

Abstract
Plasma cells and the autoreactive Abs they produce are suspected to contribute to the pathogenesis of multiple sclerosis, but recent attempts to target these components of humoral immunity have failed. MEDI551, an anti-CD19 Ab that depletes mature B cells including plasma cells may offer a compelling alternative that reduces pathogenic adaptive immune responses while sparing regulatory mechanisms. Indeed, our data demonstrate that a single dose of MEDI551, given before or during ongoing experimental autoimmune encephalomyelitis, disrupts development of the disease. Leukocyte infiltration into the spinal cord is significantly reduced, as well as short-lived and long-lived autoreactive CD138+ plasma cells in the spleen and bone marrow, respectively. In addition, potentially protective CD1dhiCD5+ regulatory B cells show resistance to depletion, and myelin-specific Foxp3+ regulatory T cells are expanded. Taken together, these results demonstrate that MEDI551 disrupts experimental autoimmune encephalomyelitis by inhibiting multiple proinflammatory components whereas preserving regulatory populations.
Multiple sclerosis (MS) is an autoimmune, demyelinating disease of the CNS that affects millions of people worldwide (1). Certainly the mechanism of this disease requires T cell involvement, but a pathological role for B cells and Abs in this devastating disease was recently established and substantiated by both human and animal studies in which B cell depletion using anti-CD20 mAbs demonstrated efficacy (2–8). However, pan CD20 depletion spares CD202 plasma cells, which contribute to immunopathology largely through generation of autoreactive Abs (9). In addition, anti-CD20 therapies potentially abolish regulatory mechanisms of B cells (10). Atacicept was proposed to demonstrate benefit by targeting Ab-secreting cells (ASCs) through sequestration of B cell survival factors BAFF and APRIL, but instead it led to a counterintuitive worsening of MS in clinical trials (11, 12). Several explanations have been described, all of which include sparing of memory B cells and targeting regulatory B cells (Bregs) (4, 11, 13). In this context, it is apparent that not all B cell targeting biologics will offer benefit in MS, and it leaves the community with an urgent need to find a safe way to deplete a broad spectrum of pathogenic B cell subsets including plasma cells without adversely affecting potential regulatory mechanisms in MS patients.
One such promising candidate is MEDI551, an affinity-optimized and humanized mAb originated from the mouse HB12 anti-human CD19 mAb (14, 15), that depletes mature B cells including plasmablasts and plasma cells with the added benefit of reducing serum Ab levels as shown in our study. In addition, a single dose of MEDI551 is sufficient to render these effects because this anti-CD19 biologic has an enhanced Ab-dependent, cellular cytotoxicity-mediated, B cell–depleting activity compared with rituximab (15).
Because MEDI551 is currently being tested in a phase I trial in relapsing-remitting MS (ClinicalTrials.gov: NCT01585766), our goal for this study was to investigate the mechanism of action of MEDI551 in a B cell–dependent model of experimental autoimmune encephalomyelitis (EAE). We found that a single dose of MEDI551 given before or after EAE induction preferentially inhibits leukocyte infiltration into the spinal cord and disrupts EAE development. Specifically, MEDI551 efficiently depletes both short-lived and long-lived plasma cells, which results in a significant reduction of total and Ag-specific Abs in both the periphery and the CNS. Interestingly, Ag-specific regulatory T cells (Tregs) were promoted by MEDI551 treatment, and potentially protective CD1dhiCD5+ Bregs showed resistance to MEDI551 depletion. These results indicate that MEDI551 effectively disrupts EAE by reducing adaptive immune responses known to participate in the disease pathogenesis while sparing regulatory mechanisms shown to suppress the disease.
Materials and Methods
Mice
Human CD19 transgenic (hCD19Tg) male mice, 6–8 wk of age, were used for MEDI551 (anti-human CD19)-mediated B cell depletion (14). Age-matched C57BL/6 male mice were purchased from Jackson Laboratories (Bar Harbor, ME). Animal protocols were approved by the Institutional Animal Care and Research Advisory Committee (University of Texas Southwestern Medical Center).
Induction of EAE
Recombinant human myelin oligodendrocyte glycoprotein (rhMOG) 1–125 (rhMOG1–125) was generated as previously described (5). EAE was induced by s.c. immunization at four sites on the back with 100 mg rhMOG emulsified in CFA containing 5 mg/ml mycobacteria (BD Biosciences, San Diego, CA). On days 0 and 2, mice were injected i.p. with 300 ng pertussis toxin (List Biological Laboratories). Clinical disease was assessed as follows: 0, no disease; 1, loss of tail tone; 2, weakness of hind limbs; 3, partial hind-limb paralysis; 4, total hind-limb paralysis with or without front-limb paralysis; 5, moribund or death.
