

Abstract
Mounting evidence has been provided regarding the crucial role of leukocyte extravasation and subsequent inflammatory response in several central nervous system (CNS) disorders. The infiltrated leukocytes release pro-inflammatory mediators and activate resident cells, leading to tissue injury. Leukocyte-endothelia interaction is critical for leukocyte extravasation and migration from the intravascular space into the tissue during inflammation. The basic physiology of leukocyte-endothelia interaction has been investigated extensively. Traditionally, three kinds of adhesion molecules, selectin, integrin, and immunoglobulin families, are responsible for this multiple-step interaction. Furthermore, blocking adhesion molecule function by genetic knockout, antagonizing antibodies, or inhibitory pharmacological drugs provides neuroprotection, which is associated with a reduction in leukocyte accumulation with in the tissue. Detection of the soluble form of adhesion molecules has also been proven to predict outcomes in CNS disorders. Lately, vascular adhesion protein-1 (VAP-1), a novel adhesion molecule and endothelial cell surface enzyme, has been implicated as a brake during leukocyte extravasation. In this review, we summarize the functions of traditional adhesion molecules as well as VAP-1 in the leukocyte adhesion cascade. We also discuss the diagnostic and therapeutic potential of adhesion molecules in CNS disorders.
Keywords: adhesion molecule, CNS disorders, leukocyte adhesion cascade, vascular adhesion protein-1
1, Introduction
Inflammation is a common response in the progression of central nervous system (CNS) diseases, including stroke, neurodegenerative diseases, traumatic brain injury (TBI), etc. For decades, it was assumed that the CNS enjoyed the privilege of being protected by the blood-brain barrier (BBB) [1]. Under normal physiological conditions, an intact BBB prevents cell trafficking into the CNS. Trans- and paracellular movement of molecules and cells across the BBB are limited by endothelial cells [2]. Unfortunately, under most pathophysiological conditions, structural integrity of the BBB collapses and leukocytes accumulate into CNS [3], which is a key stage in neuroinflammation development.
Initially, adhesion molecules were attributed to being cell surface structures that mediate cell–cell and cell-extracellular matrix (ECM) interactions. They are also the key inflammatory mediators by promoting blood-borne leukocyte crossing of the BBB and aggregating in the injury site [4, 5]. Infiltration of systemic immune cells into the injured sites by sequential interactions between leukocytes and endothelial cells is an initial step of the inflammatory response. After tissue injury, circulating immune cells will release inflammatory chemokines, recognize endothelial cells around the injured tissue, stop on the luminal surface of the blood vessel, transmigrate paracellularly across the endothelial layer, and reach the injured site [6–8]. This whole process is called the leukocyte adhesion cascade, which has multiple steps, including tethering, rolling, activation, firm adhesion, and transmigration. Different adhesion molecule superfamilies mediate each step (Figure 1). Chemokines stimulate leukocyte and endothelial cell activation, selectin promotes tethering and rolling, integrin mediates firm adhesion, and integrin and immunoglobulin facilitate transmigration.[9] In addition, some special adhesion molecules also assist in rolling, firm adhesion, and transmigration during leukocyte recruitment, such as vascular adhesion protein 1 (VAP-1), a cell surface molecule displaying amine oxidase activity [10, 11]. All steps are required for effective leukocyte recruitment, therefore, blocking any step may reduce leukocyte aggregation in the inflamed sites. To date, numerous preclinical models and clinical trials have determined the pathophysiological role of leukocyte accumulation during neuroinflammation in CNS disorders [12–17]. Better understanding of adhesion molecule function for modulating leukocyte trafficking into the brain may provide further insight into novel or alternative therapeutic approaches to treat inflammatory CNS disorders.
Figure 1. Adhesion molecules and the leukocyte adhesion cascade.
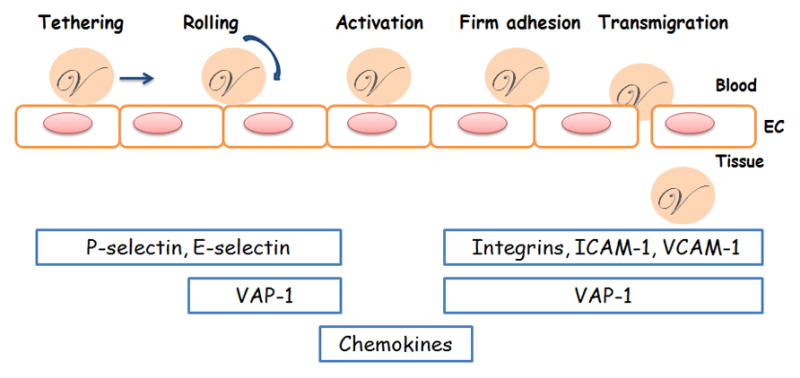
The leukocyte adhesion cascade is a multi-step process that includes tethering, rolling, activation, firm adhesion, and transmigration steps. Classical adhesion molecules, including selectin, integrin, and immunoglobulin, are involved in different phases by supporting leukocyte crossing of the endothelial layer from blood into tissue. Chemokines stimulate leukocyte activation. One novel ectoenzyme, VAP-1, contributes to the rolling, firm adhesion, and transmigration steps. EC: endothelial cell.
This review focuses on adhesion molecule mediated leukocyte-endothelial cell interaction during inflammation in multiple forms of CNS diseases in both preclinical models and clinical trials. Understanding the role of adhesion molecules in the leukocyte adhesion cascade will elucidate their involvement in neuroinflammation and present exciting therapeutic and diagnostic opportunities for all kinds of CNS diseases.
2, Adhesion Molecules
2.1 Selectin
Selectins are a group of transmembrane molecules expressed on the outer cell surfaces of leukocytes and endothelial cells following activation [18]. There are three kinds of selectins, P, E, and L. P-selectin and E-selectin are expressed on activated endothelial cells, whereas L-selectin is mainly expressed on leukocytes. P-selectin is also expressed by activated platelets.
P-selectin, also known as CD62P, is a membrane-bound glycoprotein identified primarily in the secretory granules of platelets and Weibel-Palade bodies of endothelial cells [19, 20]. P-selectin is the most important selectin for modulating leukocyte rolling by interacting with P-selectin glycoprotein ligand 1 (PSGL1) [21] through binding to its amino terminal region [22]. Under inflammatory conditions, P-selectin translocates from the storage granules to the outer surface of the cell and interacts with leukocytes. Pro-inflammatory agents stimulate P-selectin translocation, including lipopolysaccharide (LPS), tumor necrosis factor alpha (TNF-α), interleukin-1 (IL-1), and thrombin by phosphorylating tyrosine, threonine, and serine residues on P-selectin’s cytoplasmic tail [23]. P-selectin is also shed from the endothelial cell membrane and released into blood. Blood concentrations of soluble P-selectin are high in many pathophysiological conditions, such as thrombotic thrombocytopenic purpura, hemolytic-uremic syndrome, chronic obstructive pulmonary disease, myocardial infarction, hypercholesterolemia, and rheumatoid arthritis [20]. High soluble P-selectin blood concentration may be associated with excessive leukocyte infiltration in those pathophysiological conditions. E-selectin (CD62E), also called endothelial leukocyte adhesion molecule-1 (ELAM-1), is restrictedly expressed by activated endothelial cells and mediates slow leukocyte rolling. Similar to P-selectin, E-selectin is induced by pro-inflammatory agents, such as LPS, IL-1 and TNF-α. E-selectin plays a major role in leukocyte recruitment to inflamed sites through its close collaboration with P-selectin. E-selectin binds to glycosylated CD44 and E-selectin ligand 1 (ESL1) [24]. Additionally, E-selectin interacts with PSGL1, which is not only expressed by endothelial cell but also leukocytes[9]. L-selectin, also known as CD62L or leukocyte endothelial cell adhesion molecule-1 (LECAM-1), is a glycoprotein constitutively expressed by all leukocytes [25, 26]. It interacts reversibly with its endothelial cell ligands and mediates neutrophil rolling on the endothelium during inflammation [27]. Its ligands are mostly expressed on the endothelial cell surface, including GLYCAM-1, MAdCAM-1, CD34 [28], and PSGL-1 [29]. When leukocytes are activated by pro-inflammatory agents, the conformation of L-selectin will change to rapidly and transiently enhance its binding affinity to its ligands[30, 31]. Because of the proteloytic cleavage of membrane-binding L-selectin, soluble L-selectin is observed in the plasma after L-selectin mediated rolling of leukocytes [32, 33].
2.2 Integrins
Integrins are a family of adhesion molecules responsible for cellular attachment to extracellular matrix (ECM) and cell-cell interactions. They are cell-surface heterodimeric glycoproteins and consist of α and β subunits [34, 35]. The sub-classification of integrins is denominated by the β subunit. Ligand specificity is also determined by the complex of α and β subunits [8]. The β2 (CD18) integrin families are regarded as the most important for leukocyte migration during inflammation. The β2 integrins (CD11/CD18), including CD11a/CD18 (LFA-1), CD11b/CD18 (Mac-1), and CD11c/CD18, mainly modulate firm adhesion during leukocyte transmigration. LFA-1 is expressed by leukocytes, while Mac-1 is mainly expressed by neutrophils, monocytes, and natural killer cells [36]. The β2 integrins are critical in innate immunity. An encoding gene mutation will lead to defective synthesis of LFA-1, Mac-1, and CD11c/CD18 causing leukocyte adhesion deficiency (LAD). LAD patients often suffer from repeated bacterial infections because of the ineffective transmigration of leukocytes [37]. LFA-1 plays a critical role in the leukocyte adhesion cascade by interacting with ICAM-1 and ICAM-2 on the endothelial cell surface [38, 39]. Generally, integrins only mediate leukocyte firm adhesion, but β2 integrins may also be necessary for slow rolling of the leukocytes [40]. Leukocyte rolling velocities were increased in LFA-1 or Mac-1 deficient mice, but they were still lower than in CD18 deficient mice [41].
2.3 Immunoglobulin
Some members of the immunoglobulin superfamily are adhesion molecules that mediate interaction between leukocytes and endothelial cells during the inflammatory response. They are ICAM-1, ICAM-2, VCAM-1, and PECAM-1. They have similar structural and genetic features, including two to seven immunoglobulin domains that interact with leukocyte integrins[16]. ICAMs are homodimers mainly expressed by endothelial cells, and they bind to β2 integrins, LFA-1, and Mac-1. ICAM-1 (CD54) is one of the most important ligands for β2 integrins [42]. It is a glycoprotein composed of a transmembrane domain and a short cytoplasmic sequence. ICAM-1 mediates leukocyte passage across the vascular endothelial cell layer to the sites of tissue injury by promoting leukocyte firm adhesion and transmigration [43]. Under normal conditions, ICAM-1 is expressed at low concentrations on the resting endothelium. In addition, ICAM-1 is also expressed on Schwann cells and plays a role during inflammation of the peripheral nerves. ICAM-1 expression increases rapidly following stimulation by pro-inflammatory cytokines, such as IL-8, IL-1, and TNF-α, and by other pro-inflammatory agents, such as LPS. Mice with deficient ICAM-1 expression have impaired leukocyte migration during inflammation. A soluble form of ICAM-1 (sICAM-1) plays a role in neutrophil adhesion [44]. Similar to ICAM-1, ICAM-2 is a homodimeric molecule, but it does not respond to pro-inflammatory factors [16, 39]. VCAM-1 is expressed at very low levels by resting endothelial cells, but it can be upregulated under inflammatory conditions [45, 46]. It promotes leukocyte adhesion by interacting with very late antigen-4 (VLA-4) [16].
