

Abstract
Background
Hereditary medullary thyroid carcinoma (MTC) is caused by germ-line gain of function mutations in the RET proto-oncogene, and a phenotypic variability among carriers of the same mutation has been reported. We recently observed this phenomenon in a large familial MTC (FMTC) family carrying the RET-S891A mutation. Among genetic modifiers affecting RET-driven MTC, a role has been hypothesized for RET-G691S non-synonymous polymorphism, though the issue remains controversial. Aim of this study was to define the in vitro contribution of RET-G691S to the oncogenic potential of the RET-S891A, previously shown to harbour low transforming activity.
Methods
The RET-S891A and RET-G691S/S891A mutants were generated by site-directed mutagenesis, transiently transfected in HEK293T cells and stably expressed in NIH3T3 cells. Their oncogenic potential was defined by assessing the migration ability by wound healing assay and the anchorage-independent growth by soft agar assay in NIH3T3 cells stably expressing either the single or the double mutants. Two RET-S891A families were characterised for the presence of RET-G691S.
Results
The functional studies demonstrated that RET-G691S/S891A double mutant displays a higher oncogenic potential than RET-S891A single mutant, assessed by focus formation and migration ability. Moreover, among the 25 RET-S891A carriers, a trend towards an earlier age of diagnosis was found in the MTC patients harboring RET-S891A in association with RET-G691S.
Conclusions
We demonstrate that the RET-G691S non-synonymous polymorphism enhances in vitro the oncogenic activity of RET-S891A. Moreover, an effect on the phenotype was observed in the RET-G691S/S891A patients, thus suggesting that the analysis of this polymorphism could contribute to the decision on the more appropriate clinical and follow-up management.
Electronic supplementary material
The online version of this article (doi:10.1186/s13023-015-0231-z) contains supplementary material, which is available to authorized users.
Keywords: RET, Medullary thyroid cancer, G691S, S891A, Polymorphism
Background
Medullary Thyroid Cancer (MTC) is a neuroendocrine neoplasia arising from thyroid parafollicular C cells. Hereditary forms account for 25% of cases and include multiple endocrine neoplasia syndromes type 2A (MEN2A), type 2B (MEN2B) and familial MTC (FMTC), caused by mutations in the rearranged during transfection (RET) proto-oncogene. The RET gene, which encodes a tyrosine kinase receptor with a crucial role in development, comprises 21 exons and generates a transcript subjected to alternative splicing leading to two main isoforms: a protein of 1114 residues displaying 51 C-terminal-specific amino acids (RET51) and a shorter protein of 1072 residues displaying nine unrelated C-terminal-specific amino acids (RET9) [1].
Beyond the known role of RET gain-of-function mutations in MTC, in recent years several authors investigated whether the presence of single nucleotide polymorphisms (SNPs) could be associated with susceptibility for the development or progression of MTC [2-5]. Among RET polymorphisms, RET-G691S, localized in exon 11 and reported in general population with an allele frequency of 11–33% (ARUP database “MEN2 and RET”, displaying sequence variation and clinical information [6]), is a non-synonymous variant [7-9], and could potentially affect protein function. Moreover, RET-G691S has been suggested to be a genetic modifier in MEN2A, related to an earlier age at presentation [7,9,10] and has been associated to the susceptibility to sporadic MTC [8,11-13], though controversial data have been reported about this issue [14].
Interestingly, RET-G691S has been also hypothesized to play a functional role on tumor growth and aggressiveness in pancreatic cancers and cutaneous melanoma, where it works as a genetic modifier or even as a low penetrance gene [15,16]. In particular, RET-G691S was reported to enhance the activation of the downstream ERK pathway compared to RET-wt [15], though this variant was not found to be oncogenic per se by focus formation assays [5,17].
Consistently with the potential modifier role of RET-G691S in MTC, Vandenbosch et al. observed that this polymorphism is able to enhance RET oncogenicity of mutations affecting RET codon 666, by increasing its penetrance in the clinical onset [18]. Accordingly, we firstly demonstrated the in vitro effect of the RET-G691S variant as enhancer of the ERK1/2 activation and of the transforming activity of the MTC-associated RET-K666E mutant [17].
In the present study, we investigated more deeply the potential role of this non synonymous RET polymorphism as a modifier of the phenotypic expression in familial MTC. To this purpose, we focused on the potential contribution of RET-G691S to the oncogenicity of RET-S891A, a RET mutant previously reported to be associated with low transforming activity. In particular, this gain-of-function mutation, located in the second intracellular tyrosine kinase domain of the RET proto-oncogene, accounts for less than 5% of all RET mutated patients, and can cause FMTC or the phenotype associated to MEN2A [19-23].
Since the functional characterization of RET mutants adds useful information to the genotyping of patients/families optimizing the diagnostic and clinical management, we both evaluated the in vitro biological activity of the RET-G691S polymorphism on the oncogenic potential of RET-S891A and carried out expression/penetrance studies in two unrelated RET-S891A FMTC families.
Methods
In vitro analyses
Construction of the RET mutants
All RET mutants were obtained by mutagenesis of RET51-wt construct (pCDNA3 vector expressing the proto-RET gene long isoform). RET51-G691S, RET51-C634R (containing an MEN2A causing mutation), and RET51-M918T (containing the main MEN2B causing mutation) used as controls are described elsewhere [17].
RET51-S891A and RET51-G691S/S891A were obtained by site-directed mutagenesis of RET51-WT using an in vitro oligonucleotide mutagenesis system (Quik-Change XL site-directed mutagenesis; Stratagene, La Jolla, CA, USA). The transition c.2071G > A leading to G691S polymorphism (SNIP rs1799939) and the transversion c.2671 T > G leading to S891A mutation were verified by DNA direct sequencing and then the mutant clones were entirely sequenced to exclude possible additional mutations. Plasmid DNA was extracted using the MAXI PREP Kit (Qiagen) as suggested by the supplier.