Mouse B cell depletion
The anti-human CD19 Ab MEDI551 and a control Ab (16C4-TM) were produced at MedImmune. 16C4-TM is a variant Ab of MEDI551, which lacks the ability to elicit B cell depletion via Ab-dependent cellular cytotoxicity because of mutations at its FcgR binding site. B cells were depleted with a single dose of 250 mg MEDI551 via i.p. injection at indicated dates before or after EAE induction. Depletion was confirmed by staining circulating murine CD19+ B cells in peripheral blood taken 5–10 d after Ab administration.
Flow cytometry
Mice were perfused via the left ventricle with cold PBS supplemented with 10 U/ml heparin (Fisher Scientific, Pittsburgh, PA). Brains, spinal cords, draining lymph nodes (LNs; axillary, brachial, and inguinal), and spleens were harvested from the perfused animals. Bone marrow and peritoneal cells were isolated as described elsewhere (16). Tissues were pressed through a 70-mm cell strainer into RPMI 1640 (Corning, NY). Splenocytes were treated with RBC lysing buffer (Sigma-Aldrich, St. Louis, MO). Brain or spinal cord cells were pooled per two mice from each experimental group and processed as described. In brief, brain or spinal cord cells were pelleted by centrifugation at 390 3 g for 10 min at 4° C. The supernatants were collected and stored for the measurement of cytokine and Ab levels. The cell pellets were resuspended in the 30% Percoll, overlaid onto the 70% Percoll, and centrifuged at 390 3 g for 20 min at room temperature with brake-off. The interphase cells were collected, washed with RPMI 1640, resuspended in 1 ml EAE culture medium (RPMI 1640 supplemented with 10% FBS, L-glutamine, penicillin, streptomycin, HEPES buffer, nonessential amino acids, sodium pyruvate, and 2-ME), and counted. For FACS staining with 10-color survey panel, the following anti-mouse mAbs were used: anti-CD45 (clone 30-F11; Bio-legend, San Diego, CA), anti-CD3e (145-2C11), anti-TCRb (H57-597), anti-CD4 (RM4-5), anti-B220 (RA3-6B2), anti-CD19 (1D3), anti-CD11b (ICRF44), anti-Gr1 (RB6-8C5), and anti-NK1.1 (PK136). The LIVE/DEAD fixable yellow dead cell stain kit (Life Technologies, Grand Island, NY) was used to differentiate viable cells from dead cells. Other fluorescence-labeled mouse Abs used to define B cell subtypes were anti-IgD (11-26C), anti-CD138 (281-2), anti-CD1d (1B1), and anti-CD5 (OX-19). All the FACS Abs were purchased from eBioscience (San Diego, CA) or BD Biosciences (San Jose, CA) unless indicated. Cells for FACS staining were washed with PBS and resuspended in PBS at 1 3 106/ml. A total of 1 ml LIVE/DEAD fixable yellow dead cell dye per 1 million cells was added to the cells and incubated at 4 °C for 30 min in the dark. Cells were washed once with 1 ml PBS and resuspended in 200 ml FACS buffer (1% BSA, 1 mM EDTA, and 0.02% sodium azide in PBS) and blocked with anti-CD16/32 (clone 2.4G2; BD Biosciences) for 10 min at 4 °C before staining with a mixture of staining Abs for 30 min at 4° C. Cells were washed, resuspended in FACS buffer, and fixed in 1% paraformaldehyde.
For analyzing CD19 and CD20 expression on CD138+ plasma cells, splenocytes from EAE mice at the peak of disease were stained with a human CD19 Ab (BV421 mouse anti-human CD19, clone HIB19; BD Biosciences), an unconjugated mouse CD20 Ab (clone MB20-11, isotype mIgG2a; MedImmune) (14, 17), and other flow Abs to define plasma cells (IgD2CD32CD138+B2202) and IgD+ B cells (IgD+mCD19+B220+). A secondary Ab (PE anti-mouse IgG2a; Biolegend) was used to detect mCD20 Ab. Fluorescence Minus One controls were included for proper gating.
In some experiments, cells were stimulated for 4 h with 13 cell stimulation mixture (81 nM PMA and 1.34 mM Ionomycin; eBioscience) in the presence of 3 mg/ml brefeldin A (eBioscience) and subjected to intracellular staining of cytokines as described later. Cell events were acquired on a FACSAria or FACSCanto (BD Biosciences) and further analyzed using FlowJo software (Tree Star, Ashland, OR).