2.4 VAP-1, a Dual Role Adhesion Molecule
In 1992, VAP-1 was first discovered as a 90-kD endothelial cell protein in synovial vessels from arthritis patients using a monoclonal antibody, 1B2 [47]. The antibody markedly reduced lymphocyte binding to high endothelial venules in both frozen section adhesion assays and flow chamber assays. One year later, the localization of VAP-1 expression was determined by the same group. Their results showed that, in addition to abundant expression in lymphatic organs, VAP-1 is also extensively expressed on endothelial cells in several non-lymphatic tissues, including skin, brain, kidney, liver, and heart[48].
Further studies discovered the enzymatic nature of VAP-1 (Figure 1). VAP-1 belongs to the semicarbazide-sensitive monoamine oxidase (SSAO) family. This characteristic was identified by cDNA cloning that encoded a type II transmembrane protein with similar identity to the copper-containing amine oxidase family. VAP-1 also possessed amine oxidase activity [49] that can oxidize primary amines and release biologically active products, including aldehydes, hydrogen peroxide, and ammonium. Coinciding with the discovery of VAP-1’senzymatic nature, its adhesion molecule function was observed by Smith and colleagues. In their experiment, they conducted a VAP-1 cDNA transfection into endothelial cells and found that VAP-1 led to lymphocyte binding, which was partially inhibited with anti-VAP-1 mAbs[49].
Mounting evidence suggests that VAP-1 is an endothelial glycoprotein induced by inflammation. Studies with human samples using confocal microscopes showed that VAP-1 exists both on the luminal surface and in the intracellular granules of human endothelial vessels (HEVs). In samples from patients with inflammatory bowel diseases or chronic dermatoses, VAP-1 levels were also found to be upregulated at inflamed sites[48]. Furthermore, the mechanism controlling VAP-1 function has been explored. In normal endothelial cells, VAP-1 is stored in intracellular granules within the cytoplasm. Under inflammatory conditions, VAP-1 was upregulated and translocated to the endothelial surface [11]. The translocation of VAP-1 to the cell surface was observed directly after inflammation in experimental canine and pig inflammatory models. VAP-1 increased on the endothelial cell surface 60 minutes after induction by inflammation, peaked at 8 hours, and lasted until 48 hours [50]. Similar to P-selectin, VAP-1 translocates from intracellular granules to the vascular luminal surface at the site of inflammation. However, the underlying mechanism and mediators inducing VAP-1 expression and translocation remains unclear. Thirteen inflammatory factors, including TNF-α and thrombin, were used to determine the mediators leading to the VAP-1 induction in human endothelial cells. Unfortunately, none increased cell surface expression of VAP-1 [48]. It is very rare to have only a single mediator involved in inflammation in vivo, therefore, this study suggested VAP-1 might be induced by a combination of multiple mediators.
The signaling functions of VAP-1’s catalytic activity have been investigated. The potent biological products of VAP-1, such as aldehyde, H2O2, and NH3, may induce expression of other adhesion molecules or pro-inflammatory cytokines. They are cytotoxic at high concentrations and potentially trigger VAP-1 downstream signals [11, 51]. Reactive oxygen species and H2O2 help regulate gene expression in vascular endothelial cells [52]. Mounting evidence showed H2O2 formation is associated with leukocyte infiltration by inducing P-selectin expression and leukocyte rolling [53]. In human endothelial cells, both P- and E-selectin are induced on transcriptional and translational levels by VAP-1 enzymatic activity. In VAP-1 transgenic mice, P-selectin induction is dependent on VAP-1 enzymatic activity [54]. In an age-related mouse macular degeneration (AMD) model, VAP-1 suppression diminished expression of pro-inflammatory cytokines, such as TNF-α and MCP-1, as well as adhesion molecules, such as ICAM-1 [55]. Lalor and colleagues provided specific substrates of VAP-1 directly to liver endothelial cells, which lead to endothelial cell activation. They also found VAP-1 mediated activation was dependent on NFkB, PI3K, and MAP kinase pathway. Additionally VAP-1 activation upregulated E-selectin, ICAM-1, VCAM-1, as well as CXCL8, and intensified the inflammatory response [56]. Taken together, these findings suggestVAP-1 is a novel type of adhesion molecule with SSAO enzymatic activity that may also have signaling functions.
3, Adhesion Cascade in CNS Disorders
3.1 Stroke
3.1.1 Ischemic Stroke
The inflammatory response is initiated in the vascular system after brain ischemia and reperfusion (I/R) occurs. Mounting evidence indicates an accumulation of systemic immune cells, specifically blood-borne leukocytes, occurs in the ischemic region and is partly associated with ischemic tissue injury [57–59]. The infiltrated leukocytes activate resident cells in the brain, including endothelial cells, microglia, and astrocytes [17, 60]. As a consequence, blood-borne leukocytes, in addition to the aforementioned cells, amplify leukocyte recruitment signals by producing adhesion molecules and releasing pro-inflammatory cytokines [17, 36, 61]. As described above, adhesion molecules mediate leukocyte transmigration into the brain tissue after stroke and regulate the transmigration speed [36]. Therefore, restriction of adhesion molecule function may have potential therapeutic effects on cerebral injury after ischemia/reperfusion [62].
P- and E-selectin expression profiles have been determined in suture, thrombotic, and embolic middle cerebral artery (MCA) occlusion models of stroke. The results indicated P-selectin was upregulated as early as 15 min while E-selectin increased at 2 hours after the MCA occlusion. Both peaked at 6 hours after MCA occlusion [63]. Accordingly, P-selectin−/− or P-selectin monoclonal antibody (mAb) reduced recruitment of leukocytes and platelets after cerebral I/R [64]. Pharmacological compounds have also been used to inhibit early leukocyte interaction with endothelial cells. Fucoidin (FCN), a competitive inhibitor of P- and L-selectin, reduced cerebral infarction size and improved neurological function in a focal ischemia model [65]. However, another report showed HuDREG200, a humanized antibody to L-selectin, did not effectively reduce infarction size, neutrophil accumulation, or cerebral blood flow in a rabbit model of cerebral I/R, although this antibody reduced myocardial neutrophil infiltration in a feline model of myocardial I/R [66]. This evidence indicates L-selectin might not be crucial in the leukocyte adhesion cascade. CY-1503, an analog of sialylLewis(x) (SLe(x)), significantly reduced infarct volume and myeloperoxidase positive cells in the ischemic area by blocking E-selectin function [67]. Additionally, in a transient intraluminal middle cerebral artery occlusion (MCAO) model, anti-E-selectin monoclonal antibody was applied by intravenous injection [68], which attenuated cerebral neutrophil accumulation and provided dose-dependent neuroprotection. Therefore, blockage of selectin-mediated adhesion may limit immune cell transmigration and reduce reperfusion injury in the brain.
The significance of β2 integrins in ischemic injury has been demonstrated. In a cerebral I/R model, leukocyte adhesion to cerebral venules was reduced in both LFA-1 and Mac-1 deficient mice 24 hours after reperfusion. In line with this result, LFA-1 or Mac-1-deficient animals also presented significantly reduced infarct volume and improved neurological functions [14]. Prior work also showed Hu23F2G, an antibody of the CD11/CD18 integrin, reduced neutrophil infiltration as well as ischemic injury in a rabbit model of transient focal ischemia [69]. Similarly, anti-Mac-1 antibody lead to reductions in intraparenchymal neutrophil accumulation and ischemic brain injury [70]. Furthermore, individual inhibition of CD11b, CD18, or ICAM-1 also reduced brain injury by decreasing leukocyte accumulation after ischemic stroke [71–73]. In addition to β2 integrins, α integrins may also affect leukocyte infiltration following ischemia. Antibodies to α4 integrins attenuated infarction and improved neurological function by blocking lymphocyte and monocyte transmigration in a transient focal cerebral ischemia rat model [74].
ICAM-1 and VCAM-1 have been broadly studied in ischemia and hypoxia. ICAM-1 levels were highly increased in the brain at 3 hours following ischemic stroke, peaked between 6–12 hours, and remained high until 5 days later [75]. Whether VCAM-1 levels are upregulated after cerebral ischemia is still controversial. Blann et al found intense expression of VCAM-1 by astrocytes and endothelial cells in the infracted area from a stroke patient [76]. However, a study from Vemuganti et al showed VCAM-1 mRNA expression was never changed in the ipsilateral cortex between 3 and 72 hours after reperfusion [77]. ICAM-1 is one of the most important targets in anti-adhesion strategies. In ICAM-1-knockout mice, the size of ischemic lesions was reduced [78], and they had a lower mortality rate compared to wild-type [79]. Other treatment approaches, such as ICAM-1 antibodies, also reduced infarction volume and improved neurological outcomes after ischemia [80, 81]. ICAM-1 antisense oligonucleotides were also applied in an ischemic stroke model, since ICAM-1 mRNA levels were upregulated within hours after ischemia. Their results showed ICAM-1 knockdown reduced infarct volume and improved neurological function [77].
Because anti-adhesion treatments were effective in preclinical studies, they have been applied in clinical trials. A recombinant neutrophil inhibiting factor (rNIF) was administered to patients within 6 hours after symptom onset (Acute Stroke Therapy by Inhibition of Neutrophils) [82]. In addition, an anti-CD11/CD18 antibody was employed within 12 hours after symptom onset [12, 83] in ischemia patients. However, both clinical trials failed, although they inhibited neutrophil aggregation and improved functional outcomes in animal studies [69, 84, 85]. The different expression profiles of integrin between human and animal stroke models may explain these failures. Enlimomab, a murine anti-ICAM-1 antibody, has been investigated in patients with ischemic stroke [86]. Unfortunately, more adverse effects were observed with enlimomab treatment than placebo, including infections and fever. Treated patients also had a higher fatality rate than in the placebo group. The murine origin of this antibody may induce an immune response in patients [87], which may explain its failure. It has also been reported enlimomab activates neutrophilic granulocytes in the whole blood of human volunteers [88].
3.1.2 Hemorrhagic Stroke
The inflammatory response plays an important role in brain injury after intracerebral hemorrhage (ICH). Mounting evidence indicates systemic immune cells, specifically blood-derived leukocytes, accumulating at the injured site are the primary orchestrators of brain injury following ICH [89].
Neutrophils are a major type of leukocyte that mediates secondary brain injury following ICH. In a collagenase-injection mouse model, Wang and colleague found neutrophils infiltrated into and around the hematoma 4 hours after injury and peaked at 3 days [90]. Other studies conducted using an autologous blood-injection model showed neutrophils appeared within 1 day and disappeared at 3 to 7 days [91–93]. Microglia are a type of macrophage in the CNS and account for 5–20% of the total glial population [94]. Under normal conditions, they exist in a resting state. When activated by an inflammatory response following brain injury, they undergo a series of morphological changes, such as enlargement in size with stout processes, activation of phagocytic function, and upregulation of specific genes [95]. In a collagenase-injection model, studies showed microglia activation occurs earlier than neutrophil infiltration. Microglia activation occurred in the peri-hematomal region around 1–2 hours after injection, markedly increased at 1 day, peaked at 7 days, and declined to base level at 3 weeks [96, 97]. A similar time course was observed in an autologous blood model. Microglia activation appeared 1–4 hours after injection, peaked 3–7 days, and lasted as long as 4 weeks [91, 92, 98]. Prior studies suggested leukocytes, particularly neutrophils, might be major contributors to the inflammatory response and brain injury following ICH. However, a few studies addressed the effects of adhesion molecules in hemorrhagic stroke. Gong and colleagues found that ICAM-1 immunoreactivity was observed in blood vessels around the hematoma in a rat blood-injection ICH model [91]. In a collagenase model, CD18(−/−) knockout mice had reduced brain edema and mortality, which were associated with decreased MPO immunoreactivity in their brains [99].