Cell culture, transfections and focus formation assay
Human HEK293T cells were maintained in DMEM with 10% FCS. The recombinant plasmids were transiently transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions.
NIH3T3 cells were cultured in DMEM supplemented with 10% serum (Colorado Company, CO). Stable transfection was performed by the CaPO4 method [24] using 100 ng plasmid DNA together with 10 μg NIH3T3-derived DNA carrier. Transfected cells were grown in DMEM with 10% serum and selected in the presence of G418 antibiotic (650 μg/ml) to obtain G418-resistant colonies indicative of transfection efficiency; transformed foci were obtained in DMEM with 5% serum in the presence of chronic stimulation with the RET ligand GDNF at a concentration of 10 ng/ml (AlomoneLabs, Jerusalem, Israel). Both G418-resistant colonies and transformed foci were fixed and counted to determine the transforming activity, calculated as foci number/colonies number ratio. NIH3T3 cell lines stably expressing RET mutants were derived from transformation foci picked from parallel original plates of NIH3T3 transfected cells and cultured in the presence of chronic stimulation of GDNF (10 ng/ml).
Western blot analysis
Cells were lysed in ice-cold RIPA buffer (20 mM Tris pH 7.4, 137 mM NaCl, 10% glycerol, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100, 2 mM EDTA pH 8.0) containing protease and phosphatase inhibitors.
Proteins were quantified using a modified Bradford assay (Bio-Rad). Protein samples were boiled in NuPAGE LDS sample buffer (Invitrogen), separated by NuPAGE Novex Gels with the appropriate running buffer (Invitrogen), then transferred onto nitrocellulose filters, and immunoblotted with the indicated antibodies. Anti-RET (C-20), anti-RET (H300) and anti-phospho-RET are from Santa Cruz Biotechnology (Santa Cruz, CA, USA); anti-MAP kinase (ERK1/2), anti-MAP kinase activated (pERK1/2), anti-vinculin and anti-β-tubulin are from Sigma Aldrich (Saint Louis, Missouri, USA).
Densitometric analyses were performed by the Quantity One 4.6.6 software (Bio-Rad, Hercules, CA).
Wound healing assay
NIH3T3 cells stably expressing RET mutants were seeded at 30000 cells/chamber in a culture insert (Ibidi #80209) placed in 24-well plate and incubated overnight. Inserts were then removed to generate a 500 μm gap between cells. Following PBS wash and fresh medium supplement, plates were placed in the Cell-IQ SLF instrument (CM Technology Oy, Tampere, Finland) and cultured at 37°C, 5% CO2. Two images for well were taken each hour for 24 h using Cell-IQ Imagen software (CM Technology Oy) to monitor the gap closure. Images were then analyzed with the Scratch wound measurement tool of the Cell-IQ Analyser software (CM Technology Oy), to evaluate the % of closed area calculated with the equation: (Start wound - wound [μm2])/Start wound × 100. Data and graphs were analyzed using GraphPad Prism 5.02.
Soft-agar assay
For anchorage-independent growth assay, NIH3T3 cells stably expressing RET mutants were suspended in DMEM containing 0.33% agar, 10% serum and GDFN 10 ng/ml (50000 cells/1.5 ml medium) and added into a layer of medium containing 0.5% agar and 10% serum in 60 mm dishes. After 3 weeks incubation, plates were analyzed for colonies number and size. Colony number was determined in plates stained with p-iodonitrotetrazolium chloride violet (Sigma Aldrich).
Agar colonies size, calculated as mean diameter, was determined by the count/size tool in Image-Pro Plus 7.0.1 software analyzing the agar colonies images the day before staining. A cutoff area of 250 pixel was manually set on NIH3T3 control cells. Only foci surpassing the cut-off area were automatically selected and scored by the software. Diameter data relative to the total foci analyzed per each field were exported and the mean diameter for each sample was calculated.
Patients: clinical and molecular characterization
RET-G691S polymorphism expression/penetrance was assessed in two FMTC families carrying the germline activating mutation RET-S891A. Family 1 is an extremely large family with members distributed in several cities of the North of Italy, with a common ancestor [25]. Data related to some members of Family 1 have been partially reported in a previous study [23]. Family 2, from Southern Italy, is composed by 2 generations for a total of 3 subjects. Clinical and molecular features, including age at diagnosis, gender, TNM staging [26] and the disease’s outcome after a mean follow-up time of 60 months were obtained for both families. Patients were diagnosed and treated according to the American guidelines for the management of MTC [27,28]; in particular, RET gene carriers were screened by means of yearly basal and stimulated CT measurement and neck ultrasound. Prophylactic surgery was performed at the first evidence of a positive provocative test.
Genomic DNA was extracted from peripheral blood leucocytes of FMTC families members and the presence of RET-G691S polymorphism was assessed by PCR and direct sequencing as previously described [23]. All patients gave their informed consent, approved by the local Ethical Committee, to the genetic characterization and to the analysis of data.
Statistical analysis
Statistical analysis and graphs were generated using GraphPad Prism version 5.02. Comparisons between two groups were performed by two-tailed Student’s t-test with unequal variance. A value of P < 0.05 was considered statistically significant.
Results
Transforming activity of RET-S891A and RET-G691S/S891A mutants
To assess the contribution of the RET-G691S polymorphism to the in vitro oncogenic potential of RET-S891A mutant, both localized in the intracellular domains of RET protein (Figure 1A), we generated RET-S891A single and RET-G691S/S891A double mutants by site-directed mutagenesis of recombinant plasmids carrying the RET-wt long isoform.
