this study, we de novo assembled transcriptomes of three wild Chinese Vitis accessions. Distinct genes and genomic variations between Chinese Vitis and the reference PN40024 were extensively investigated. We identified a total of more than 1,000 distinct protein-coding genes, as well as 600–700 non-coding transcripts from each of the three Chinese wild Vitis. Almost half of the distinct protein-coding genes were functionally related to stimulus responses, and ~30% were involved in defense responses. The enrichment of stress-related processes in distinct genes is consistent with the hypotheses that wild plants, which are often exposed to an adverse environment, such as extreme temperatures, aberrant pH, toxic chemicals and pathogens, tend to have accelerated adaptive evolution of stress genes [52], while during domestication, a large number of stress and defense-related genes are lost in the cultivated species, which might be due to the many years of cultivation and selection that have focused on desirable fruit qualities at the expense of disease resistance [6].
Genomic variations, such as SNPs and indels, are important sources of genetic diversity. They are functionally significant and can cause phenotypic changes. Genetic analysis of plant disease resistance has shown that resistance is dominated by multiple loci and alleles at each locus are often highly polymorphic [53-55]. In the present study, we obtained 87,000-110,000 homozygous SNPs and ~2,000 small indels between each of the three wild Chinese Vitis and V. vinifera, genotype PN40024. This number is distinguished from a previous report on the identification of many fewer SNPs (~59,000) between V. vinifera cv. Corvina and PN40024 transcriptomes [24], probably due to both species belong to V. vinifera.
Cis-NATs are derived from the same genomic loci as their sense counterparts, but from the opposite strand. Cis-NAT pairs can be classified into three groups based on their overlapping patterns, including 3’ end overlap, 5’ end overlap and one entirely contained within the other [49]. In our study, across all three Vitis spp., the majority of cis-NAT pairs were those entirely contained within the other transcripts. This is consistent with the finding in soybean [56] but different from that in Arabidopsis [49], where most cis-NAT pairs have 3’ end overlap. Cis-NATs were reported to participate in plant responses to a wide range of abiotic stresses, such as cold [57] and salt [50] stresses. Consistent with these findings, we found that cis-NATs identified from wild Chinese grapes were significantly enriched with transcripts related to stress responses. In addition, we also observed high enrichment of secondary metabolism-related genes in the wild grape cis-NATs, implying the functional diversity of cis-NATs in mediating important biological processes in plants.
In the present study, we found 6-10% distinct genes were surface-localized receptor like kinases (RLKs). Some RLKs, as well as receptor like proteins (RLPs), are pattern-recognition receptors (PRRs) [58]. They contain various ligand-binding ectodomains that can perceive pathogen-associated molecular patterns (PAMPs), e.g. flagellin, peptidoglycans (PGNs) and chitins, or damage-associated molecular patterns (DAMPs). RLKs (e.g. LYK1) or RLPs (e.g. LYM2) can function independently or cohesively [59]. In this study, we discovered five distinct genes (four in S including one LYK1 and one LYM2 gene, and one in HN) containing the LysM motif, a common unit in both RLKs and RLPs that are responsible for binding to various types of PGNs and chitins. Unlike typical PRRs, Pep1 receptor (PEPR2) acts as a receptor for PEP defense peptides and senses an endogenous elicitor that potentiates PAMP-inducible plant responses [60]. One PEPR2 was found in HN distinct genes. PEPR2 and its closest homolog PEPR1 can interact with BAK1 [61], which is a co-receptor for several PAMPs and regulates their function [58]. In addition, other RLKs that do not belong to PRRs, such as wall-associated kinase (WAK) also play important roles in plant immunity signaling pathway. In this study, we found 7–10 WAK family members in each distinct gene set. WAKs have an extracellular domain and they can interact with pectin and other proteins located in the cell wall. Induction of WAK1 is required for plants to survive during P. syringae infection. Furthermore, the increase of WAK1 mRNA levels is part of the defense response caused by exposure to jasmonic acid (JA), ethylene, or fungi [62]. A previous study has unraveled adaptive evolution of extracellular domains of RLKs [63], which may explain why many RLKs and RLPs were observed in distinct genes. The discovery of these genes may also imply the functional conservation in response to pathogen invasion.