Few studies explored the role of VAP-1 in the stroke-induced inflammatory response. VAP-1 may be involved in immune cell infiltration after stroke since leukocyte accumulation has been observed at the injured site. In myocardial samples from ischemic hearts of human patients, VAP-1 expression was markedly upregulated on endothelial cells, and VAP-1 neutralizing antibody reduced the number of adherent granulocytes by 60% [50]. In a transient forebrain ischemic rat model, a small molecule VAP-1 inhibitor was administered either at onset or 6 hours after reperfusion. The VAP-1 inhibitor limited neutrophil adhesion and infiltration, even when treated 6 hours after reperfusion, as well as provided subsequent neuroprotection[100]. We studied the role of VAP-1 in ICH and found VAP-1 inhibition can reduce brain edema and improve neurological function in both collagenase and blood-injection mouse models. Accordingly, P-selectin, E-seletin, and ICAM levels as well as infiltrated neutrophils were diminished [101].
Soluble VAP-1 (sVAP-1) may also play an important role after stroke. Airas and colleagues found sVAP-1 was markedly increased in the serum of acute stroke patients less than 6 hours after ischemia and may promote vasculopathy[102]. Hernandez-Guillamon and colleagues reported the baseline VAP-1/SSAO activity is a predictor forintracranial bleeding after tissue plasminogen activator (tPA) treatment [103]. They found elevated plasma VAP-1 activity in patients who suffered from a hemorrhage and had subsequent deleterious neurological outcomes. Additionally, in a rat model, they found the VAP-1 inhibitor prevented the side effects associated with delayed tPA treatment. The authors also elucidated potential mechanisms. In vitro, tPA promoted neutrophil degranulation and MMP-9 release, which mediate BBB disruption. VAP-1 is an adhesion molecule that mediates leukocyte infiltration. Therefore, VAP-1 inhibition may protect against tPA-induced vascular damage by inhibiting leukocyte infiltration. Recently, the same group found plasma VAP-1/SSAO activity is increased in hemorrhagic stroke patients and may be a predictor for neurological outcome after ICH [104]. All of these clinical reports suggest VAP-1 is a potential therapeutic target for ICH-induced brain injury.
Cerebral vasospasm is a major complication in subarachnoid hemorrhage (SAH) patients, and its mechanisms have not been elucidated. Increasing evidence showed inflammation, especially leukocyte-endothelial interaction, plays a crucial role in the pathogenesis of vasospasm following SAH. It has been reported increased leukocyte levels increase the risk of vasospasm [105–108] and were associated with a 90% rate of poor outcome [105].
In an SAH canine model, Aihara et al quantitatively measured the expression levels of inflammatory genes and found ICAM-1 mRNA levels were highest at day 7, which may be associated with the persistent contraction of the basilar artery [109]. Before this study, in an SAH rat model, Handa et al observed increased ICAM-1 expression in both the endothelial and medial layer of the basilar artery following SAH [110]. To examine the role of ICAM-1 and CD18 in vasospasm after SAH, Bavbek et al administered anti-ICAM-1 antibody or anti-CD18 antibody, alone or combination, in a rabbit SAH model. They found both antibodies alone inhibit vasospasm, and their effects are enhanced when used in combination [111]. Recently, in a blood-injection SAH rat model, ICAM-1 and E-selectin, but not VCAM-1, were significantly elevated in the basilar arteries at 72 hours after SAH, while 6-mercaptopurine administration repeated at 24 and 48 hours markedly reduced their expression levels compared to the control group [112]. Adhesion molecule expression has also been examined in a femoral artery model of vasospasm. Sills et al found exposing large intracranial arteries to blood extensively increased ICAM-1 immunoreactivity in endothelial cells starting at 3 hours after blood exposure and lasting until 24 hours [113]. Using an anti-ICAM-1 antibody in this model significantly inhibited vasospasm, which was associated with decreased inflammatory cell infiltration, including macrophages and granulocytes, into the peri-adventitial region of blood-exposed arteries [114]. Similar results were also found when administering anti-LFA-1 monoclonal antibody systemically in the rat femoral artery model [115]. Lin et al examined the efficacy of anti-E-selectin monoclonal antibody on cerebral vasospasm in a murine SAH model. Results showed diameters of anterior cerebral arteries were reduced at 24 hours after anti-E-selectin antibody treatment in addition to a reduction of infiltrated leukocytes[116].
In patients, the levels of soluble adhesion molecules were detected in cerebrospinal fluid (CSF) after subarachnoid hemorrhage (SAH). Polin et al found soluble forms of E-selectin, ICAM-1, and VCAM-1, but not L-selectin, were markedly increased in the CSF of patients suffering from recent aneurysmal SAH [117]. However, the levels of adhesion molecules in SAH patients are not consistent. In a report from Nissen et al, they observed serum concentrations of P- and L-selectin, but not E-selectin, ICAM-1, VCAM-1 and PECAM, were altered in patients with and without ischemic neurological deficit (DIND) after aneurysmal subarachnoid haemorrhage[118, 119]. Statins are a class of drugs that induce anti-inflammtory effects by interacting with LFA-1 and Mac-1 [120, 121]. In a clinical study, statins were used to treat vasospasm after SAH. Kramer et al found statin treatment was not associated with any reduced vasospasm development or improved outcomes after aneurysmal SAH [122]. However, Tseng et al reported acute pravastatin therapy improved vasospasm-related delayed ischemic deficits and functional outcomes after aneurysmal SAH [123, 124].
Based on the aforementioned evidence, therapeutic targeting of adhesion molecules may protect against inflammation and reverse vasospasm as well as improve functional outcomes in SAH patients.
3.2 Brain Trauma
Traumatic brain injury (TBI) is a fatal condition and most common cause of death worldwide, especially in children and young adults below 40 years old[125]. Traumatic brain injury (TBI) occurs when sudden trauma damages the brain. The injury is divided into primary and secondary injury [126]. The primary injury is the direct insult that may lead to blood vessel disruption and bleeding into or around brain tissue. The secondary injury is an indirect result of the traumatic insult, which is a series of complex cellular and/or biochemical events leading to delayed brain damage, mostly by inflammation and reactive oxygen species (ROS) [126, 127].
TBI-induced disturbances in the cerebrovasculature may cause endothelial cell activation that alters expression of cell surface markers. Balabanov and colleagues observed E-selectin and ICAM-1 were upregulated as early as 2 hours and was sustained through 24 hours, but VCAM-1 was not induced, in a percussion model of moderate TBI in rats[128]. Similarly, ICAM-1 levels were significantly upregulated on the endothelium of the traumatized hemisphere at 4 hours and remained elevated until 72 hours, while PECAM-1 was not changed. Administration of an ICAM-1 monoclonal antibody significantly reduced neutrophil accumulation (about 40%) at 24 hours following TBI [129]. Another study observed ICAM-1 levels were significantly increased in the choroid plexus and cerebral endothelium at 24 hours after TBI, but ICAM-1 deficient mice did not reduce neutrophil accumulation compared to the wild-type group [130]. Knoblach and colleagues found anti-ICAM-1 antibody administration with multiple treatments at 1, 10, and 24 hours significantly improved motor function in a rat TBI model, which was associated with reduced MPO activity, an indicator of neutrophil invasion [13]. These results indicated ICAM-1 blockage might reduce leukocyte accumulation and improve neurological function after TBI.
In TBI patients, sICAM-1 levels in their CSF correlate with tissue injury as well as BBB disruption and maybe a predictor for TBI outcomes. Patients with elevated sICAM-1 levels in their CSF had severe cerebral injuries and BBB impairment while patients with normal sICAM-1 levels had minor injuries [131, 132]. Similarly, both P-selectin and ICAM-1 CSF levels increased in children with severe TBI and were associated with the greater risk for poor outcomes [133]. As a novel marker in neurological disorders, sVAP-1 has also been investigated following TBI. Lin and colleagues found sVAP-1 activity was significantly reduced in TBI patients compared to control groups, which negatively correlated with circulating leucocytes. Patients with high plasma sVAP-1 activity have a much higher mortality rate than patients with lower sVAP-1 activity [134].
3.3 Neurodegenerative Disease
3.3.1 Alzheimer Disease
Alzheimer’s disease (AD) is the most common neurodegenerative disease that worsens as it progresses and eventually leads to death. There is still no effective cure for this disease. The pathology of AD involves formation of plaques and tangles in the brain composed of amyloid and tau proteins. In particular, inflammation has been shown to directly affect the pathogenesis of AD [135].
Adhesion molecules have been associated with AD. Vascular functions are altered in the brains of AD patients, which indicate a common genetic link between acute myocardial infarction and AD [136]. Several brain and peripheral blood vessel abnormalities in AD have been described. Previous studies showed platelets could release amyloid-β in the circulatory system and play a critically important pro-inflammatory role. Upon activation, platelets secrete inflammatory mediators and adhere to leukocytes and endothelial cells by means of adhesive proteins, such as P-selectin, platelet endothelial cell adhesion molecule-1 (PECAM), and intercellular adhesion molecule-1 and -2 (ICAM-1 and -2)[137]. Endothelial markers change during the pre-dementia stage of mild cognitive impairment and mild AD [138]. Interestingly, in a preliminary clinical study, P-selectin and L-selectin plasma levels were decreased in 12 AD patients, and L-selectin was also very low in AD patients with the highest cognitive deterioration [139]. The reduction in P-selectin and, particularly, L-selectin levels in AD patients reflected regulatory impairment of both endothelial function and leukocyte migration [140]. However, E-selectin was not significant different between AD patients and control subjects in a small sample from a case-control study[141]. Determination of selectin blood concentration could is useful in diagnosing and monitoring of the activity of AD. Other adhesion molecules, such as soluble PECAM and ICAM-1, were also increased in AD patient plasma [142, 143]. More importantly, in brain senile plaques from AD patients, ICAM-1 and integrin accumulation was observed by immunostaining[144]. In one study, AD patients had higher serum levels of sICAM-1 than control subjects [141]. In AD patient CSF, sICAM and sVCAM-1 were strongly correlated with each other and with BBB impairment [142]. Zuliani and colleagues found increased plasma VCAM-1 levels in 60 patients with AD as well as 80 patients with vascular dementia when compared to normal elderly controls independent of small or large vessel disease [145]. In summary, data derived from the above studies suggest the promise of using those adhesion molecules as biomarkers for early diagnosis and assessment of AD progression. Future study will further establish the sensitivity and specificity of adhesion molecules for early detection and diagnosis of AD.