Figure 1.
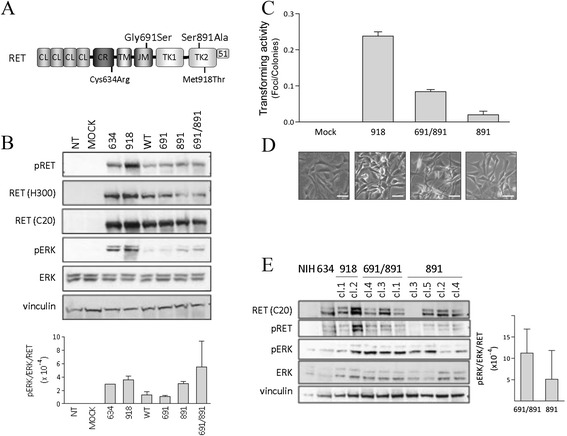
RET-S891A and RET -G691S/S891A mutants biochemical characterization and transforming activity. A. RET protein domains (CL, cadherin-like; CR, Cysteine-rich; TM, transmembrane; JM, juxtamembrane; TK, tyrosine kinase) and the RET51 isoform C-terminal tail are indicated. B. Western blot analysis of RET protein and associated ERK signaling in HEK293T cells transiently transfected. RET was detected by the antibody RET H300, specific for the RET extracellular portion, and by RET C20, specific for the intracellular portion of the RET long isoform. Vinculin is shown as protein loading control. Protein levels were quantified by densitometric analysis by Quantity One 4.6.6 software. Data are shown as mean expression value ± SD for 2 independent transfections. C. RET-S891A and RET-G691S/S891A mutants transforming activity by focus formation assay in NIH3T3 cells. pCDNA3 empty vector (Mock) and RET-M918T are included as negative and positive controls, respectively. The transforming activity, calculated as foci number/colonies number ratio, is reported as mean ± SD of triplicate counts. Data from one representative of two independent transfections are shown. D. Representative images of foci-derived clones. Scale bar 50um. E. Western blot analysis of RET protein and associated ERK signaling in NIH3T3 cells stably expressing the indicated RET variants: 634, RET-C634R; 918, RET-M918T; 691/891, RET-G691S/S891A; 891, RET-S891A. NIH, NIH3T3 untransfected cells. The partially and fully glycosylated forms of RET are identified in NIH3T3 cells. Vinculin is shown as protein loading control. Three RET-G691S/S891A clones and four RET-S891A clones were analysed. The ratio pERK/ERK/RET was calculated for each clone and the mean among clones ± SD was reported.
The obtained mutants, the control mutants (MEN2A-associated RET-C634R, MEN2B-associated RET-M918T, RET-wt and RET-G691S) and the empty vector were transiently transfected into HEK293T cells and the RET proteins and the associated downstream signaling on ERK1/2 were analyzed (Figure 1B). Although slightly heterogeneous, all the constructs express detectable levels of RET protein. Interestingly, the RET-G691S/S891A double mutant appears to activate ERK1/2 pathway more than the RET-S891A single mutant, as suggested by densitometric quantification (Figure 1B, lower panel).
Then, in order to analyze the biological activity of both mutants, we performed focus formation assay. The RET-S891A single and RET-G691S/S891A double mutants, the empty vector (negative control) and RET-M918T mutant (positive control), were stably transfected in NIH3T3 cells and the transforming activity was assessed (Figure 1C).
The RET-S891A single mutant shows transforming activity lower than the well known RET-M918T mutant, confirming previous report that indicates RET-S891A mutation associated to low transforming activity [21]. Interestingly, RET-G691S/S891A double mutant displays a threefold higher transforming activity than RET-S891A single mutant (0.09 vs. 0.03 respectively), suggesting for this polymorphism a role of transforming potential enhancer. This is also suggested by the cells morphology (Figure 1D). Indeed, whereas RET-S891A-derived cells display an apparently more flat morphology, RET-G691S/S891A-derived cells show a spindle shape morphology with contact inhibition loss, more similar to RET-M918T-derived control cells.
Western blot analyses performed on stable clones derived from transfection foci show that cells expressing the RET-G691S/S891A double mutant display on average higher levels of ERK1/2 phosphorylation, indicative of ERK1/2 pathway activation, than RET-S891A single mutant (Figure 1E), similarly to what shown in transient transfections (Figure 1B, lower panel).
Cells expressing either RET-S891A or RET-G691S/S891A display different migratory and anchorage-independent growth abilities
To better characterize the tumorigenic properties of RET-S891A single and RET-G691S/S891A double mutants, we performed the wound healing assay (Figure 2). The wound closure capacity, indicative of migration ability, was assessed for both mutants in two representative clones expressing different levels of RET protein (Figure 2A). Data indicate that RET-S891A single mutants display a statistically significant reduced capacity to close the wound compared with the RET-G691S/S891A double mutants. Of note, the presence of the 691S variant enhances the wound closure ability in clones expressing both high levels of RET (691/891 clone 3 vs. 891 clone 5) and low levels of RET (691/891 clone 1 vs. 891 clone 4).
Figure 2.