Detection of total IgG and anti-MOG IgG
Serum was obtained from mice at peak of the disease (days 14–16) or at dates indicated. Brain and spinal cord supernatants were collected while isolating single cells for FACS analysis as described earlier. For detecting total IgG or MOG-specific IgG, 96-Immulon 2HB plates (Thermo Scientific, Waltham, MA) were coated with unlabeled goat anti-mouse Ig (Southern Biotech, Birmingham, AL) or rhMOG at 10 mg/ml and then blocked with 1% BSA (Sigma). Diluted serum or supernatants were added to the wells and incubated at room temperature for 2 h. Plate-bound total or MOG-specific IgG Abs were detected with horseradish peroxide–conjugated anti-mouse IgG (1:5000; SouthernBiotech, Birmingham, AL). For detecting total IgM, a horseradish peroxide–conjugated goat anti-mouse IgM (1:5000; SouthernBiotech) was used as a secondary Ab in the ELISA. The total Ig titers were quantified using the commercially available mouse IgG (Thermo Scientific) or mouse IgM (Santa Cruz, Dallas, TX). Signal was developed using 3,39,5,59-tetramethylbenzidine solution (eBioscience), and the reaction was stopped with 1 M HCL. The plates were read at 450-nm wavelength on an Epic Plate Reader (BioTek, Winooski, VT).
Cytokine analysis
For cytokine quantification of IFN-g, IL-17A, IL-1b, IL-6, TNF-a, and IL-10, serum, brain, or spinal cord supernatants, and in vitro culture supernatants were analyzed using the mouse cytokine TH17 6-plex immunoassay (Bio-Rad, Hercules, CA) according to the manufacturer’s instruction. Data were acquired on a Luminex Magpix system (Millipore, Billerica, MA) at the Microarray core (University of Texas Southwestern Medical Center).
Cell culture and intracellular cytokine staining
Draining LN cells (axillary, brachial, and inguinal) were incubated in 0.2 mM CFSE for 8 min at room temperature (Life Technologies). CFSE-labeled cells were washed and cultured in medium only, with rhMOG, or with OVA protein (Sigma, St. Louis, MO) at 10 or 30 mg/ml for 96 h. Culture supernatants were collected for cytokine measurement, and cells were restimulated with 13 cell stimulation mixture (eBioscience) in the presence of 3 mg/ml brefeldin A (eBioscience) for 4 h. Surface Ags were stained with anti-TCRb, anti-CD4, and anti-CD19, and followed by the intracellular staining. For the detection of Foxp3 and intracellular cytokines, cells were treated with Foxp3 Fixation/Permeabilization buffer (eBioscience) and stained with anti-Foxp3 (NRRF-30; eBioscience) and anti–IL-17A (TC11-18H10.1; Biolegend) as an Ab pair or anti–IFN-g (XMG1.2; BD Biosciences) and anti–IL-17A as a pair in the FACS buffer.
ELISPOT assay for detecting ASCs
ELISPOT was performed to assess ASCs. In brief, 96-well Immobilon-P MultiScreen plates (Millipore) were coated with 30 mg/ml rhMOG1–125 to detect MOG-specific ASCs. Plates were coated with goat anti-mouse IgG or IgM (R&D Systems, Minneapolis, MN) to detect total IgG or IgM ASCs. Cell suspensions from spleens and bone marrows were added to individual wells at different dilutions (4 3 103 to 4 3 105 cells/well). Cells were incubated for 48 h at 37° C in a 5% CO2 atmosphere. After incubation, plates were washed several times with 0.05% Tween in PBS, incubated with mouse IgG or IgM detection Abs (R&D Systems) overnight at 4° C. The plates were finally developed using ELISPOT Blue Color Module (R&D Systems). ASCs were enumerated under microscope.
Statistical analyses
Statistical analyses were performed with GraphPad Prism (GraphPad Software, La Jolla, CA). Data were presented as mean 6 SEM. The p values were calculated by two-tailed unpaired Student t test, and significance was defined as *p, 0.05, **p, 0.01, ***p, 0.001, and ****p, 0.0001.
Results
EAE response is similar in hCD19Tg and wild-type mice
Susceptibility to rhMOG1–125 (rhMOG)–induced EAE (rhMOG-EAE) was assessed in male hCD19Tg mice (14) in comparison with age and sex-matched wild-type (WT) mice. EAE symptoms appeared on day10 postimmunization for both groups. After onset, EAE progressed very rapidly, and within 4–5 d both groups reached the peak of the disease (Fig. 1A). hCD19Tg and WT mice were statistically indistinguishable by the peak severity (peak of the disease at day 15 with maximal severity at 4 for both groups; p . 0.9999) or mean cumulative disease score (hCD19Tg 34 6 4.3 versus WT 33.5 6 6.3; p = 0.7965). Thus, the expression of human CD19 on mouse B cells has no significant effect on the course of the disease in our rhMOG-EAE model.