3.3.2 Multiple Sclerosis
Multiple sclerosis (MS) is an immune-mediated disorder of the CNS. MS consists of multifocal areas of leukocyte infiltration into the CNS parenchyma, causing in ammation, BBB breakdown, and demyelination, which develop into the clinical manifestations of this disabling disease. Adhesion molecules regulate adhesive interactions between leukocytes and endothelial cells. Circulating immune cells gain access to the CNS and induce inflammation in MS. The animal model for MS, experimental autoimmune encephalomyelitis (EAE), implicates adhesion molecules as critical for EAE pathogenesis. The initial goal for MS research, regarding adhesion molecules, was to identify CNS-specific adhesion molecule expression. Studies were initially performed on brain microvessel endothelial cells because adhesion molecules have an indisputable role in leukocyte extravasation. It has been well documented that adhesion molecules, such as E-selectin, ICAM-1, and VCAM-1, are present on brain microvessels in active lesions of MS patients[146]. Washington et al found temporal and topographical expression of ICAM-1 is correlated with leukocyte infiltration [147]. Accumulating data shows glial cells, astrocytes, and microglia are sources of those adhesion molecules when activated [148]. Moreover, soluble forms of adhesion molecules have been found in the serum and CSF of MS patients, and they are considered useful diagnostically tools. Doerck et al addressed the important question of temporal ICAM-1 and VCAM-1 expression in a serial manner. They found brain ICAM-1 and VCAM-1 upregulation (quantitative analysis by immunocytochemistry) parallels disease onset and remains elevated in the clinical remission phase in the rat model of adoptive transfer experimental autoimmune encephalomyelitis[149]. The important role for P-selectin glycoprotein ligand-1(PSGL-1) or its endothelial ligand, P-selectin, in MS pathogenesis has been largely discussed in EAE models ([150]). Blocking PSGL-1 or P-selectin did not have any impact on disease development.[151]. Furthermore, E-/P-selectin-deficient [152] or PSGL-1-deficient C57BL/6 mice [153, 154] failed to demonstrate slowed development of actively induced EAE by immunization with myelin peptides in multiple laboratories. Firategrast, a novel antagonist of α4β integrins, reduced inflammatory immune cell trafficking into the nervous system in patients with relapsing-remitting MS[155]. Piraino and colleagues concluded α4β1-integrin mediates leukocyte entry through the BBB into the CNS during EAE[156]. Subsequent studies confirmed the predominant role of α4-integrin and vascular cell adhesion molecule (VCAM)-1 in inflammatory cell accumulation in the brain using various EAE animal models [15]. Natalizumab inhibits the influx of leukocytes into the CNS via blockade of the 3–4 subunit of very late activation antigen (VLA)-4. Recently, Natalizumab effectively inhibited EAE development and clearly reduced inflammatory cell recruitment into the CNS [157]. The development of a humanized anti-α4-integrin antibody might be a novel treatment for relapsing-remitting multiple sclerosis [158]. Although accurate function of VAP-1 is still unknown, it is supposed to bind to its putative ligand on the leukocyte surface to hinder infiltration into brain tissues. In a case-controlled study, Airas L et al reported significantly higher VAP-1 serum concentrations in multiple sclerosis patients with ongoing inflammatory activity when compared to MS patients without inflammatory activity [159]. Their study indicated soluble VAP-1 is crucial for MS pathogenesis.
The collective data lends credence to a potential association between adhesion molecules and MS. On a global perspective, many further studies remain to be done to address the therapeutic effects of inhibiting adhesion molecules for MS. Because poor outcomes of the disease are still unacceptably high, prospective, randomized, double-blind, placebo-controlled, and crossover methods must be employed.
4. Concluding perspectives
Inflammation is one of the key contributors exacerbating and driving pathological mechanisms behind several forms of CNS disorders. Adhesion molecules are groups of cell surface molecules controlling the multiple-step leukocyte adhesion cascade. Due to their critical role in leukocyte-endothelial interaction, adhesion molecules, especially novel ectoenzyme, VAP-1, have been extensively investigated in preclinical research and in clinical trials. Therapeutic targeting of adhesion molecules prevented the inflammatory response in multiple forms of CNS disorders. Our review discussed one of the ectoenzymes, VAP-1 in CNS disorders since it is a dual function molecule regulating leukocyte extravasation and a promising target for reducing inflammation in CNS disorders.
Although understanding of the leukocyte adhesion cascade has advanced over the past several decades, several gaps exist in our knowledge that slow successful translation of anti-adhesion strategies in clinic. First, inflammatory response differences between patients and animal models need more attention. Further study should discern the optimal adhesion molecule intervention time in both animal models and patients. Additionally, inflammation is a protective defense system against pathogens, but it is also involved in tissue repair and functional recovery from CNS disorders. Therefore, whether inflammation is detrimental or protective depends on the phase or severity of CNS disorders. Furthermore, the extent of the effects from blockage of a single adhesion molecule on the entire leukocyte adhesion cascade must be determined. Prior studies reported some novel multiple-function cell surface enzymes [10, 160, 161], such as VAP-1, that act as adhesion cascade brakes by regulating other adhesion molecules and cytokine expression [55, 162]. Future study should enhance understanding of ectoenzyme functioning in leukocyte extravasation and identify more possible ectoenzyme mediators. Concentrating on discerning new ectoenzyme functions involved in leukocyte extravasation may help develop new therapeutic strategies for treating neurological diseases.
Figure 2. Enzymatic function of VAP-1.
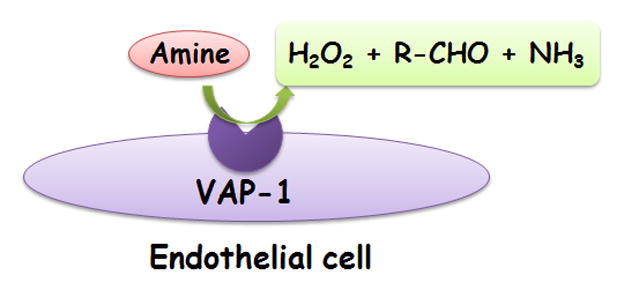
The figure shows VAP-1 is an endothelial cell semi-carbazide sensitive amine oxidase (SSAO). VAP-1 catalyzes oxidative deamination of primary amines and releases biologically active products, including NH3, aldehydes, and H2O2.
References
- 1.Wraith DC, Nicholson LB. The adaptive immune system in diseases of the central nervous system. J Clin Invest. 2012;122(4):1172–1179. doi: 10.1172/JCI58648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Man S, Ubogu EE, Ransohoff RM. Inflammatory cell migration into the central nervous system: a few new twists on an old tale. Brain Pathol. 2007;17(2):243–250. doi: 10.1111/j.1750-3639.2007.00067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37(1):13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 4.Vestweber D. Adhesion and signaling molecules controlling the transmigration of leukocytes through endothelium. Immunol Rev. 2007;218:178–196. doi: 10.1111/j.1600-065X.2007.00533.x. [DOI] [PubMed] [Google Scholar]
- 5.Nourshargh S, Hordijk PL, Sixt M. Breaching multiple barriers: leukocyte motility through venular walls and the interstitium. Nat Rev Mol Cell Biol. 2010;11(5):366–378. doi: 10.1038/nrm2889. [DOI] [PubMed] [Google Scholar]
- 6.Bargatze RF, Kurk S, Butcher EC, Jutila MA. Neutrophils roll on adherent neutrophils bound to cytokine-induced endothelial cells via L-selectin on the rolling cells. J Exp Med. 1994;180(5):1785–1792. doi: 10.1084/jem.180.5.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wittchen ES. Endothelial signaling in paracellular and transcellular leukocyte transmigration. Front Biosci. 2009;14:2522–2545. doi: 10.2741/3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Penberthy TW, Jiang Y, Graves DT. Leukocyte adhesion molecules. Crit Rev Oral Biol Med. 1997;8(4):380–388. doi: 10.1177/10454411970080040201. [DOI] [PubMed] [Google Scholar]
- 9.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7(9):678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 10.Salmi M, Jalkanen S. Cell-surface enzymes in control of leukocyte trafficking. Nat Rev Immunol. 2005;5(10):760–771. doi: 10.1038/nri1705. [DOI] [PubMed] [Google Scholar]
- 11.Salmi M, Jalkanen S. VAP-1: an adhesin and an enzyme. Trends Immunol. 2001;22(4):211–216. doi: 10.1016/s1471-4906(01)01870-1. [DOI] [PubMed] [Google Scholar]
- 12.Vexler ZS, Tang XN, Yenari MA. Inflammation in adult and neonatal stroke. Clin Neurosci Res. 2006;6(5):293–313. doi: 10.1016/j.cnr.2006.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knoblach SM, Faden AI. Administration of either anti-intercellular adhesion molecule-1 or a nonspecific control antibody improves recovery after traumatic brain injury in the rat. J Neurotrauma. 2002;19(9):1039–1050. doi: 10.1089/089771502760341956. [DOI] [PubMed] [Google Scholar]
- 14.Arumugam TV, Salter JW, Chidlow JH, Ballantyne CM, Kevil CG, Granger DN. Contributions of LFA-1 and Mac-1 to brain injury and microvascular dysfunction induced by transient middle cerebral artery occlusion. Am J Physiol Heart Circ Physiol. 2004;287(6):H2555–2560. doi: 10.1152/ajpheart.00588.2004. [DOI] [PubMed] [Google Scholar]
- 15.Engelhardt B. Immune cell entry into the central nervous system: involvement of adhesion molecules and chemokines. J Neurol Sci. 2008;274(1–2):23–26. doi: 10.1016/j.jns.2008.05.019. [DOI] [PubMed] [Google Scholar]