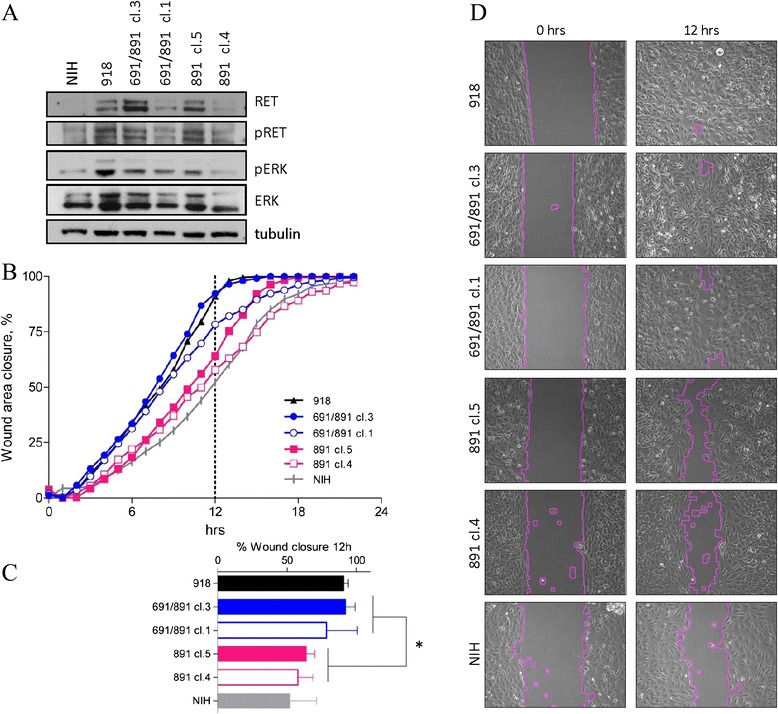
RET-S891A and RET -G691S/S891A stable mutants migration ability by wound healing assay. A. Western blot analysis of RET protein and associated ERK signaling in NIH3T3 cells stably expressing either RET-S891A or RET-G691S/S891A. RET was detected by RET C20 primary antibody. Tubulin is shown as protein loading control. Two clones expressing different levels of RET were analyzed for both variants. NIH3T3 cells and RET-M918T-expressing cells are included as negative and positive controls, respectively. B. Wound-healing assay in RET stable mutants. The wound closure percentage was quantified each hour for 24 h post-wound. Graphs are mean of two independent experiments performed in duplicate. C. Wound closure percentage at 12 h post-wound. Data are shown as mean ± SD of two independent experiments performed in duplicate. Statistical significance determined by Two-tailed Unpaired t test. *p < 0.05. D. Representative images at 0 h and 12 h post-wound (x10).
Moreover, the anchorage-independent growth ability was assessed by soft agar assay in cells expressing the single and double mutants (Figure 3). On average RET-G691S/S891A double mutants give rise to a statistically significant higher number of agar colonies than RET-S891A single mutants (Figure 3A). In addition, the size of agar colonies originated by RET-G691S/S891A double mutants results statistically significant higher than those generated by RET-S891A single mutants (Figure 3B). To be noted, RET-M918T mutant generates agar colonies bigger than RET-G691S/S891A double mutants in agreement with its reported high oncogenic potential. These results indicate that in RET mutants the presence of the G691S polymorphism enhances the oncogenic properties, such as the migration and the agar colony formation ability.
Figure 3.
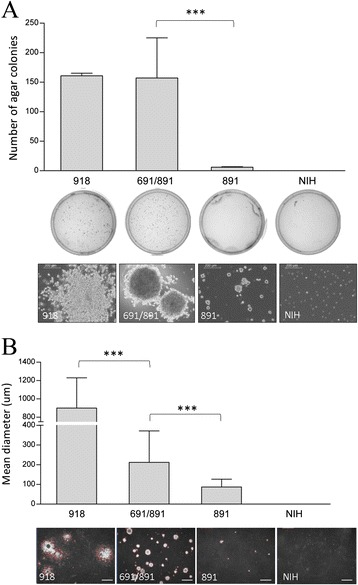
RET -S891A and RET -G691S/S891A stable mutants anchorage-independent growth ability by soft- agar assay. A. Agar colonies in NIH3T3 cells stably expressing either RET-S891A or RET-G691S/S891A. NIH3T3 cells and RET-M918T-expressing cells are included as negative and positive controls. The mean ± SD of duplicate counts is reported. For RET-S891A and RET-G691S/S891A mutants two independent clones were analyzed and the mean between clones is reported. Data from one representative of two independent soft-agar assays are shown. Below representative images of the plates scored for agar colonies number and the corresponding pictures of agar colonies the day before fixing and staining (scale bar 200 um). B. Agar colonies mean diameter determined by Image-Pro Plus 7.0.1 software. Data are presented as mean diameter ± SD for each sample. For RET-S891A and RET-G691S/S891A two independent clones were analyzed and the mean between clones is reported. ***P < 0.0001 statistical significance determined via Student’s t-test. Below representative images of agar colonies used for diameter measurement (LEICA inverted microscope, scale bar 1000 μm).
Allele frequency of RET-G691S polymorphism in FMTC cases harboring RET-S891A mutation and correlation between genetic and clinical features
The RET-S891A germline mutation was present in 39/69 members of the two FMTC families analyzed, whereas the RET-G691S polymorphism was documented in 16/69 cases (23.2%). Among the 39 RET-S891A carriers, 14 also harbored the RET-G691S polymorphism either in heterozygosis (n = 13) or in homozygosis (n = 1), showing an overall allele frequency of 19% (15 mutated alleles out of 78 alleles). Thus, in the present series, the prevalence of the co-segregation RET891/RET691 was similar to that reported in general population by the publicly available ARUP database [6]. Interestingly, RET-G691S appears to display a trans presentation with RET-S891A in Family 1, whereas a cis presentation is observed in Family 2 (Table 1). In 25/39 RET-S891A carriers, the clinical records were available, and they were thus classified according to the histologic diagnosis: 13 MTC, 6 CCH and 6 non C-cells disease (Table 1).