Pathogens can overcome PAMP-triggered immunity (PTI) by deploying effectors to interfere with the PTI activated signaling pathways. These effectors can be perceived by plant R genes (mainly NBS-LRR) and activate plant immune system, also known as effector-triggered immunity. These pathogens are usually highly specialized for specific host plants, and the interaction at the molecular level is often complicated because of the co-evolution of the host and pathogens [64]. Wild and domesticated plant species have been exposed to the natural selection forces, and show divergent R genes as they have to initiate an arms race with the pathogen effector. However, compared to clonally propagated grape cultivars, wild Vitis may have evolved new disease resistance genes during sexual propagation. Several studies have demonstrated that in plant R-gene products, LRR domains are the major determinants of recognition specificity for effectors [65] and these domains were under diversifying selection to increase amino acid variability. Mechanisms for the evolution of new specificities are flexible, such as gene conversion and unequal recombination, as well as accumulation of amino acid codon exchanges in members of anciently duplicated gene families [66]. In our distinct genes, ~8% were NBS-LRR genes. Notably, from our SNP analysis, we found an R gene, GSVIVT01032161001, which has longer protein product in the three wild Vitis compared with that in PN40024 caused by a SNP in its stop codon. The longer R gene might confer increased specificity for pathogen recognition in the three Chinese wild Vitis species. Interestingly, in a wild potato, a homolog of the Vitis longer R gene has been identified, which is heterozygous (six amino acids loss in the 18th LRR repeat for one allele) and confers broad spectrum resistance to late blight [67].
In this study, a number of flavonoid and isoprenoid biosynthesis genes were identified in the distinct gene sets. Flavonoid and isoprenoid biosynthesis was found to be up-regulated in both V. vinifera and V. pseudoreticulata that were inoculated with powdery mildew [27,68]. We also found cis-NATs were enriched with flavonoid biosynthesis-related genes in all three wild grapes, which may suggest that flavonoid metabolism is regulated by cis-NATs. In our distinct gene sets, several genes related to coumarin biosynthesis and metabolism were discovered. Coumarin, a known phytoalexin, was found to accumulate in parsley cells treated with a fungal glucan elicitor [69]. Several other phytoalexin-related genes were also found in our distinct gene sets, including stilbene synthase (STS) which is a key gene in the biosynthesis of stilbenes, and phytoalexin-deficient 4 (PAD4), a well-known resistance gene in Arabidopsis. A recent study showed that Arabidopsis transformed with Vitis enhanced disease susceptibility 1 (EDS1) and PAD4 did not display rescued resistance to powdery mildew, even though these two proteins interacted when transiently expressed in Nicotiana benthamiana. Therefore, involvement of additional interacting proteins might be necessary for resistance to occur [70].
Transcriptional regulation of stress responsive genes plays a central role in abiotic/biotic stress responses. ERF, a subfamily of AP2 transcription factor, has been reported to be involved in many stress responses [71]. Pti4, an ERF in tomato, can be phosphorylated by a disease resistance protein (Pto) and regulate GCC-box PR genes [72]. McGrath et al. [73] found that AP2/ERF genes were the predominant transcription factor family genes responsive to both JA and a fungal pathogen Alternaria brassicicola in Arabidopsis. Specifically, AtERF2 acted as a positive regulator of JA-responsive defense gene expression and resistance to A. brassicico while AtERF4 showed the opposite functions. Their results suggest that plants coordinately express multiple repressor- and activator-type AP2/ERFs during pathogen challenge to modulate defense gene expression and disease resistance [73]. Interestingly, in our distinct gene sets from each of three wild species, 70% of the transcription factors belonged to the AP2/ERF family, which suggest that in Chinese wild Vitis the distinct AP2/ERF genes might co-evolve with R genes and have a high degree of sequence variations.
Conclusions
In the present study we de novo constructed transcriptomes of three Chinese wild grapes, which showed resistances to various fungal pathogens. A comprehensive comparison between these transcriptomes and the reference grape genome unraveled a large number of distinct genes and a rich resource of genetic variations such as SNPs and small indels. Interestingly, many genetic divergences between wild and cultivated grapes were found to be highly related to many important biological processes, particularly defense associated processes, suggesting that the accelerated evolution of these genes may contribute to plant adaptation to different environments. Furthermore, the significant enrichment of cis-NAT pairs related to secondary metabolism and abiotic stress responses may shed lights on the potential regulatory roles of cis-NATs in Chinese wild Vitis.
Methods
Plant material, PM inoculation and RNA-Seq library preparation
Two Chinese wild V. pseudoreticulata accessions “Baihe-13-1” and “Hunan-1”, and one V. quinquangularis accession “Shang-24”, were maintained in the grape germplasm resource orchard at Northwest A&F University, Yangling, China (34° 20’ N, 108°24’ E). Young leaves from three separate vines of each accession were inoculated with PM [Erysiphe necator (Schw.) Burr.] as previously described [18]. E. necator as an obligate biotrophic fungus, grows and reproduces only on living grapes. The isolate we used was obtained from grape leaves showing fully developed PM symptom. To collect samples that had a good represntation for the PM infection process, which would more broadly capture disease related genes responsive to PM infection, we harvested leaves at 0, 6, 12, 24, 48, 72, 96 and 120 hpi based on the proposed PM infection cycle [26,27] and our previous studies [28-30]. The collected leaves were immediately frozen in liquid nitrogen and stored at −80°C till use. Total RNA was extracted following the method described in Guo et al. [30]. The quality and quantity of RNA were assessed by electrophoresis on 1% agarose gels and by a NanoDrop 1000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA), respectively. Strand-specific RNA-Seq libraries were constructed using the protocol described in Zhong et al. [74] and sequenced on the Illumina HiSeq 2000 platform using the single-end mode.