FIGURE 1.
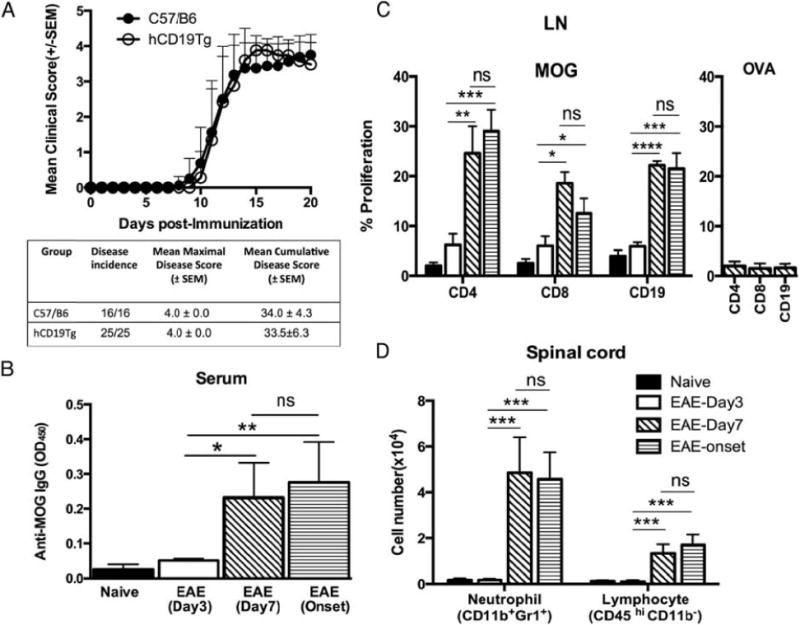
Robust MOG-specific B and T cell reactivity in the periphery and leukocyte infiltration into the CNS at day 7 after EAE induction. (A) WT and hCD19Tg mice were immunized with rhMOG on day 0 and scored daily thereafter for EAE onset and severity. Filled circles indicate the WT group (n = 16); open circles represent the hCD19Tg mice group (n = 25). Three independent experiments were done and data from two experiments are shown. (B–D) Cells and serum were collected from hCD19Tg mice at different time points (day 3, day 7, and disease onset at days 10–11) after disease induction. Data from four mice per group are shown and are representative of two independent experiments. (B) Serum MOG-specific IgG levels in naive and immunized mice were determined by ELISA. (C) Cells from LNs of naive or immunized mice were stimulated in vitro with 30 mg/ml rhMOG (left panel) or OVA, an unrelated protein (right panel), for 4 d. Proliferation of each cell subset as indicated was determined by CFSE dilution. For OVA stimulation, only the data from EAE day 7 mice were shown. (D) Mononuclear cells isolated from spinal cords were analyzed using our in-house developed 10-color survey panel. The numbers of infiltrated neutrophils (CD11b+Gr1+) and lymphocytes (CD45 hi CD11b2) were presented. *p, 0.05, **p, 0.01, ***p, 0.001, ****p, 0.0001.
Cellular autoreactivity and CNS cell infiltration are robust before EAE clinical signs
To define the time point at which cellular autoreactivity is emerging in our EAE model, we analyzed the activation of immune components at different time points (day 3, day 7, and disease onset at days 10–11) during the early phase of EAE development after disease induction. We found that at day 7 postimmunization, hCD19Tg mice developed significantly higher levels of anti-rhMOG IgG than did naive transgenic mice, indicating that autoreactive B cells had differentiated to autoantibody-secreting plasma cells at this early time point in disease (Fig. 1B). Similarly, B cells and T cells in the draining LNs at day 7 post-immunization showed a significant MOG-specific recall response when compared with naive mice, suggesting a robust B cell and T cell activation in the periphery at this early time point of disease (Fig. 1C). In addition, infiltration of CD11b+Gr1+ neutrophils and CD45hi CD11b2 lymphocytes into the spinal cord was present at day 7 postimmunization (Fig. 1D and gating strategy shown in Supplemental Fig. 1). Importantly, mice at day 7 postimmunization, before the appearance of clinical signs, had similar levels of B cell and T cell activity in the periphery and comparable cellular infiltration into the spinal cord compared with mice analyzed at EAE onset (days 10–11).