- 16.Rivera-Nieves J, Gorfu G, Ley K. Leukocyte adhesion molecules in animal models of inflammatory bowel disease. Inflamm Bowel Dis. 2008;14(12):1715–1735. doi: 10.1002/ibd.20501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ceulemans AG, Zgavc T, Kooijman R, Hachimi-Idrissi S, Sarre S, Michotte Y. The dual role of the neuroinflammatory response after ischemic stroke: modulatory effects of hypothermia. J Neuroinflammation. 2010;7:74. doi: 10.1186/1742-2094-7-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J Neuroimmunol. 2007;184(1–2):53–68. doi: 10.1016/j.jneuroim.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geng JG, Bevilacqua MP, Moore KL, McIntyre TM, Prescott SM, Kim JM, Bliss GA, Zimmerman GA, McEver RP. Rapid neutrophil adhesion to activated endothelium mediated by GMP-140. Nature. 1990;343(6260):757–760. doi: 10.1038/343757a0. [DOI] [PubMed] [Google Scholar]
- 20.Janner G. Enter the union safety representative. Occup Health (Lond) 1978;30(3):110–113. [PubMed] [Google Scholar]
- 21.McEver RP, Cummings RD. Perspectives series: cell adhesion in vascular biology. Role of PSGL-1 binding to selectins in leukocyte recruitment. J Clin Invest. 1997;100(3):485–491. doi: 10.1172/JCI119556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Setiadi H, Sedgewick G, Erlandsen SL, McEver RP. Interactions of the cytoplasmic domain of P-selectin with clathrin-coated pits enhance leukocyte adhesion under flow. J Cell Biol. 1998;142(3):859–871. doi: 10.1083/jcb.142.3.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fujimoto T, McEver RP. The cytoplasmic domain of P-selectin is phosphorylated on serine and threonine residues. Blood. 1993;82(6):1758–1766. [PubMed] [Google Scholar]
- 24.Hidalgo A, Peired AJ, Wild MK, Vestweber D, Frenette PS. Complete identification of E-selectin ligands on neutrophils reveals distinct functions of PSGL-1, ESL-1, and CD44. Immunity. 2007;26(4):477–489. doi: 10.1016/j.immuni.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Butcher EC. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 1991;67(6):1033–1036. doi: 10.1016/0092-8674(91)90279-8. [DOI] [PubMed] [Google Scholar]
- 26.Walcheck B, White M, Kurk S, Kishimoto TK, Jutila MA. Characterization of the bovine peripheral lymph node homing receptor: a lectin cell adhesion molecule (LECAM) Eur J Immunol. 1992;22(2):469–476. doi: 10.1002/eji.1830220227. [DOI] [PubMed] [Google Scholar]
- 27.Lasky LA. Selectins: interpreters of cell-specific carbohydrate information during inflammation. Science. 1992;258(5084):964–969. doi: 10.1126/science.1439808. [DOI] [PubMed] [Google Scholar]
- 28.Tedder TF, Steeber DA, Chen A, Engel P. The selectins: vascular adhesion molecules. FASEB J. 1995;9(10):866–873. [PubMed] [Google Scholar]
- 29.Ohmori K, Kanda K, Mitsuoka C, Kanamori A, Kurata-Miura K, Sasaki K, Nishi T, Tamatani T, Kannagi R. P- and E-selectins recognize sialyl 6-sulfo Lewis X, the recently identified L-selectin ligand. Biochem Biophys Res Commun. 2000;278(1):90–96. doi: 10.1006/bbrc.2000.3768. [DOI] [PubMed] [Google Scholar]
- 30.Li X, Steeber DA, Tang ML, Farrar MA, Perlmutter RM, Tedder TF. Regulation of L-selectin-mediated rolling through receptor dimerization. J Exp Med. 1998;188(7):1385–1390. doi: 10.1084/jem.188.7.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vestweber D, Blanks JE. Mechanisms that regulate the function of the selectins and their ligands. Physiol Rev. 1999;79(1):181–213. doi: 10.1152/physrev.1999.79.1.181. [DOI] [PubMed] [Google Scholar]
- 32.Schleiffenbaum B, Spertini O, Tedder TF. Soluble L-selectin is present in human plasma at high levels and retains functional activity. J Cell Biol. 1992;119(1):229–238. doi: 10.1083/jcb.119.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kishimoto TK, Jutila MA, Berg EL, Butcher EC. Neutrophil Mac-1 and MEL-14 adhesion proteins inversely regulated by chemotactic factors. Science. 1989;245(4923):1238–1241. doi: 10.1126/science.2551036. [DOI] [PubMed] [Google Scholar]
- 34.Ruoslahti E. Integrins. J Clin Invest. 1991;87(1):1–5. doi: 10.1172/JCI114957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69(1):11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 36.Yilmaz G, Granger DN. Cell adhesion molecules and ischemic stroke. Neurol Res. 2008;30(8):783–793. doi: 10.1179/174313208X341085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anderson DC, Springer TA. Leukocyte adhesion deficiency: an inherited defect in the Mac-1, LFA-1, and p150,95 glycoproteins. Annu Rev Med. 1987;38:175–194. doi: 10.1146/annurev.me.38.020187.001135. [DOI] [PubMed] [Google Scholar]
- 38.Marlin SD, Springer TA. Purified intercellular adhesion molecule-1 (ICAM-1) is a ligand for lymphocyte function-associated antigen 1 (LFA-1) Cell. 1987;51(5):813–819. doi: 10.1016/0092-8674(87)90104-8. [DOI] [PubMed] [Google Scholar]
- 39.Staunton DE, Dustin ML, Springer TA. Functional cloning of ICAM-2, a cell adhesion ligand for LFA-1 homologous to ICAM-1. Nature. 1989;339(6219):61–64. doi: 10.1038/339061a0. [DOI] [PubMed] [Google Scholar]
- 40.Jung U, Norman KE, Scharffetter-Kochanek K, Beaudet AL, Ley K. Transit time of leukocytes rolling through venules controls cytokine-induced inflammatory cell recruitment in vivo. J Clin Invest. 1998;102(8):1526–1533. doi: 10.1172/JCI119893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dunne JL, Ballantyne CM, Beaudet AL, Ley K. Control of leukocyte rolling velocity in TNF-alpha-induced inflammation by LFA-1 and Mac-1. Blood. 2002;99(1):336–341. doi: 10.1182/blood.v99.1.336. [DOI] [PubMed] [Google Scholar]
- 42.Diamond MS, Staunton DE, Marlin SD, Springer TA. Binding of the integrin Mac-1 (CD11b/CD18) to the third immunoglobulin-like domain of ICAM-1 (CD54) and its regulation by glycosylation. Cell. 1991;65(6):961–971. doi: 10.1016/0092-8674(91)90548-d. [DOI] [PubMed] [Google Scholar]
- 43.Knorr R, Dustin ML. The lymphocyte function-associated antigen 1 I domain is a transient binding module for intercellular adhesion molecule (ICAM)-1 and ICAM-3 in hydrodynamic flow. J Exp Med. 1997;186(5):719–730. doi: 10.1084/jem.186.5.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simon RH, DeHart PD, Todd RF., 3rd Neutrophil-induced injury of rat pulmonary alveolar epithelial cells. J Clin Invest. 1986;78(5):1375–1386. doi: 10.1172/JCI112724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carlos TM, Schwartz BR, Kovach NL, Yee E, Rosa M, Osborn L, Chi-Rosso G, Newman B, Lobb R, et al. Vascular cell adhesion molecule-1 mediates lymphocyte adherence to cytokine-activated cultured human endothelial cells. Blood. 1990;76(5):965–970. [PubMed] [Google Scholar]
- 46.Elices MJ, Osborn L, Takada Y, Crouse C, Luhowskyj S, Hemler ME, Lobb RR. VCAM-1 on activated endothelium interacts with the leukocyte integrin VLA-4 at a site distinct from the VLA-4/fibronectin binding site. Cell. 1990;60(4):577–584. doi: 10.1016/0092-8674(90)90661-w. [DOI] [PubMed] [Google Scholar]
- 47.Salmi M, Jalkanen S. A 90-kilodalton endothelial cell molecule mediating lymphocyte binding in humans. Science. 1992;257(5075):1407–1409. doi: 10.1126/science.1529341. [DOI] [PubMed] [Google Scholar]
- 48.Salmi M, Kalimo K, Jalkanen S. Induction and function of vascular adhesion protein-1 at sites of inflammation. J Exp Med. 1993;178(6):2255–2260. doi: 10.1084/jem.178.6.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith DJ, Salmi M, Bono P, Hellman J, Leu T, Jalkanen S. Cloning of vascular adhesion protein 1 reveals a novel multifunctional adhesion molecule. J Exp Med. 1998;188(1):17–27. doi: 10.1084/jem.188.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jaakkola K, Nikula T, Holopainen R, Vahasilta T, Matikainen MT, Laukkanen ML, Huupponen R, Halkola L, Nieminen L, Hiltunen J, Parviainen S, Clark MR, Knuuti J, Savunen T, Kaapa P, Voipio-Pulkki LM, Jalkanen S. In vivo detection of vascular adhesion protein-1 in experimental inflammation. Am J Pathol. 2000;157(2):463–471. doi: 10.1016/S0002-9440(10)64558-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu PH, Zuo DM. Aminoguanidine inhibits semicarbazide-sensitive amine oxidase activity: implications for advanced glycation and diabetic complications. Diabetologia. 1997;40(11):1243–1250. doi: 10.1007/s001250050816. [DOI] [PubMed] [Google Scholar]
- 52.Bogdan C, Rollinghoff M, Diefenbach A. Reactive oxygen and reactive nitrogen intermediates in innate and specific immunity. Curr Opin Immunol. 2000;12(1):64–76. doi: 10.1016/s0952-7915(99)00052-7. [DOI] [PubMed] [Google Scholar]
- 53.Johnston B, Kanwar S, Kubes P. Hydrogen peroxide induces leukocyte rolling: modulation by endogenous antioxidant mechanisms including NO. Am J Physiol. 1996;271(2 Pt 2):H614–621. doi: 10.1152/ajpheart.1996.271.2.H614. [DOI] [PubMed] [Google Scholar]
- 54.Jalkanen S, Karikoski M, Mercier N, Koskinen K, Henttinen T, Elima K, Salmivirta K, Salmi M. The oxidase activity of vascular adhesion protein-1 (VAP-1) induces endothelial E- and P-selectins and leukocyte binding. Blood. 2007;110(6):1864–1870. doi: 10.1182/blood-2007-01-069674. [DOI] [PubMed] [Google Scholar]
- 55.Noda K, She H, Nakazawa T, Hisatomi T, Nakao S, Almulki L, Zandi S, Miyahara S, Ito Y, Thomas KL, Garland RC, Miller JW, Gragoudas ES, Mashima Y, Hafezi-Moghadam A. Vascular adhesion protein-1 blockade suppresses choroidal neovascularization. FASEB J. 2008;22(8):2928–2935. doi: 10.1096/fj.07-105346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lalor PF, Sun PJ, Weston CJ, Martin-Santos A, Wakelam MJ, Adams DH. Activation of vascular adhesion protein-1 on liver endothelium results in an NF-kappaB-dependent increase in lymphocyte adhesion. Hepatology. 2007;45(2):465–474. doi: 10.1002/hep.21497. [DOI] [PubMed] [Google Scholar]