Data processing, de novo assembly and comparison with the reference genome
Raw reads from each of the three Chinese wild Vitis accessions were processed using Trimmomatic [75] to remove adaptor and low quality sequences. Reads shorter than 40 bp were discarded. The resulting reads were aligned to the ribosomal RNA database [76] using bowtie [77] and those aligned were discarded. The resulting high-quality cleaned reads (final reads) were subjected to Trinity [78] for de novo assembly with the minimum kmer coverage set to two. The final reads were then aligned to the assembled contigs using bowtie [77]. To remove false transcripts with antisense direction which was due to the incomplete digestion of the 2nd strand during the strand-specific RNA-Seq library construction [74], contigs with the number of reads aligned in sense direction less than 1/10 of the number of reads aligned in antisense direction were discarded. The assembled contigs were then compared against the GenBank nt database [79], and those having hits from viruses, bacteria, archaea and fungi, but not from plant species, were removed. The final set of assembled contigs were further clustered to remove redundancies using iAssembler [80] with sequence identity cutoff set to 97%. Finally, we used seqclean [81] to trim polyA tails and to remove rRNA sequences at the contig level. The final assembled transcripts were aligned to the 12 × PN40024 genome assembly [82] using BLAT [83] with sequence identity no less than 97%.
Functional annotation of assembled transcripts
To annotate the assembled transcripts, their sequences were searched against the TrEMBL and Swiss-Prot [31] protein databases using the blastx program, with the E-value cutoff of 1e-10. GO terms were assigned to the assembled transcripts based on the GO terms assigned to their hits in TrEMBL and Swiss-Prot databases [84]. The functional terms were assigned to each transcript using automated assignment of human readable descriptions (AHRD) [32]. Enzyme-encoding genes were extracted based on the AHRD result, and used to predict biochemical pathways using the Pathway Tools software [33].
Identification of distinct genes
We first clustered the assembled transcripts under the criteria, sequence identity ≥ 97% and overlap length ≥ 100 bp, to remove redundant transcripts that could be derived from the same gene locus, mainly due to alternative splicing. The longest representative transcript from each cluster was kept and then aligned to the PN40024 genome [82] using BLAT [83] with sequence identity ≥ 97%. The unmapped transcripts, which were considered as distinct transcripts, were then checked for their coding potential using Coding Potential Calculator (CPC) [34]. Transcripts with positive coding potential scores were identified as distinct protein-coding genes and those with negative scores were non-coding transcripts. GO term enrichment analysis of the distinct protein-coding genes was performed using GO::TermFinder [85]. The putative functional domains from Pfam database [86] were identified for these distinct genes using HMMER3.0 [87].
Identification of SNPs and small indels
To identify SNPs and small indels between each of the three Chinese wild accessions and the grape reference, raw RNA-Seq reads were aligned to the grape reference genome using the Burrows-Wheeler Aligner [88]. Only one of the duplicated RNA-Seq reads was kept to minimize the artifacts of PCR amplification, and only reads uniquely mapped to the genome were kept. Following mapping, SNPs and small indels were identified based on the mpileup files generated by SAMtools [89]. The identified SNPs and small indels were supported by at least four distinct RNA-Seq reads and had an allele frequency > 70%. The effects of detected SNPs and small indels were analyzed based on the 12 × V0 annotation of the PN40024 genome.
Identification of cis-NATs
To identify cis-NAT pairs, we first compared the sequences of assembled transcripts from each wild Vitis accession against themselves. Transcript pairs which were aligned in reverse direction and had the overlap length greater than 50 bp were kept. The resulting transcript pairs were then aligned to the reference grape genome and those aligned to the same genome regions and showing distinct splicing patterns were identified as cis-NAT pairs.
Availability of supporting data
The raw sequencing data has been deposited in NBCI SRA under the accession numbers SRP051051, SRP051054 and SRP051078: http://www.ncbi.nlm.nih.gov/sra/?term=SRP051051, http://www.ncbi.nlm.nih.gov/sra/?term=SRP051054, http://www.ncbi.nlm.nih.gov/sra/?term=SRP051078.
Acknowledgements
The authors would like to thank Monica Franciscus for proofreading. This work was supported by grants from National Science Foundation (IOS-0923312 and IOS-1313887) to ZF, National Natural Science Foundation of China (grant no. 31272136) and the Program for Innovative Research Team of Grape Germplasm Resources and Breeding (grant no. 2013KCT-25) to XW. CJ was partially supported by a fellowship from China Scholarship Council.