Table 1.
Features and 691 polymorphic status of patients harboring the RET-S891A germline mutation
| Patient | Gender | Age at Dx (years) | RET 691 | 691 variant | Diagnosis | TNM | Stage | |
|---|---|---|---|---|---|---|---|---|
| Family 1 | 6 TL | M | 15 | G/G | wt | Normal | - | - |
| 7 RA | M | 18 | G/G | wt | Normal | - | - | |
| 1 BS | M | 5 | G/S | polymorphic | Normal | - | - | |
| 2 RF | M | 7 | G/S | polymorphic | Normal | - | - | |
| 4 RM | M | 9 | G/S | polymorphic | Normal | - | - | |
| 10 RE | F | 27 | G/S | polymorphic | Normal | - | - | |
| 3 TM | M | 8 | G/G | wt | CCH | - | - | |
| 8 MM | M | 24 | G/G | wt | CCH | - | - | |
| 21 RP | M | 56 | G/G | wt | CCH | - | - | |
| 5 BM | M | 10 | G/S | polymorphic | CCH | - | - | |
| 9 RL | M | 24 | G/S | polymorphic | CCH | - | - | |
| 11 RI | F | 27 | G/S | polymorphic | CCH | - | - | |
| 16 FE | F | 42 | G/G | wt | MTC | pT1N0M0 | I | |
| 17 BA | F | 43 | G/G | wt | MTC | pT1N0M0 | I | |
| 18 FA | M | 43 | G/G | wt | MTC | pT1N0M0 | I | |
| 19 BC | F | 46 | G/G | wt | MTC | pT3N1bM0 | IVA | |
| 20 MC | F | 54 | G/G | wt | MTC | pT2N1aM0 | III | |
| 22 RU | M | 62 | G/G | wt | MTC | pT1NXM0 | I | |
| 25 TG | F | 76 | G/G | wt | MTC | pT1N1aM0 | III | |
| 12 RS | F | 29 | G/S | polymorphic | MTC | pT1NXM0 | I | |
| 23 RL | F | 71 | G/S | polymorphic | MTC | pT2NXM0 | II | |
| 13 RF | M | 30 | G/S | polymorphic | MTC | pT1N1aM0 | I | |
| Family 2 | 14 VV | F | 35 | G/S | polymorphic | MTC | pT1NXM0 | I |
| 15VN* | M | 38 | G/S | polymorphic | MTC | pT1NXM0 | I | |
| 24 SV* | F | 74 | S/S | polymorphic† | MTC | pT2N1aM0 | III |
Age at Dx, age at diagnosis; G/G, Glycine/Glycine; G/S, Glycine/Serine; S/S, Serine/Serine; wt, wild type; Normal, absence of C-cells disease; CCH, C cells hyperplasia; MTC, medullary thyroid cancer;
*elevated urinary normetanephrine levels.
†RET-G691S polymorphism in homozygosis.
The Table includes the 25 patients for whom clinical data are available.
Interestingly, even though not statistically significant, a trend towards an earlier age at diagnosis was found in MTC patients harboring the RET-G691S polymorphism either considering MTC patients of Family 1 (median age 30 vs. 46 years; Additional file 1: Figure S1A), or the two families all together (median age 36.5 vs. 46 years; Additional file 1: Figure S1B) thus confirming in vivo the role of this polymorphism in the enhancement of the oncogenic activity of RET-S891A. Consistently, 2/3 members of Family 2 carrying RET-G691S/S891A had elevated urinary normetanephrine levels (#VN 445.3 mcg/24 h, nv <390 μg/24 h and #SV 578 mcg/24 h, nv <354 μg/24 h), though no lesion suspicious for pheochromocytoma (PHEO) have been yet identified using specific imaging screening (CT scan, FDG PET and MIBG scintiscan). Finally, no significant differences were noted in the outcome of patients with or without RET-G691S, likely as a consequence of the bias introduced by prophylactic thyroidectomy in gene carriers.
Discussion
In this study we demonstrated for the first time that the functional polymorphic variant RET-G691S, not oncogenic per se, enhances the in vitro oncogenic potential of the RET-S891A, a mutant causing hereditary MTC. Furthermore, we showed in two FMTC families that carriers of RET-S891A mutation associated with the RET-G691S polymorphic variant display a trend towards an earlier age at diagnosis of MTC and the adrenal medulla involvement.
RET mutations in hereditary MTC are a paradigmatic example of clinical decision based on molecular diagnosis. Nevertheless, even within families and patients with the same RET mutation, the age of disease onset and the phenotypes are unpredictable, suggesting the existence of modifier loci influencing the expression and severity of the disease whose identification could drive the diagnostic and therapeutic management. Accordingly, though RET-S891A mutation has been associated to isolated FMTC for several years after its first identification in 1997 [29], a MEN2 clinical spectrum has been more recently reported in around 4% of cases [19]. Interestingly, a recent case report paper identify bilateral pheochromocytoma as the first manifestation of MEN2A disease in a patient of a family carrying the RET-S891A mutation [30].
Among the potential genetic modifiers, RET gene polymorphisms have been associated with susceptibility and/or disease progression [2-5], and the attention has been mainly focused on the non-synonymous variant RET-G691S. Although debated, RET-G691S has been suggested to have a role in MTC susceptibility and in the modulation of the age of onset [7-9,13,31]. In this context we recently showed that RET-G691S, not oncogenic per se, enhances in vitro the oncogenic potential of the rare germline mutation RET-K666E [5,17], accordingly with the observation of Vandenbosch et al. that this polymorphism is able to increase the penetrance in the clinical onset of mutations affecting RET 666 codon [18].