- 57.Hallenbeck JM, Dutka AJ, Tanishima T, Kochanek PM, Kumaroo KK, Thompson CB, Obrenovitch TP, Contreras TJ. Polymorphonuclear leukocyte accumulation in brain regions with low blood flow during the early postischemic period. Stroke. 1986;17(2):246–253. doi: 10.1161/01.str.17.2.246. [DOI] [PubMed] [Google Scholar]
- 58.Zhang RL, Chopp M, Chen H, Garcia JH. Temporal profile of ischemic tissue damage, neutrophil response, and vascular plugging following permanent and transient (2H) middle cerebral artery occlusion in the rat. J Neurol Sci. 1994;125(1):3–10. doi: 10.1016/0022-510x(94)90234-8. [DOI] [PubMed] [Google Scholar]
- 59.Campanella M, Sciorati C, Tarozzo G, Beltramo M. Flow cytometric analysis of inflammatory cells in ischemic rat brain. Stroke. 2002;33(2):586–592. doi: 10.1161/hs0202.103399. [DOI] [PubMed] [Google Scholar]
- 60.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22(9):391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 61.Carvalho-Tavares J, Hickey MJ, Hutchison J, Michaud J, Sutcliffe IT, Kubes P. A role for platelets and endothelial selectins in tumor necrosis factor-alpha-induced leukocyte recruitment in the brain microvasculature. Circ Res. 2000;87(12):1141–1148. doi: 10.1161/01.res.87.12.1141. [DOI] [PubMed] [Google Scholar]
- 62.Sughrue ME, Mehra A, Connolly ES, Jr, D’Ambrosio AL. Anti-adhesion molecule strategies as potential neuroprotective agents in cerebral ischemia: a critical review of the literature. Inflamm Res. 2004;53(10):497–508. doi: 10.1007/s00011-004-1282-0. [DOI] [PubMed] [Google Scholar]
- 63.Zhang R, Chopp M, Zhang Z, Jiang N, Powers C. The expression of P- and E-selectins in three models of middle cerebral artery occlusion. Brain Res. 1998;785(2):207–214. doi: 10.1016/s0006-8993(97)01343-7. [DOI] [PubMed] [Google Scholar]
- 64.Ishikawa M, Cooper D, Arumugam TV, Zhang JH, Nanda A, Granger DN. Platelet-leukocyte-endothelial cell interactions after middle cerebral artery occlusion and reperfusion. J Cereb Blood Flow Metab. 2004;24(8):907–915. doi: 10.1097/01.WCB.0000132690.96836.7F. [DOI] [PubMed] [Google Scholar]
- 65.Ruehl ML, Orozco JA, Stoker MB, McDonagh PF, Coull BM, Ritter LS. Protective effects of inhibiting both blood and vascular selectins after stroke and reperfusion. Neurol Res. 2002;24(3):226–232. doi: 10.1179/016164102101199738. [DOI] [PubMed] [Google Scholar]
- 66.Ma XL, Weyrich AS, Lefer DJ, Buerke M, Albertine KH, Kishimoto TK, Lefer AM. Monoclonal antibody to L-selectin attenuates neutrophil accumulation and protects ischemic reperfused cat myocardium. Circulation. 1993;88(2):649–658. doi: 10.1161/01.cir.88.2.649. [DOI] [PubMed] [Google Scholar]
- 67.Zhang RL, Chopp M, Zhang ZG, Phillips ML, Rosenbloom CL, Cruz R, Manning A. E-selectin in focal cerebral ischemia and reperfusion in the rat. J Cereb Blood Flow Metab. 1996;16(6):1126–1136. doi: 10.1097/00004647-199611000-00006. [DOI] [PubMed] [Google Scholar]
- 68.Huang J, Choudhri TF, Winfree CJ, McTaggart RA, Kiss S, Mocco J, Kim LJ, Protopsaltis TS, Zhang Y, Pinsky DJ, Connolly ES., Jr Postischemic cerebrovascular E-selectin expression mediates tissue injury in murine stroke. Stroke. 2000;31(12):3047–3053. [PubMed] [Google Scholar]
- 69.Yenari MA, Kunis D, Sun GH, Onley D, Watson L, Turner S, Whitaker S, Steinberg GK. Hu23F2G, an antibody recognizing the leukocyte CD11/CD18 integrin, reduces injury in a rabbit model of transient focal cerebral ischemia. Exp Neurol. 1998;153(2):223–233. doi: 10.1006/exnr.1998.6876. [DOI] [PubMed] [Google Scholar]
- 70.Chopp M, Zhang RL, Chen H, Li Y, Jiang N, Rusche JR. Postischemic administration of an anti-Mac-1 antibody reduces ischemic cell damage after transient middle cerebral artery occlusion in rats. Stroke. 1994;25(4):869–875. doi: 10.1161/01.str.25.4.869. discussion 875–866. [DOI] [PubMed] [Google Scholar]
- 71.Chen H, Chopp M, Zhang RL, Bodzin G, Chen Q, Rusche JR, Todd RF., 3rd Anti-CD11b monoclonal antibody reduces ischemic cell damage after transient focal cerebral ischemia in rat. Ann Neurol. 1994;35(4):458–463. doi: 10.1002/ana.410350414. [DOI] [PubMed] [Google Scholar]
- 72.Bowes MP, Rothlein R, Fagan SC, Zivin JA. Monoclonal antibodies preventing leukocyte activation reduce experimental neurologic injury and enhance efficacy of thrombolytic therapy. Neurology. 1995;45(4):815–819. doi: 10.1212/wnl.45.4.815. [DOI] [PubMed] [Google Scholar]
- 73.Bednar MM, Wright SD, Raymond-Russell SJ, Kohut JJ, Gross CE. IB4, a monoclonal antibody against the CD18 leukocyte adhesion protein, reduces intracranial pressure following thromboembolic stroke in the rabbit. Neurol Res. 1996;18(2):171–175. doi: 10.1080/01616412.1996.11740398. [DOI] [PubMed] [Google Scholar]
- 74.Becker K, Kindrick D, Relton J, Harlan J, Winn R. Antibody to the alpha4 integrin decreases infarct size in transient focal cerebral ischemia in rats. Stroke. 2001;32(1):206–211. doi: 10.1161/01.str.32.1.206. [DOI] [PubMed] [Google Scholar]
- 75.Wang X, Feuerstein GZ. Induced expression of adhesion molecules following focal brain ischemia. J Neurotrauma. 1995;12(5):825–832. doi: 10.1089/neu.1995.12.825. [DOI] [PubMed] [Google Scholar]
- 76.Blann A, Kumar P, Krupinski J, McCollum C, Beevers DG, Lip GY. Soluble intercelluar adhesion molecule-1, E-selectin, vascular cell adhesion molecule-1 and von Willebrand factor in stroke. Blood Coagul Fibrinolysis. 1999;10(5):277–284. doi: 10.1097/00001721-199907000-00009. [DOI] [PubMed] [Google Scholar]
- 77.Vemuganti R, Dempsey RJ, Bowen KK. Inhibition of intercellular adhesion molecule-1 protein expression by antisense oligonucleotides is neuroprotective after transient middle cerebral artery occlusion in rat. Stroke. 2004;35(1):179–184. doi: 10.1161/01.STR.0000106479.53235.3E. [DOI] [PubMed] [Google Scholar]
- 78.Kitagawa K, Matsumoto M, Mabuchi T, Yagita Y, Ohtsuki T, Hori M, Yanagihara T. Deficiency of intercellular adhesion molecule 1 attenuates microcirculatory disturbance and infarction size in focal cerebral ischemia. J Cereb Blood Flow Metab. 1998;18(12):1336–1345. doi: 10.1097/00004647-199812000-00008. [DOI] [PubMed] [Google Scholar]
- 79.Connolly ES, Jr, Winfree CJ, Springer TA, Naka Y, Liao H, Yan SD, Stern DM, Solomon RA, Gutierrez-Ramos JC, Pinsky DJ. Cerebral protection in homozygous null ICAM-1 mice after middle cerebral artery occlusion. Role of neutrophil adhesion in the pathogenesis of stroke. J Clin Invest. 1996;97(1):209–216. doi: 10.1172/JCI118392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chopp M, Li Y, Jiang N, Zhang RL, Prostak J. Antibodies against adhesion molecules reduce apoptosis after transient middle cerebral artery occlusion in rat brain. J Cereb Blood Flow Metab. 1996;16(4):578–584. doi: 10.1097/00004647-199607000-00007. [DOI] [PubMed] [Google Scholar]
- 81.Kanemoto Y, Nakase H, Akita N, Sakaki T. Effects of anti-intercellular adhesion molecule-1 antibody on reperfusion injury induced by late reperfusion in the rat middle cerebral artery occlusion model. Neurosurgery. 2002;51(4):1034–1041. doi: 10.1097/00006123-200210000-00033. discussion 1041–1032. [DOI] [PubMed] [Google Scholar]
- 82.Krams M, Lees KR, Hacke W, Grieve AP, Orgogozo JM, Ford GA. Acute Stroke Therapy by Inhibition of Neutrophils (ASTIN): an adaptive dose-response study of UK-279,276 in acute ischemic stroke. Stroke. 2003;34(11):2543–2548. doi: 10.1161/01.STR.0000092527.33910.89. [DOI] [PubMed] [Google Scholar]
- 83.Becker KJ. Anti-leukocyte antibodies: LeukArrest (Hu23F2G) and Enlimomab (R6.5) in acute stroke. Curr Med Res Opin. 2002;18(Suppl 2):s18–22. doi: 10.1185/030079902125000688. [DOI] [PubMed] [Google Scholar]
- 84.Jiang N, Moyle M, Soule HR, Rote WE, Chopp M. Neutrophil inhibitory factor is neuroprotective after focal ischemia in rats. Ann Neurol. 1995;38(6):935–942. doi: 10.1002/ana.410380615. [DOI] [PubMed] [Google Scholar]
- 85.Jean WC, Spellman SR, Nussbaum ES, Low WC. Reperfusion injury after focal cerebral ischemia: the role of inflammation and the therapeutic horizon. Neurosurgery. 1998;43(6):1382–1396. doi: 10.1097/00006123-199812000-00076. discussion 1396–1387. [DOI] [PubMed] [Google Scholar]
- 86.Use of anti-ICAM-1 therapy in ischemic stroke: results of the Enlimomab Acute Stroke Trial. Neurology. 2001;57(8):1428–1434. doi: 10.1212/wnl.57.8.1428. [DOI] [PubMed] [Google Scholar]
- 87.DeGraba TJ. The role of inflammation after acute stroke: utility of pursuing anti-adhesion molecule therapy. Neurology. 1998;51(3 Suppl 3):S62–68. doi: 10.1212/wnl.51.3_suppl_3.s62. [DOI] [PubMed] [Google Scholar]
- 88.Vuorte J, Lindsberg PJ, Kaste M, Meri S, Jansson SE, Rothlein R, Repo H. Anti-ICAM-1 monoclonal antibody R6.5 (Enlimomab) promotes activation of neutrophils in whole blood. J Immunol. 1999;162(4):2353–2357. [PubMed] [Google Scholar]
- 89.Wang J, Dore S. Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2007;27(5):894–908. doi: 10.1038/sj.jcbfm.9600403. [DOI] [PubMed] [Google Scholar]
- 90.Wang J, Tsirka SE. Neuroprotection by inhibition of matrix metalloproteinases in a mouse model of intracerebral haemorrhage. Brain. 2005;128(Pt 7):1622–1633. doi: 10.1093/brain/awh489. [DOI] [PubMed] [Google Scholar]
- 91.Gong C, Hoff JT, Keep RF. Acute inflammatory reaction following experimental intracerebral hemorrhage in rat. Brain Res. 2000;871(1):57–65. doi: 10.1016/s0006-8993(00)02427-6. [DOI] [PubMed] [Google Scholar]
- 92.Xue M, Del Bigio MR. Intracerebral injection of autologous whole blood in rats: time course of inflammation and cell death. Neurosci Lett. 2000;283(3):230–232. doi: 10.1016/s0304-3940(00)00971-x. [DOI] [PubMed] [Google Scholar]
- 93.Xue M, Del Bigio MR. Intracortical hemorrhage injury in rats : relationship between blood fractions and brain cell death. Stroke. 2000;31(7):1721–1727. doi: 10.1161/01.str.31.7.1721. [DOI] [PubMed] [Google Scholar]