Additional files
GO classification of assembled transcriptomes of the three Chinese wild Vitis . The figure shows GO term classification of assembled transcripts of the three Chinese wild Vitis, V. pseudoreticulata accession “Baihe-13-1” (BH), V. pseudoreticulata accession “Hunan-1” (HN) and V. quinquangularis accession “Shang-24” (S).
Distinct genes identified in the three Chinese wild Vitis . The table provides the lists of distinct genes identified in the three Chinese wild Vitis, V. pseudoreticulata accession “Baihe-13-1” (BH), V. pseudoreticulata accession “Hunan-1” (HN) and V. quinquangularis accession “Shang-24” (S).
GO terms enriched in the distinct genes from the three Chinese wild Vitis . The table provides the lists of GO terms that were enriched in the distinct genes from the three Chinese wild Vitis, V. pseudoreticulata accession “Baihe-13-1” (BH), V. pseudoreticulata accession “Hunan-1” (HN) and V. quinquangularis accession “Shang-24” (S).
SNPs and small indels between each of the three Chinese wild Vitis . The table provides the lists of SNPs and small indels between the three Chinese wild Vitis, V. pseudoreticulata accession “Baihe-13-1” (BH), V. pseudoreticulata accession “Hunan-1” (HN), V. quinquangularis accession “Shang-24” (S), and V. vinifera PN40024.
cis -NAT pairs identified in the three Chinese wild Vitis . The table provides the lists of cis-NAT pairs identified in the three Chinese wild Vitis, V. pseudoreticulata accession “Baihe-13-1” (BH), V. pseudoreticulata accession “Hunan-1” (HN) and V. quinquangularis accession “Shang-24” (S).
GO terms enriched in cis -NATs from the three Chinese wild Vitis . The table provides the lists of GO terms that were enriched in cis-NATs from the three Chinese wild Vitis, V. pseudoreticulata accession “Baihe-13-1” (BH), V. pseudoreticulata accession “Hunan-1” (HN) and V. quinquangularis accession “Shang-24” (S).
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CJ and MG prepared samples for Illumina sequencing. CJ performed data analysis. CJ and ZF wrote the manuscript. ZF and XW designed the experiment and provided guidance. All authors have read and approved the manuscript.
Contributor Information
Chen Jiao, Email: cj334@cornell.edu.
Min Gao, Email: gaomin.001@163.com.
Xiping Wang, Email: wangxiping@nwsuaf.edu.cn.
Zhangjun Fei, Email: zf25@cornell.edu.
References
- 1.FAOSTAT. http://faostat3.fao.org/
- 2.Owens CL. Grapes. In: Hancock J, editor. Temperate Fruit Crop Breeding. Netherlands: Springer; 2008. pp. 197–233. [Google Scholar]
- 3.Doster MA, Schnathorst WC. Comparative susceptibility of various grapevine cultivars to the powdery mildew fungus Uncinula necator. Am J Enol Viticult. 1985;36:101–104. [Google Scholar]
- 4.Langcake P, Lovell PA. Light and electron microscopical studies of the infection of Vitis spp by Plasmopara viticola, the downy mildew pathogen. Vitis. 1980;19:321–337. [Google Scholar]
- 5.Mortensen JA. Sources and inheritance of resistance to anthracnose in Vitis. J Hered. 1981;72:423–426. [Google Scholar]
- 6.Guo S, Zhang J, Sun H, Salse J, Lucas WJ, Zhang H, et al. The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat Genet. 2013;45:51–58. doi: 10.1038/ng.2470. [DOI] [PubMed] [Google Scholar]
- 7.Yang X, Cheng YF, Deng C, Ma Y, Wang ZW, Chen XH, et al. Comparative transcriptome analysis of eggplant (Solanum melongena L) and turkey berry (Solanum torvum Sw): phylogenomics and disease resistance analysis. BMC Genomics. 2014;15:412. doi: 10.1186/1471-2164-15-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Myles S, Boyko AR, Owens CL, Brown PJ, Grassi F, Aradhya MK, et al. Genetic structure and domestication history of the grape. Proc Natl Acad Sci U S A. 2011;108:3530–3535. doi: 10.1073/pnas.1009363108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barker CL, Donald T, Pauquet J, Ratnaparkhe MB, Bouquet A, Adam-Blondon AF, et al. Genetic and physical mapping of the grapevine powdery mildew resistance gene, Run1, using a bacterial artificial chromosome library. Theor Appl Genet. 2005;111:370–377. doi: 10.1007/s00122-005-2030-8. [DOI] [PubMed] [Google Scholar]