In the present work, to get more insights and to confirm the possible role of RET-G691S in the modulation of the oncogenic capacity and phenotype expression of RET mutants, we focused on the RET-S891A. This intermediated risk mutation displays in vitro low transforming activity [21] that may be enhanced by GDNF treatment (data not shown), as previously reported for other RET mutations localized in the tyrosine kinase domain, such as RET-M918T causally related to MEN2B [32]. In agreement with our initial hypothesis, we showed in this study that also for RET-S891A mutation the association with RET-G691S polymorphism correlates in vitro with enhanced tumorigenic properties such as transforming activity and migratory and clonogenic ability. These findings, indicating a possible role of genetic modifier for this polymorphism, are concordant with the expression/penetrance pattern of the two analyzed RET-S891A FMTC families, that include members harboring the RET-S891A mutation associated or not with the RET-G691S polymorphism. Though not statistically significant, an earlier age at diagnosis of MTC was observed in RET-G691S/S891A carriers compared with RET-S891A carriers, suggesting a possible role in the modulation of the age at disease onset.
Our in vitro studies, mimicking the cis presentation of RET-S891A mutation and RET-G691S polymorphism observed in the Family 2, demonstrate the oncogenic enhancer role of the polymorphism in the case of cis presentation. Moreover, the clinical data from both families suggest a trend towards an earlier age at diagnosis of MTC in the RET-G691S/S891A carriers irrespectively of the cis or trans presentation of the RET-S891A mutation and the RET-G691S polymorphism (Additional file 1: Figure S1B). Therefore, it is conceivable to hypothesize that the RET-G691S polymorphism may act as genetic modifier also in the case of trans presentation with RET-S891A mutation. However, further studies are necessary to demonstrate that RET-G691S polymorphism may enhance the oncogenic activity of RET-S891A, or of other RET mutants, also in trans.
Moreover, clinical data from Family 2 indicate that two out of three RET-G691/S891A patients had elevated normetanephrine levels, suggesting that the full expression of MEN2A phenotype could be related to the association with this non synonymous polymporphism, especially in the case of cis presentation with RET-S891A mutation.
Conclusion
In conclusion, RET-G691S polymorphism has been shown in vitro to enhance the oncogenic potential of the RET-S891A mutation and in vivo to modify the clinical expression of the disease.
The genetic analysis of RET-G691S in other series with RET-S891A or other RET mutations is desirable to confirm the role of this polymorphism in the modulation of the phenotype, particularly in intermediate risk forms. If confirmed, present data suggest that the evaluation of RET-G691S in RET carriers should be always recommended in order to select the more appropriate clinical and follow-up management.
Acknowledgements
This work was supported by Fondazione Berlucchi, by Associazione Italiana per la Ricerca sul Cancro (AIRC), and by Institutional Strategic Projects ‘contribution 5 per mille’ Fondazione IRCCS Istituto Nazionale Tumori. The authors wish to thank Dr Italia Bongarzone and Dr Antonella Aiello for helpful methodological advices and discussion and Silvia Grassi for secretarial assistance.
Abbreviations
- MTC
Medullary Thyroid Cancer
- RET
REarranged during Transfection
- MEN
Multiple Endocrine Neoplasia
- FMTC
Familial Medullary Thyroid Cancer
- ERK
Extracellular signal-Regulated Kinase
- DMEM
Dulbecco’s Modified Eagle Medium
- G418
Geneticin selective antibiotic
- GDNF
Glial cell line-Derived Neurotrophic Factor
- TNM
Tumor-Nodes-Metastasis
- PHEO
Pheochromocytoma
Additional file
Age distribution of RET-S891A MTC patients harboring or not the polymorphism. The age at the diagnosis (age at DX) and the median are reported for MTC patient from Family 1 (A) and for MTC patient from both families analyzed together (B). The comparison between groups, performed by Wilcoxon rank sum test, results not statistically significant. The two RET-G691S/S891A MTC patients of family 2 displaying elevated urinary normetanephrine levels are marked (red asterisks).
Footnotes
Emanuela Minna, Maria Grazia Rizzetti and Paola Romeo contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CC, LF, MGB conceived the study and participated in its design and coordination. CC, MGR, EM, PR performed in vitro studies, analyzed the data and carried out statistical analysis. DL performed and analyzed wound healing assay. CC, LP, LF, MGB contributed to clinical and genetic evaluation of the patients. CC, LF, MGB wrote the manuscript. EM, PR, LP, AG participated in manuscript writing. All authors read and approved the final manuscript.
Contributor Information
Carla Colombo, Email: carla.colombo1@unimi.it.
Emanuela Minna, Email: emanuela.minna@istitutotumori.mi.it.
Maria Grazia Rizzetti, Email: mariagrazia.rizzetti@istitutotumori.mi.it.
Paola Romeo, Email: paola.romeo@istitutotumori.mi.it.
Daniele Lecis, Email: daniele.lecis@istitutotumori.mi.it.
Luca Persani, Email: luca.persani@unimi.it.
Piera Mondellini, Email: piermonde@alice.it.
Marco A Pierotti, Email: marco.pierotti@istitutotumori.mi.it.
Angela Greco, Email: angela.greco@istitutotumori.mi.it.
Laura Fugazzola, Email: laura.fugazzola@policlinico.mi.it.
Maria Grazia Borrello, Email: mariagrazia.borrello@istitutotumori.mi.it.