- 94.Lawson LJ, Perry VH, Gordon S. Turnover of resident microglia in the normal adult mouse brain. Neuroscience. 1992;48(2):405–415. doi: 10.1016/0306-4522(92)90500-2. [DOI] [PubMed] [Google Scholar]
- 95.Wang J, Tsirka SE. Contribution of extracellular proteolysis and microglia to intracerebral hemorrhage. Neurocrit Care. 2005;3(1):77–85. doi: 10.1385/NCC:3:1:077. [DOI] [PubMed] [Google Scholar]
- 96.Wang J, Rogove AD, Tsirka AE, Tsirka SE. Protective role of tuftsin fragment 1-3 in an animal model of intracerebral hemorrhage. Ann Neurol. 2003;54(5):655–664. doi: 10.1002/ana.10750. [DOI] [PubMed] [Google Scholar]
- 97.Wang J, Tsirka SE. Tuftsin fragment 1-3 is beneficial when delivered after the induction of intracerebral hemorrhage. Stroke. 2005;36(3):613–618. doi: 10.1161/01.STR.0000155729.12931.8f. [DOI] [PubMed] [Google Scholar]
- 98.Hickenbottom SL, Grotta JC, Strong R, Denner LA, Aronowski J. Nuclear factor-kappaB and cell death after experimental intracerebral hemorrhage in rats. Stroke. 1999;30(11):2472–2477. doi: 10.1161/01.str.30.11.2472. discussion 2477–2478. [DOI] [PubMed] [Google Scholar]
- 99.Titova E, Ostrowski RP, Kevil CG, Tong W, Rojas H, Sowers LC, Zhang JH, Tang J. Reduced brain injury in CD18-deficient mice after experimental intracerebral hemorrhage. J Neurosci Res. 2008;86(14):3240–3245. doi: 10.1002/jnr.21762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Xu HL, Salter-Cid L, Linnik MD, Wang EY, Paisansathan C, Pelligrino DA. Vascular adhesion protein-1 plays an important role in postischemic inflammation and neuropathology in diabetic, estrogen-treated ovariectomized female rats subjected to transient forebrain ischemia. J Pharmacol Exp Ther. 2006;317(1):19–29. doi: 10.1124/jpet.105.096958. [DOI] [PubMed] [Google Scholar]
- 101.Ma Q, Manaenko A, Khatibi NH, Chen W, Zhang JH, Tang J. Vascular adhesion protein-1 inhibition provides antiinflammatory protection after an intracerebral hemorrhagic stroke in mice. J Cereb Blood Flow Metab. 2011;31(3):881–893. doi: 10.1038/jcbfm.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Airas L, Lindsberg PJ, Karjalainen-Lindsberg ML, Mononen I, Kotisaari K, Smith DJ, Jalkanen S. Vascular adhesion protein-1 in human ischaemic stroke. Neuropathol Appl Neurobiol. 2008;34(4):394–402. doi: 10.1111/j.1365-2990.2007.00911.x. [DOI] [PubMed] [Google Scholar]
- 103.Hernandez-Guillamon M, Garcia-Bonilla L, Sole M, Sosti V, Pares M, Campos M, Ortega-Aznar A, Dominguez C, Rubiera M, Ribo M, Quintana M, Molina CA, Alvarez-Sabin J, Rosell A, Unzeta M, Montaner J. Plasma VAP-1/SSAO activity predicts intracranial hemorrhages and adverse neurological outcome after tissue plasminogen activator treatment in stroke. Stroke. 2010;41(7):1528–1535. doi: 10.1161/STROKEAHA.110.584623. [DOI] [PubMed] [Google Scholar]
- 104.Hernandez-Guillamon M, Sole M, Delgado P, Garcia-Bonilla L, Giralt D, Boada C, Penalba A, Garcia S, Flores A, Ribo M, Alvarez-Sabin J, Ortega-Aznar A, Unzeta M, Montaner J. VAP-1/SSAO Plasma Activity and Brain Expression in Human Hemorrhagic Stroke. Cerebrovasc Dis. 2011;33(1):55–63. doi: 10.1159/000333370. [DOI] [PubMed] [Google Scholar]
- 105.Maiuri F, Gallicchio B, Donati P, Carandente M. The blood leukocyte count and its prognostic significance in subarachnoid hemorrhage. J Neurosurg Sci. 1987;31(2):45–48. [PubMed] [Google Scholar]
- 106.McGirt MJ, Mavropoulos JC, McGirt LY, Alexander MJ, Friedman AH, Laskowitz DT, Lynch JR. Leukocytosis as an independent risk factor for cerebral vasospasm following aneurysmal subarachnoid hemorrhage. J Neurosurg. 2003;98(6):1222–1226. doi: 10.3171/jns.2003.98.6.1222. [DOI] [PubMed] [Google Scholar]
- 107.Neil-Dwyer G, Cruickshank J. The blood leucocyte count and its prognostic significance in subarachnoid haemorrhage. Brain. 1974;97(1):79–86. doi: 10.1093/brain/97.1.79. [DOI] [PubMed] [Google Scholar]
- 108.Chaichana KL, Pradilla G, Huang J, Tamargo RJ. Role of inflammation (leukocyte-endothelial cell interactions) in vasospasm after subarachnoid hemorrhage. World Neurosurg. 2010;73(1):22–41. doi: 10.1016/j.surneu.2009.05.027. [DOI] [PubMed] [Google Scholar]
- 109.Aihara Y, Kasuya H, Onda H, Hori T, Takeda J. Quantitative analysis of gene expressions related to inflammation in canine spastic artery after subarachnoid hemorrhage. Stroke. 2001;32(1):212–217. doi: 10.1161/01.str.32.1.212. [DOI] [PubMed] [Google Scholar]
- 110.Handa Y, Kubota T, Kaneko M, Tsuchida A, Kobayashi H, Kawano H. Expression of intercellular adhesion molecule 1 (ICAM-1) on the cerebral artery following subarachnoid haemorrhage in rats. Acta Neurochir (Wien) 1995;132(1–3):92–97. doi: 10.1007/BF01404854. [DOI] [PubMed] [Google Scholar]
- 111.Bavbek M, Polin R, Kwan AL, Arthur AS, Kassell NF, Lee KS. Monoclonal antibodies against ICAM-1 and CD18 attenuate cerebral vasospasm after experimental subarachnoid hemorrhage in rabbits. Stroke. 1998;29(9):1930–1935. doi: 10.1161/01.str.29.9.1930. discussion 1935–1936. [DOI] [PubMed] [Google Scholar]
- 112.Chang CZ, Lin CL, Kassel NF, Kwan AL, Howng SL. 6-Mercaptopurine attenuates adhesive molecules in experimental vasospasm. Acta Neurochir (Wien) 2010;152(5):861–867. doi: 10.1007/s00701-010-0602-0. [DOI] [PubMed] [Google Scholar]
- 113.Sills AK, Jr, Clatterbuck RE, Thompson RC, Cohen PL, Tamargo RJ. Endothelial cell expression of intercellular adhesion molecule 1 in experimental posthemorrhagic vasospasm. Neurosurgery. 1997;41(2):453–460. doi: 10.1097/00006123-199708000-00025. discussion 460–451. [DOI] [PubMed] [Google Scholar]
- 114.Oshiro EM, Hoffman PA, Dietsch GN, Watts MC, Pardoll DM, Tamargo RJ. Inhibition of experimental vasospasm with anti-intercellular adhesion molecule-1 monoclonal antibody in rats. Stroke. 1997;28(10):2031–2037. doi: 10.1161/01.str.28.10.2031. discussion 2037–2038. [DOI] [PubMed] [Google Scholar]
- 115.Clatterbuck RE, Oshiro EM, Hoffman PA, Dietsch GN, Pardoll DM, Tamargo RJ. Inhibition of vasospasm with lymphocyte function-associated antigen-1 monoclonal antibody in a femoral artery model in rats. J Neurosurg. 2002;97(3):676–682. doi: 10.3171/jns.2002.97.3.0676. [DOI] [PubMed] [Google Scholar]
- 116.Lin CL, Dumont AS, Calisaneller T, Kwan AL, Hwong SL, Lee KS. Monoclonal antibody against E selectin attenuates subarachnoid hemorrhage-induced cerebral vasospasm. Surg Neurol. 2005;64(3):201–205. doi: 10.1016/j.surneu.2005.04.038. discussion 205–206. [DOI] [PubMed] [Google Scholar]
- 117.Polin RS, Bavbek M, Shaffrey ME, Billups K, Bogaev CA, Kassell NF, Lee KS. Detection of soluble E-selectin, ICAM-1, VCAM-1, and L-selectin in the cerebrospinal fluid of patients after subarachnoid hemorrhage. J Neurosurg. 1998;89(4):559–567. doi: 10.3171/jns.1998.89.4.0559. [DOI] [PubMed] [Google Scholar]
- 118.Nissen JJ, Mantle D, Gregson B, Mendelow AD. Serum concentration of adhesion molecules in patients with delayed ischaemic neurological deficit after aneurysmal subarachnoid haemorrhage: the immunoglobulin and selectin superfamilies. J Neurol Neurosurg Psychiatry. 2001;71(3):329–333. doi: 10.1136/jnnp.71.3.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nissen JJ, Mantle D, Blackburn A, Barnes J, Wooldridge T, Gregson B, Mendelow AD. The selectin superfamily: the role of selectin adhesion molecules in delayed cerebral ischaemia after aneurysmal subarachnoid haemorrhage. Acta Neurochir Suppl. 2000;76:55–60. doi: 10.1007/978-3-7091-6346-7_11. [DOI] [PubMed] [Google Scholar]
- 120.Weber C, Erl W, Weber KS, Weber PC. HMG-CoA reductase inhibitors decrease CD11b expression and CD11b-dependent adhesion of monocytes to endothelium and reduce increased adhesiveness of monocytes isolated from patients with hypercholesterolemia. J Am Coll Cardiol. 1997;30(5):1212–1217. doi: 10.1016/s0735-1097(97)00324-0. [DOI] [PubMed] [Google Scholar]
- 121.Kallen J, Welzenbach K, Ramage P, Geyl D, Kriwacki R, Legge G, Cottens S, Weitz-Schmidt G, Hommel U. Structural basis for LFA-1 inhibition upon lovastatin binding to the CD11a I-domain. J Mol Biol. 1999;292(1):1–9. doi: 10.1006/jmbi.1999.3047. [DOI] [PubMed] [Google Scholar]
- 122.Kramer AH, Gurka MJ, Nathan B, Dumont AS, Kassell NF, Bleck TP. Statin use was not associated with less vasospasm or improved outcome after subarachnoid hemorrhage. Neurosurgery. 2008;62(2):422–427. doi: 10.1227/01.neu.0000316009.19012.e3. discussion 427–430. [DOI] [PubMed] [Google Scholar]
- 123.Tseng MY, Czosnyka M, Richards H, Pickard JD, Kirkpatrick PJ. Effects of acute treatment with pravastatin on cerebral vasospasm, autoregulation, and delayed ischemic deficits after aneurysmal subarachnoid hemorrhage: a phase II randomized placebo-controlled trial. Stroke. 2005;36(8):1627–1632. doi: 10.1161/01.STR.0000176743.67564.5d. [DOI] [PubMed] [Google Scholar]
- 124.Tseng MY, Hutchinson PJ, Czosnyka M, Richards H, Pickard JD, Kirkpatrick PJ. Effects of acute pravastatin treatment on intensity of rescue therapy, length of inpatient stay, and 6-month outcome in patients after aneurysmal subarachnoid hemorrhage. Stroke. 2007;38(5):1545–1550. doi: 10.1161/STROKEAHA.106.475905. [DOI] [PubMed] [Google Scholar]
- 125.Helmy A, De Simoni MG, Guilfoyle MR, Carpenter KL, Hutchinson PJ. Cytokines and innate inflammation in the pathogenesis of human traumatic brain injury. Prog Neurobiol. 2011;95(3):352–372. doi: 10.1016/j.pneurobio.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 126.Marklund N, Bakshi A, Castelbuono DJ, Conte V, McIntosh TK. Evaluation of pharmacological treatment strategies in traumatic brain injury. Curr Pharm Des. 2006;12(13):1645–1680. doi: 10.2174/138161206776843340. [DOI] [PubMed] [Google Scholar]
- 127.Schouten JW. Neuroprotection in traumatic brain injury: a complex struggle against the biology of nature. Curr Opin Crit Care. 2007;13(2):134–142. doi: 10.1097/MCC.0b013e3280895d5c. [DOI] [PubMed] [Google Scholar]