- 10.Hoffmann S, Di Gaspero G, Kovacs L, Howard S, Kiss E, Galbacs Z, et al. Resistance to Erysiphe necator in the grapevine ‘Kishmish vatkana’ is controlled by a single locus through restriction of hyphal growth. Theor Appl Genet. 2008;116:427–438. doi: 10.1007/s00122-007-0680-4. [DOI] [PubMed] [Google Scholar]
- 11.Dalbó MA, Ye GN, Weeden NF, Wilcox WF, Reisch BI. Marker-assisted selection for powdery mildew resistance in grapes. J Am Soc Hortic Sci. 2001;126:83–89. [Google Scholar]
- 12.Fischer BM, Salakhutdinov I, Akkurt M, Eibach R, Edwards KJ, Topfer R, et al. Quantitative trait locus analysis of fungal disease resistance factors on a molecular map of grapevine. Theor Appl Genet. 2004;108:501–515. doi: 10.1007/s00122-003-1445-3. [DOI] [PubMed] [Google Scholar]
- 13.Merdinoglu D, Wiedeman-Merdinoglu S, Coste P, Dumas V, Haetty S, Butterlin G, et al. Genetic analysis of downy mildew resistance derived from Muscadinia rotundifolia. Acta Hort. 2003;603:451–456. [Google Scholar]
- 14.Peressotti E, Wiedemann-Merdinoglu S, Delmotte F, Bellin D, Di Gaspero G, Testolin R, et al. Breakdown of resistance to grapevine downy mildew upon limited deployment of a resistant variety. BMC Plant Biol. 2010;10:147. doi: 10.1186/1471-2229-10-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Gaspero G, Copetti D, Coleman C, Castellarin SD, Eibach R, Kozma P, et al. Selective sweep at the Rpv3 locus during grapevine breeding for downy mildew resistance. Theor Appl Genet. 2012;124:277–286. doi: 10.1007/s00122-011-1703-8. [DOI] [PubMed] [Google Scholar]
- 16.Wan YZ, Schwaninger H, Li D, Simon CJ, Wang YJ, Zhang CH. A review of taxonomic research on Chinese wild grapes. Vitis. 2008;47:81–88. [Google Scholar]
- 17.Carroll JE, Wilcox WF. Effects of humidity on the development of grapevine powdery mildew. Phytopathology. 2003;93:1137–1144. doi: 10.1094/PHYTO.2003.93.9.1137. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Liu Y, He P, Chen J, Lamikanra O, Lu J. Evaluation of foliar resistance to Uncinula necator in Chinese wild Vitis species. Vitis. 1995;34:159–164. [Google Scholar]
- 19.Wan YZ, Schwaninger H, He PC, Wang YJ. Comparison of resistance to powdery mildew and downy mildew in Chinese wild grapes. Vitis. 2007;46:132–136. [Google Scholar]
- 20.Wang Y, Liu Y, He P, Lamikanra O, Lu J. Resistance of Chinese Vitis species to Elsinoe ampelina (de Bary) Shear. Hortscience. 1998;33:123–126. [Google Scholar]
- 21.Li D, Wan Y, Wang Y, He P. Relatedness of resistance to anthracnose and to white rot in Chinese wild grapes. Vitis. 2008;47:213–215. [Google Scholar]
- 22.Zhu L, Wen XY, Li WW. Studies on photosynthetic characteristic of a Chinese wild species grapevine. Acta Hortic Sin. 1994;21:31–34. [Google Scholar]
- 23.Shi J, He M, Cao J, Wang H, Ding J, Jiao Y, et al. The comparative analysis of the potential relationship between resveratrol and stilbene synthase gene family in the development stages of grapes (Vitis quinquangularis and Vitis vinifera) Plant Physiol Biochem. 2014;74:24–32. doi: 10.1016/j.plaphy.2013.10.021. [DOI] [PubMed] [Google Scholar]
- 24.Venturini L, Ferrarini A, Zenoni S, Tornielli GB, Fasoli M, Dal Santo S, et al. De novo transcriptome characterization of Vitis vinifera cv. Corvina unveils varietal diversity. BMC Genomics. 2013;14:41. doi: 10.1186/1471-2164-14-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Da Silva C, Zamperin G, Ferrarini A, Minio A, Dal Molin A, Venturini L, et al. The high polyphenol content of grapevine cultivar tannat berries is conferred primarily by genes that are not shared with the reference genome. Plant Cell. 2013;25:4777–4788. doi: 10.1105/tpc.113.118810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fekete C, Fung RW, Szabo Z, Qiu W, Chang L, Schachtman DP, et al. Up-regulated transcripts in a compatible powdery mildew-grapevine interaction. Plant Physiol Biochem. 2009;47:732–738. doi: 10.1016/j.plaphy.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 27.Fung RW, Gonzalo M, Fekete C, Kovacs LG, He Y, Marsh E, et al. Powdery mildew induces defense-oriented reprogramming of the transcriptome in a susceptible but not in a resistant grapevine. Plant Physiol. 2008;146:236–249. doi: 10.1104/pp.107.108712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Xu Y, Xiao Y, Zhu Z, Xie X, Zhao H, et al. Expression and functional analysis of two genes encoding transcription factors, VpWRKY1 and VpWRKY2, isolated from Chinese wild Vitis pseudoreticulata. Planta. 2010;232:1325–1337. doi: 10.1007/s00425-010-1258-y. [DOI] [PubMed] [Google Scholar]