References
- 1.Arighi E, Borrello MG, Sariola H. RET tyrosine kinase signaling in development and cancer. Cytokine Growth Factor Rev. 2005;16(4–5):441–67. doi: 10.1016/j.cytogfr.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 2.Cebrian A, Lesueur F, Martin S, Leyland J, Ahmed S, Luccarini C, et al. Polymorphisms in the initiators of RET (rearranged during transfection) signaling pathway and susceptibility to sporadic medullary thyroid carcinoma. J Clin Endocrinol Metab. 2005;90(11):6268–74. doi: 10.1210/jc.2004-2449. [DOI] [PubMed] [Google Scholar]
- 3.Baumgartner-Parzer SM, Lang R, Wagner L, Heinze G, Niederle B, Kaserer K, et al. Polymorphisms in exon 13 and intron 14 of the RET protooncogene: genetic modifiers of medullary thyroid carcinoma? J Clin Endocrinol Metab. 2005;90:6232–6. doi: 10.1210/jc.2005-1278. [DOI] [PubMed] [Google Scholar]
- 4.Machens A, Frank-Raue K, Lorenz K, Rondot S, Raue F, Dralle H. Clinical relevance of RET variants G691S, L769L, S836S and S904S to sporadic medullary thyroid cancer. Clin Endocrinol (Oxf) 2012;76(5):691–7. doi: 10.1111/j.1365-2265.2011.04293.x. [DOI] [PubMed] [Google Scholar]
- 5.Fugazzola L, Muzza M, Mian C, Cordella D, Barollo S, Alberti L, et al. RET genotypes in sporadic medullary thyroid cancer: studies in a large Italian series. Clin Endocrinol (Oxf) 2008;69(3):418–25. doi: 10.1111/j.1365-2265.2008.03218.x. [DOI] [PubMed] [Google Scholar]
- 6.Margraf RL, Crockett DK, Krautscheid PM, Seamons R, Calderon FR, Wittwer CT, et al. Multiple endocrine neoplasia type 2 RET protooncogene database: repository of MEN2-associated RET sequence variation and reference for genotype/phenotype correlations. Hum Mutat. 2009;30(4):548–56. doi: 10.1002/humu.20928. [DOI] [PubMed] [Google Scholar]
- 7.Robledo M, Gil L, Pollan M, Cebrian A, Ruiz S, Azanedo M, et al. Polymorphisms G691S/S904S of RET as genetic modifiers of MEN 2A. Cancer Res. 2003;63:1814–7. [PubMed] [Google Scholar]
- 8.Elisei R, Cosci B, Romei C, Bottici V, Sculli M, Lari R, et al. RET exon 11 (G691S) polymorphism is significantly more frequent in sporadic medullary thyroid carcinoma than in the general population. J Clin Endocrinol Metab. 2004;89:3579–84. doi: 10.1210/jc.2003-031898. [DOI] [PubMed] [Google Scholar]
- 9.Cardot-Bauters C, Leteurtre E, Leclerc L, Vantyghem MC, Do CC, Wemeau JL, et al. Does the RET variant G691S influence the features of sporadic medullary thyroid carcinoma? Clin Endocrinol (Oxf) 2008;69(3):506–10. doi: 10.1111/j.1365-2265.2008.03230.x. [DOI] [PubMed] [Google Scholar]
- 10.Gil L, Azanedo M, Pollan M, Cristobal E, Arribas B, Garcia-Albert L, et al. Genetic analysis of RET, GFR alpha 1 and GDNF genes in Spanish families with multiple endocrine neoplasia type 2A. Int J Cancer. 2002;99:299–304. doi: 10.1002/ijc.10298. [DOI] [PubMed] [Google Scholar]
- 11.Costa P, Domingues R, Sobrinho LG, Bugalho MJ. RET polymorphisms and sporadic medullary thyroid carcinoma in a Portuguese population. Endocrine. 2005;27(3):239–43. doi: 10.1385/ENDO:27:3:239. [DOI] [PubMed] [Google Scholar]
- 12.Figlioli G, Landi S, Romei C, Elisei R, Gemignani F. Medullary thyroid carcinoma (MTC) and RET proto-oncogene: mutation spectrum in the familial cases and a meta-analysis of studies on the sporadic form. Mutat Res. 2013;752(1):36–44. doi: 10.1016/j.mrrev.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 13.Lantieri F, Caroli F, Ceccherini I, Griseri P. The involvement of the RET variant G691S in medullary thyroid carcinoma: conflicting results of meta-analyses need to be reconciled. Int J Cancer. 2013;133(7):1760–1. doi: 10.1002/ijc.28177. [DOI] [PubMed] [Google Scholar]
- 14.Lesueur F, Cebrian A, Robledo M, Niccoli-Sire P, Svensson KA, Pinson S, et al. Polymorphisms in RET and its coreceptors and ligands as genetic modifiers of multiple endocrine neoplasia type 2A. Cancer Res. 2006;66(2):1177–80. doi: 10.1158/0008-5472.CAN-05-2995. [DOI] [PubMed] [Google Scholar]
- 15.Sawai H, Okada Y, Kazanjian K, Kim J, Hasan S, Hines OJ, et al. The G691S RET polymorphism increases glial cell line-derived neurotrophic factor-induced pancreatic cancer cell invasion by amplifying mitogen-activated protein kinase signaling. Cancer Res. 2005;65(24):11536–44. doi: 10.1158/0008-5472.CAN-05-2843. [DOI] [PubMed] [Google Scholar]
- 16.Narita N, Tanemura A, Murali R, Scolyer RA, Huang S, Arigami T, et al. Functional RET G691S polymorphism in cutaneous malignant melanoma. Oncogene. 2009;28(34):3058–68. doi: 10.1038/onc.2009.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Borrello MG, Aiello A, Peissel B, Rizzetti MG, Mondellini P, Degl’Innocenti D, et al. Functional characterization of the MTC-associated germline RET-K666E mutation: evidence of oncogenic potential enhanced by the G691S polymorphism. Endocr Relat Cancer. 2011;18(4):519–27. doi: 10.1530/ERC-10-0306. [DOI] [PubMed] [Google Scholar]