- 128.Balabanov R, Goldman H, Murphy S, Pellizon G, Owen C, Rafols J, Dore-Duffy P. Endothelial cell activation following moderate traumatic brain injury. Neurol Res. 2001;23(2–3):175–182. doi: 10.1179/016164101101198514. [DOI] [PubMed] [Google Scholar]
- 129.Carlos TM, Clark RS, Franicola-Higgins D, Schiding JK, Kochanek PM. Expression of endothelial adhesion molecules and recruitment of neutrophils after traumatic brain injury in rats. J Leukoc Biol. 1997;61(3):279–285. doi: 10.1002/jlb.61.3.279. [DOI] [PubMed] [Google Scholar]
- 130.Whalen MJ, Carlos TM, Dixon CE, Schiding JK, Clark RS, Baum E, Yan HQ, Marion DW, Kochanek PM. Effect of traumatic brain injury in mice deficient in intercellular adhesion molecule-1: assessment of histopathologic and functional outcome. J Neurotrauma. 1999;16(4):299–309. doi: 10.1089/neu.1999.16.299. [DOI] [PubMed] [Google Scholar]
- 131.Pleines UE, Stover JF, Kossmann T, Trentz O, Morganti-Kossmann MC. Soluble ICAM-1 in CSF coincides with the extent of cerebral damage in patients with severe traumatic brain injury. J Neurotrauma. 1998;15(6):399–409. doi: 10.1089/neu.1998.15.399. [DOI] [PubMed] [Google Scholar]
- 132.Pleines UE, Morganti-Kossmann MC, Rancan M, Joller H, Trentz O, Kossmann T. S-100 beta reflects the extent of injury and outcome, whereas neuronal specific enolase is a better indicator of neuroinflammation in patients with severe traumatic brain injury. J Neurotrauma. 2001;18(5):491–498. doi: 10.1089/089771501300227297. [DOI] [PubMed] [Google Scholar]
- 133.Whalen MJ, Carlos TM, Kochanek PM, Wisniewski SR, Bell MJ, Carcillo JA, Clark RS, DeKosky ST, Adelson PD. Soluble adhesion molecules in CSF are increased in children with severe head injury. J Neurotrauma. 1998;15(10):777–787. doi: 10.1089/neu.1998.15.777. [DOI] [PubMed] [Google Scholar]
- 134.Lin Z, Han M, Li H, Luo H, Zhang Y, Luo W. Soluble vascular adhesion protein-1: decreased activity in the plasma of trauma victims and predictive marker for severity of traumatic brain injury. Clin Chim Acta. 2011;412(17–18):1678–1682. doi: 10.1016/j.cca.2011.05.031. [DOI] [PubMed] [Google Scholar]
- 135.Engelhardt B, Ransohoff RM. The ins and outs of T-lymphocyte trafficking to the CNS: anatomical sites and molecular mechanisms. Trends Immunol. 2005;26(9):485–495. doi: 10.1016/j.it.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 136.Licastro F, Chiappelli M, Caldarera CM, Porcellini E, Carbone I, Caruso C, Lio D, Corder EH. Sharing pathogenetic mechanisms between acute myocardial infarction and Alzheimer’s disease as shown by partially overlapping of gene variant profiles. J Alzheimers Dis. 2011;23(3):421–431. doi: 10.3233/JAD-2010-090871. [DOI] [PubMed] [Google Scholar]
- 137.Casoli T, Di Stefano G, Balietti M, Solazzi M, Giorgetti B, Fattoretti P. Peripheral inflammatory biomarkers of Alzheimer’s disease: the role of platelets. Biogerontology. 2010;11(5):627–633. doi: 10.1007/s10522-010-9281-8. [DOI] [PubMed] [Google Scholar]
- 138.Ewers M, Mielke MM, Hampel H. Blood-based biomarkers of microvascular pathology in Alzheimer’s disease. Exp Gerontol. 2010;45(1):75–79. doi: 10.1016/j.exger.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kozubski W, Swiderek M, Kloszewska I, Gwozdzinski K, Watala C. Blood platelet membrane fluidity and the exposition of membrane protein receptors in Alzheimer disease (AD) patients--preliminary Study. Alzheimer Dis Assoc Disord. 2002;16(1):52–54. doi: 10.1097/00002093-200201000-00009. [DOI] [PubMed] [Google Scholar]
- 140.Gamble JR, Skinner MP, Berndt MC, Vadas MA. Prevention of activated neutrophil adhesion to endothelium by soluble adhesion protein GMP140. Science. 1990;249(4967):414–417. doi: 10.1126/science.1696029. [DOI] [PubMed] [Google Scholar]
- 141.Rentzos M, Michalopoulou M, Nikolaou C, Cambouri C, Rombos A, Dimitrakopoulos A, Kapaki E, Vassilopoulos D. Serum levels of soluble intercellular adhesion molecule-1 and soluble endothelial leukocyte adhesion molecule-1 in Alzheimer’s disease. J Geriatr Psychiatry Neurol. 2004;17(4):225–231. doi: 10.1177/0891988704269822. [DOI] [PubMed] [Google Scholar]
- 142.Nielsen HM, Londos E, Minthon L, Janciauskiene SM. Soluble adhesion molecules and angiotensin-converting enzyme in dementia. Neurobiol Dis. 2007;26(1):27–35. doi: 10.1016/j.nbd.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 143.Rentzos M, Michalopoulou M, Nikolaou C, Cambouri C, Rombos A, Dimitrakopoulos A, Vassilopoulos D. The role of soluble intercellular adhesion molecules in neurodegenerative disorders. J Neurol Sci. 2005;228(2):129–135. doi: 10.1016/j.jns.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 144.Eikelenboom P, Zhan SS, Kamphorst W, van der Valk P, Rozemuller JM. Cellular and substrate adhesion molecules (integrins) and their ligands in cerebral amyloid plaques in Alzheimer’s disease. Virchows Arch. 1994;424(4):421–427. doi: 10.1007/BF00190565. [DOI] [PubMed] [Google Scholar]
- 145.Zuliani G, Cavalieri M, Galvani M, Passaro A, Munari MR, Bosi C, Zurlo A, Fellin R. Markers of endothelial dysfunction in older subjects with late onset Alzheimer’s disease or vascular dementia. J Neurol Sci. 2008;272(1–2):164–170. doi: 10.1016/j.jns.2008.05.020. [DOI] [PubMed] [Google Scholar]
- 146.Sobel RA, Mitchell ME, Fondren G. Intercellular adhesion molecule-1 (ICAM-1) in cellular immune reactions in the human central nervous system. Am J Pathol. 1990;136(6):1309–1316. [PMC free article] [PubMed] [Google Scholar]
- 147.Washington R, Burton J, Todd RF, 3rd, Newman W, Dragovic L, Dore-Duffy P. Expression of immunologically relevant endothelial cell activation antigens on isolated central nervous system microvessels from patients with multiple sclerosis. Ann Neurol. 1994;35(1):89–97. doi: 10.1002/ana.410350114. [DOI] [PubMed] [Google Scholar]
- 148.Jha MK, Jeon S, Suk K. Glia as a Link between Neuroinflammation and Neuropathic Pain. Immune Netw. 2012;12(2):41–47. doi: 10.4110/in.2012.12.2.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Doerck S, Gobel K, Weise G, Schneider-Hohendorf T, Reinhardt M, Hauff P, Schwab N, Linker R, Maurer M, Meuth SG, Wiendl H. Temporal pattern of ICAM-I mediated regulatory T cell recruitment to sites of inflammation in adoptive transfer model of multiple sclerosis. PLoS One. 2010;5(11):e15478. doi: 10.1371/journal.pone.0015478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bill R, Doring A, Deutsch U, Engelhardt B. PSGL-1 is dispensible for the development of active experimental autoimmune encephalomyelitis in SJL/J mice. J Neuroimmunol. 2011;232(1–2):207–208. doi: 10.1016/j.jneuroim.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 151.Engelhardt B, Vestweber D, Hallmann R, Schulz M. E- and P-selectin are not involved in the recruitment of inflammatory cells across the blood-brain barrier in experimental autoimmune encephalomyelitis. Blood. 1997;90(11):4459–4472. [PubMed] [Google Scholar]
- 152.Doring A, Wild M, Vestweber D, Deutsch U, Engelhardt B. E- and P-selectin are not required for the development of experimental autoimmune encephalomyelitis in C57BL/6 and SJL mice. J Immunol. 2007;179(12):8470–8479. doi: 10.4049/jimmunol.179.12.8470. [DOI] [PubMed] [Google Scholar]
- 153.Kerfoot SM, Norman MU, Lapointe BM, Bonder CS, Zbytnuik L, Kubes P. Reevaluation of P-selectin and alpha 4 integrin as targets for the treatment of experimental autoimmune encephalomyelitis. J Immunol. 2006;176(10):6225–6234. doi: 10.4049/jimmunol.176.10.6225. [DOI] [PubMed] [Google Scholar]
- 154.Osmers I, Bullard DC, Barnum SR. PSGL-1 is not required for development of experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;166(1–2):193–196. doi: 10.1016/j.jneuroim.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 155.Crunkhorn S. Trial watch: integrin antagonist shows promise in MS. Nat Rev Drug Discov. 2012;11(3):178. doi: 10.1038/nrd3676. [DOI] [PubMed] [Google Scholar]
- 156.Piraino PS, Yednock TA, Freedman SB, Messersmith EK, Pleiss MA, Vandevert C, Thorsett ED, Karlik SJ. Prolonged reversal of chronic experimental allergic encephalomyelitis using a small molecule inhibitor of alpha4 integrin. J Neuroimmunol. 2002;131(1–2):147–159. doi: 10.1016/s0165-5728(02)00273-4. [DOI] [PubMed] [Google Scholar]
- 157.Gan Y, Liu R, Wu W, Bomprezzi R, Shi FD. Antibody to alpha4 integrin suppresses natural killer cells infiltration in central nervous system in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2012;247(1–2):9–15. doi: 10.1016/j.jneuroim.2012.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Engelhardt B, Kappos L. Natalizumab: targeting alpha4-integrins in multiple sclerosis. Neurodegener Dis. 2008;5(1):16–22. doi: 10.1159/000109933. [DOI] [PubMed] [Google Scholar]
- 159.Airas L, Mikkola J, Vainio JM, Elovaara I, Smith DJ. Elevated serum soluble vascular adhesion protein-1 (VAP-1) in patients with active relapsing remitting multiple sclerosis. J Neuroimmunol. 2006;177(1–2):132–135. doi: 10.1016/j.jneuroim.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 160.Jalkanen S, Salmi M. VAP-1 and CD73, endothelial cell surface enzymes in leukocyte extravasation. Arterioscler Thromb Vasc Biol. 2008;28(1):18–26. doi: 10.1161/ATVBAHA.107.153130. [DOI] [PubMed] [Google Scholar]
- 161.Salmi M, Jalkanen S. Ectoenzymes controlling leukocyte traffic. Eur J Immunol. 2012;42(2):284–292. doi: 10.1002/eji.201142223. [DOI] [PubMed] [Google Scholar]
- 162.Tohka S, Laukkanen M, Jalkanen S, Salmi M. Vascular adhesion protein 1 (VAP-1) functions as a molecular brake during granulocyte rolling and mediates recruitment in vivo. FASEB J. 2001;15(2):373–382. doi: 10.1096/fj.00-0240com. [DOI] [PubMed] [Google Scholar]