- 29.Wen Y, Wang X, Xiao S, Wang Y. Ectopic expression of VpALDH2B4, a novel aldehyde dehydrogenase gene from Chinese wild grapevine (Vitis pseudoreticulata), enhances resistance to mildew pathogens and salt stress in Arabidopsis. Planta. 2012;236:525–539. doi: 10.1007/s00425-012-1624-z. [DOI] [PubMed] [Google Scholar]
- 30.Guo R, Xu X, Carole B, Li X, Gao M, Zheng Y, et al. Genome-wide identification, evolutionary and expression analysis of the aspartic protease gene superfamily in grape. BMC Genomics. 2013;14:554. doi: 10.1186/1471-2164-14-554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Consortium UP. Ongoing and future developments at the universal protein resource. Nucleic Acids Res. 2011;39:D214–219. doi: 10.1093/nar/gkq1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.AHRD (Automated Assignment of Human Readable Descriptions). https://github.com/asishallab/AHRD-1
- 33.Karp PD, Paley S, Romero P. The Pathway Tools software. Bioinformatics. 2002;18(Suppl 1):S225–232. doi: 10.1093/bioinformatics/18.suppl_1.S225. [DOI] [PubMed] [Google Scholar]
- 34.Kong L, Zhang Y, Ye ZQ, Liu XQ, Zhao SQ, Wei L, et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35:W345–349. doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dixon RA. Natural products and plant disease resistance. Nature. 2001;411:843–847. doi: 10.1038/35081178. [DOI] [PubMed] [Google Scholar]
- 36.Mithofer A, Boland W. Plant defense against herbivores: chemical aspects. Annu Rev Plant Biol. 2012;63:431–450. doi: 10.1146/annurev-arplant-042110-103854. [DOI] [PubMed] [Google Scholar]
- 37.Pieterse CM, Van der Does D, Zamioudis C, Leon-Reyes A, Van Wees SC. Hormonal modulation of plant immunity. Annu Rev Cell Dev Biol. 2012;28:489–521. doi: 10.1146/annurev-cellbio-092910-154055. [DOI] [PubMed] [Google Scholar]
- 38.Santner A, Calderon-Villalobos LI, Estelle M. Plant hormones are versatile chemical regulators of plant growth. Nat Chem Biol. 2009;5:301–307. doi: 10.1038/nchembio.165. [DOI] [PubMed] [Google Scholar]
- 39.DeYoung BJ, Innes RW. Plant NBS-LRR proteins in pathogen sensing and host defense. Nat Immunol. 2006;7:1243–1249. doi: 10.1038/ni1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van der Biezen EA, Jones JD. The NB-ARC domain: a novel signalling motif shared by plant resistance gene products and regulators of cell death in animals. Curr Biol. 1998;8:R226–227. doi: 10.1016/S0960-9822(98)70145-9. [DOI] [PubMed] [Google Scholar]
- 41.Ralph S, Park JY, Bohlmann J, Mansfield SD. Dirigent proteins in conifer defense: gene discovery, phylogeny, and differential wound- and insect-induced expression of a family of DIR and DIR-like genes in spruce (Picea spp.) Plant Mol Biol. 2006;60:21–40. doi: 10.1007/s11103-005-2226-y. [DOI] [PubMed] [Google Scholar]
- 42.Glazebrook J, Zook M, Mert F, Kagan I, Rogers EE, Crute IR, et al. Phytoalexin-deficient mutants of Arabidopsis reveal that PAD4 encodes a regulatory factor and that four PAD genes contribute to downy mildew resistance. Genetics. 1997;146:381–392. doi: 10.1093/genetics/146.1.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aarts N, Metz M, Holub E, Staskawicz BJ, Daniels MJ, Parker JE. Different requirements for EDS1 and NDR1 by disease resistance genes define at least two R gene-mediated signaling pathways in Arabidopsis. Proc Natl Acad Sci U S A. 1998;95:10306–10311. doi: 10.1073/pnas.95.17.10306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xiao S, Ellwood S, Calis O, Patrick E, Li T, Coleman M, et al. Broad-spectrum mildew resistance in Arabidopsis thaliana mediated by RPW8. Science. 2001;291:118–120. doi: 10.1126/science.291.5501.118. [DOI] [PubMed] [Google Scholar]
- 45.Buschges R, Hollricher K, Panstruga R, Simons G, Wolter M, Frijters A, et al. The barley Mlo gene: a novel control element of plant pathogen resistance. Cell. 1997;88:695–705. doi: 10.1016/S0092-8674(00)81912-1. [DOI] [PubMed] [Google Scholar]