- 18.Vandenbosch K, Renard M, Uyttebroeck A, Sciot R, Matthijs G, Legius E. Medullary thyroid carcinoma in a child with a new RET mutation and a RET polymorphism. Genet Couns. 2005;16(1):95–100. [PubMed] [Google Scholar]
- 19.Schulte KM, Machens A, Fugazzola L. McGregor A, az-Cano S, Izatt L, Aylwin S, Talat N, Beck-Peccoz P, Dralle H: The clinical spectrum of multiple endocrine neoplasia type 2a caused by the rare intracellular RET mutation S891A. J Clin Endocrinol Metab. 2010;95(9):E92–7. doi: 10.1210/jc.2010-0375. [DOI] [PubMed] [Google Scholar]
- 20.Plaza MI, Koster R, van der Sloot AM, Quax WJ, Osinga J, Van Der ST, et al. RET-familial medullary thyroid carcinoma mutants Y791F and S891A activate a Src/JAK/STAT3 pathway, independent of glial cell line-derived neurotrophic factor. Cancer Res. 2005;65(5):1729–37. doi: 10.1158/0008-5472.CAN-04-2363. [DOI] [PubMed] [Google Scholar]
- 21.Iwashita T, Kato M, Murakami H, Asai N, Ishiguro Y, Ito S, et al. Biological and biochemical properties of Ret with kinase domain mutations identified in multiple endocrine neoplasia type 2B and familial medullary thyroid carcinoma. Oncogene. 1999;18:3919–22. doi: 10.1038/sj.onc.1202742. [DOI] [PubMed] [Google Scholar]
- 22.Romei C, Mariotti S, Fugazzola L, Taccaliti A, Pacini F, Opocher G, et al. Multiple endocrine neoplasia type 2 syndromes (MEN 2): results from the ItaMEN network analysis on the prevalence of different genotypes and phenotypes. Eur J Endocrinol. 2010;163(2):301–8. doi: 10.1530/EJE-10-0333. [DOI] [PubMed] [Google Scholar]
- 23.Fugazzola L, Cerutti N, Mannavola D, Ghilardi G, Alberti L, Romoli R, et al. Multigenerational familial medullary thyroid cancer (FMTC): evidence for FMTC phenocopies and association with papillary thyroid cancer. Clin Endocrinol (Oxf) 2002;56(1):53–63. doi: 10.1046/j.0300-0664.2001.01434.x. [DOI] [PubMed] [Google Scholar]
- 24.Bongarzone I, Pierotti MA, Monzini N, Mondellini P, Manenti G, Donghi R, et al. High frequency of activation of tyrosine kinase oncogenes in human papillary thyroid carcinoma. Oncogene. 1989;4:1457–62. [PubMed] [Google Scholar]
- 25.Giacchè M, Panarotto A, Tosini R, Mori L, Orsini A, Cherubini L, et al. Evidence of a founder effect in thirteen families with medullary thyroid cancer carrying RET Ser891Ala mutation. [Abstract PP346] XXXIII National Congress of the Italian Society of Endocrinology. J Endocr Invest. 2009;32(2 Suppl):193. [Google Scholar]
- 26.Compton CC, Byrd DR, Garcia-Aguilar J, Kurup P, Kurtzman SH, Olawaiye A, et al. AJCC Cancer Staging Handbook. 7. New York: Springer Verlag; 2012. TNM classification of malignat tumors. [Google Scholar]
- 27.Pacini F, Schlumberger M, Dralle H, Elisei R, Smit JW, Wiersinga W. European consensus for the management of patients with differentiated thyroid carcinoma of the follicular epithelium. Eur J Endocrinol. 2006;154(6):787–803. doi: 10.1530/eje.1.02158. [DOI] [PubMed] [Google Scholar]
- 28.Kloos RT, Eng C, Evans DB, Francis GL, Gagel RF, Gharib H, et al. Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid. 2009;19(6):565–612. doi: 10.1089/thy.2008.0403. [DOI] [PubMed] [Google Scholar]
- 29.Hofstra RM, Fattoruso O, Quadro L, Wu Y, Libroia A, Verga U, et al. A novel point mutation in the intracellular domain of the ret protooncogene in a family with medullary thyroid carcinoma. J Clin Endocrinol Metab. 1997;82:4176–8. doi: 10.1210/jcem.82.12.4439. [DOI] [PubMed] [Google Scholar]
- 30.Hibi Y, Ohye T, Ogawa K, Shimizu Y, Shibata M, Kagawa C, et al. Pheochromocytoma as the first manifestation of MEN2A with RET mutation S891A: report of a case. Surg Today. 2014;44(11):2195–200. doi: 10.1007/s00595-013-0826-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lesueur F, Cebrian A, Cranston A, Leyland J, Faid TM, Clements MR, et al. Germline homozygous mutations at codon 804 in the RET protooncogene in medullary thyroid carcinoma/multiple endocrine neoplasia type 2A patients. J Clin Endocrinol Metab. 2005;90(6):3454–7. doi: 10.1210/jc.2004-1622. [DOI] [PubMed] [Google Scholar]
- 32.Bongarzone I, Vigano’ E, Alberti L, Borrello MG, Pasini B, Greco A, et al. Full activation of MEN2B mutant RET by an additional MEN2A mutation or by ligand (GDNF) stimulation. Oncogene. 1998;16:2295–301. doi: 10.1038/sj.onc.1201759. [DOI] [PubMed] [Google Scholar]