- 46.Licausi F, Ohme-Takagi M, Perata P. APETALA2/Ethylene Responsive Factor (AP2/ERF) transcription factors: mediators of stress responses and developmental programs. New Phytol. 2013;199:639–649. doi: 10.1111/nph.12291. [DOI] [PubMed] [Google Scholar]
- 47.Jiao WB, Huang D, Xing F, Hu Y, Deng XX, Xu Q, et al. Genome-wide characterization and expression analysis of genetic variants in sweet orange. Plant J. 2013;75:954–964. doi: 10.1111/tpj.12254. [DOI] [PubMed] [Google Scholar]
- 48.Lai J, Li R, Xu X, Jin W, Xu M, Zhao H, et al. Genome-wide patterns of genetic variation among elite maize inbred lines. Nat Genet. 2010;42:1027–1030. doi: 10.1038/ng.684. [DOI] [PubMed] [Google Scholar]
- 49.Wang XJ, Gaasterland T, Chua NH. Genome-wide prediction and identification of cis-natural antisense transcripts in Arabidopsis thaliana. Genome Biol. 2005;6:R30. doi: 10.1186/gb-2005-6-4-r30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Borsani O, Zhu J, Verslues PE, Sunkar R, Zhu JK. Endogenous siRNAs derived from a pair of natural cis-antisense transcripts regulate salt tolerance in Arabidopsis. Cell. 2005;123:1279–1291. doi: 10.1016/j.cell.2005.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Katiyar-Agarwal S, Morgan R, Dahlbeck D, Borsani O, Villegas A, Jr, Zhu JK, et al. A pathogen-inducible endogenous siRNA in plant immunity. Proc Natl Acad Sci U S A. 2006;103:18002–18007. doi: 10.1073/pnas.0608258103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen S, Krinsky BH, Long M. New genes as drivers of phenotypic evolution. Nat Rev Genet. 2013;14:645–660. doi: 10.1038/nrg3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ellis J, Dodds P, Pryor T. The generation of plant disease resistance gene specificities. Trends Plant Sci. 2000;5:373–379. doi: 10.1016/S1360-1385(00)01694-0. [DOI] [PubMed] [Google Scholar]
- 54.Noel L, Moores TL, van Der Biezen EA, Parniske M, Daniels MJ, Parker JE, et al. Pronounced intraspecific haplotype divergence at the RPP5 complex disease resistance locus of Arabidopsis. Plant Cell. 1999;11:2099–2112. doi: 10.1105/tpc.11.11.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Parniske M, Hammond-Kosack KE, Golstein C, Thomas CM, Jones DA, Harrison K, et al. Novel disease resistance specificities result from sequence exchange between tandemly repeated genes at the Cf-4/9 locus of tomato. Cell. 1997;91:821–832. doi: 10.1016/S0092-8674(00)80470-5. [DOI] [PubMed] [Google Scholar]
- 56.Hu Z, Jiang QY, Ni ZY, Zhang H. Prediction and identification of natural antisense transcripts and their small RNAs in soybean (Glycine max) BMC Genomics. 2013;14:230. doi: 10.1186/1471-2164-14-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Swiezewski S, Liu F, Magusin A, Dean C. Cold-induced silencing by long antisense transcripts of an Arabidopsis Polycomb target. Nature. 2009;462:799–802. doi: 10.1038/nature08618. [DOI] [PubMed] [Google Scholar]
- 58.Zipfel C. Plant pattern-recognition receptors. Trends Immunol. 2014;35:345–351. doi: 10.1016/j.it.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 59.Hayafune M, Berisio R, Marchetti R, Silipo A, Kayama M, Desaki Y, et al. Chitin-induced activation of immune signaling by the rice receptor CEBiP relies on a unique sandwich-type dimerization. Proc Natl Acad Sci U S A. 2014;111:E404–413. doi: 10.1073/pnas.1312099111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yamaguchi Y, Huffaker A, Bryan AC, Tax FE, Ryan CA. PEPR2 is a second receptor for the Pep1 and Pep2 peptides and contributes to defense responses in Arabidopsis. Plant Cell. 2010;22:508–522. doi: 10.1105/tpc.109.068874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Postel S, Kufner I, Beuter C, Mazzotta S, Schwedt A, Borlotti A, et al. The multifunctional leucine-rich repeat receptor kinase BAK1 is implicated in Arabidopsis development and immunity. Eur J Cell Biol. 2010;89:169–174. doi: 10.1016/j.ejcb.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 62.Verica JA, He ZH. The cell wall-associated kinase (WAK) and WAK-like kinase gene family. Plant Physiol. 2002;129:455–459. doi: 10.1104/pp.011028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shiu SH, Karlowski WM, Pan R, Tzeng YH, Mayer KF, Li WH. Comparative analysis of the receptor-like kinase family in Arabidopsis and rice. Plant Cell. 2004;16:1220–1234. doi: 10.1105/tpc.020834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meyers BC, Kaushik S, Nandety RS. Evolving disease resistance genes. Curr Opin Plant Biol. 2005;8:129–134. doi: 10.1016/j.pbi.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 65.Ellis J, Dodds P, Pryor T. Structure, function and evolution of plant disease resistance genes. Curr Opin Plant Biol. 2000;3:278–284. doi: 10.1016/S1369-5266(00)00080-7. [DOI] [PubMed] [Google Scholar]
- 66.Dangl JL, Jones JD. Plant pathogens and integrated defence responses to infection. Nature. 2001;411:826–833. doi: 10.1038/35081161. [DOI] [PubMed] [Google Scholar]
- 67.Song J, Bradeen JM, Naess SK, Raasch JA, Wielgus SM, Haberlach GT, et al. Gene RB cloned from Solanum bulbocastanum confers broad spectrum resistance to potato late blight. Proc Natl Acad Sci U S A. 2003;100:9128–9133. doi: 10.1073/pnas.1533501100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weng K, Li Z-Q, Liu R-Q, Wang L, Wang Y-J, Xu Y. Transcriptome of Erysiphe necator-infected Vitis pseudoreticulata leaves provides insight into grapevine resistance to powdery mildew. Horti Res. 2014;1:14049. doi: 10.1038/hortres.2014.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Davis KR, Hahlbrock K. Induction of defense responses in cultured parsley cells by plant cell wall fragments. Plant Physiol. 1987;84:1286–1290. doi: 10.1104/pp.84.4.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gao F, Dai R, Pike SM, Qiu W, Gassmann W. Functions of EDS1-like and PAD4 genes in grapevine defenses against powdery mildew. Plant Mol Biol. 2014;86:381–393. doi: 10.1007/s11103-014-0235-4. [DOI] [PubMed] [Google Scholar]
- 71.Singh K, Foley RC, Onate-Sanchez L. Transcription factors in plant defense and stress responses. Curr Opin Plant Biol. 2002;5:430–436. doi: 10.1016/S1369-5266(02)00289-3. [DOI] [PubMed] [Google Scholar]
- 72.Gu YQ, Yang C, Thara VK, Zhou J, Martin GB. Pti4 is induced by ethylene and salicylic acid, and its product is phosphorylated by the Pto kinase. Plant Cell. 2000;12:771–785. doi: 10.1105/tpc.12.5.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McGrath KC, Dombrecht B, Manners JM, Schenk PM, Edgar CI, Maclean DJ, et al. Repressor- and activator-type ethylene response factors functioning in jasmonate signaling and disease resistance identified via a genome-wide screen of Arabidopsis transcription factor gene expression. Plant Physiol. 2005;139:949–959. doi: 10.1104/pp.105.068544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhong S, Joung JG, Zheng Y, Chen YR, Liu B, Shao Y, et al. High-throughput Illumina strand-specific RNA sequencing library preparation. Cold Spring Harb Protoc. 2011;2011:940–949. doi: 10.1101/pdb.prot5652. [DOI] [PubMed] [Google Scholar]
- 75.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, et al. GenBank. Nucleic Acids Res. 2013;41:D36–42. doi: 10.1093/nar/gks1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zheng Y, Zhao L, Gao J, Fei Z. iAssembler: a package for de novo assembly of Roche-454/Sanger transcriptome sequences. BMC Bioinformatics. 2011;12:453. doi: 10.1186/1471-2105-12-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Seqclean program. http://sourceforge.net/projects/seqclean/files/
- 82.Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449:463–467. doi: 10.1038/nature06148. [DOI] [PubMed] [Google Scholar]
- 83.Kent WJ. BLAT - The BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202.ArticlepublishedonlinebeforeMarch2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Barrell D, Dimmer E, Huntley RP, Binns D, O’Donovan C, Apweiler R. The GOA database in 2009–an integrated Gene Ontology Annotation resource. Nucleic Acids Res. 2009;37:D396–403. doi: 10.1093/nar/gkn803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Boyle EI, Weng S, Gollub J, Jin H, Botstein D, Cherry JM, et al. GO::TermFinder–open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics. 2004;20:3710–3715. doi: 10.1093/bioinformatics/bth456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. 2011;7:e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. Genome Project Data Processing S: The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]