MEDI551 d7 treatment during development of EAE suppresses disease severity and duration
Although MEDI551 treatment before EAE induction prevents disease development (Supplemental Fig. 2), we chose to focus on MEDI551 treatment at day 7 postinduction because autoreactive plasma cell differentiation was emerging at this time point. In contrast with the control Ab-treated group, in which 100% of the mice developed severe EAE with mean maximal score of 4.0 6 0.1, we found that the majority of mice in the MEDI551-treated group (13/15) experienced development of EAE, with a same mean onset at day 11, but the disease was much less severe with mean maximal score of 2.3 6 0.4 (Fig. 2). In fact, mice treated with a single dose of MEDI551 showed a continuous decline of EAE severity with almost full recovery by day 23 as evidenced by decreased incidence (control Ab 100% versus MEDI551 66.7%) and decreased severity (control Ab 2.94 6 0.24 versus MEDI551 0.56 6 0.13, p, 0.0001).
FIGURE 2.
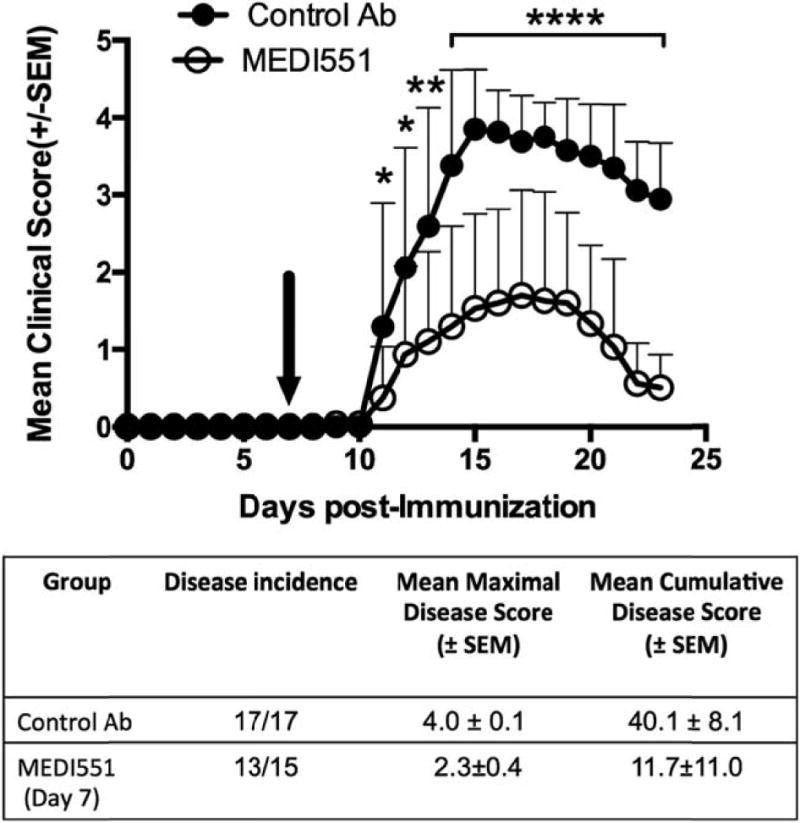
MEDI551 day 7 treatment suppresses EAE progression. hCD19Tg mice received a single dose of 250 mg MEDI551 or control Ab i.p. at day 7 after EAE induction. Filled circles indicate the control Ab-treated group (n = 17); open circles represent the MEDI551-treated group (n = 15). Black arrows indicate the day of Ab treatment. Three independent experiments were done and data from two experiments are shown. *p, 0.1, **p, 0.01, ****p, 0.0001.
Alleviation of EAE by MEDI551 is dependent on reduction of inflammatory T cell subsets in the spinal cord, but not in the brain
Because immune cell infiltration of the CNS is considered a hallmark of EAE, we investigated the effect of MEDI551 on leukocyte dynamics in the CNS. Mononuclear cells were isolated from brain and spinal cords separately of animals at peak of the disease and analyzed by multiparameter flow cytometry (Fig. 3). CD45+ leukocytes including neutrophils (CD11b+Gr1+), activated monocytes/microglial cells (CD11b+Gr12), and lymphocytes (CD45hi CD11b2) were major populations of immune cells identified in the brain and spinal cords of animals with EAE. A small population of NK cells (NK1.1+ CD3e2), which are proposed to play a regulatory role in EAE (18), was also identified in both CNS compartments (gating strategy shown in Supplemental Fig. 1).
FIGURE 3.
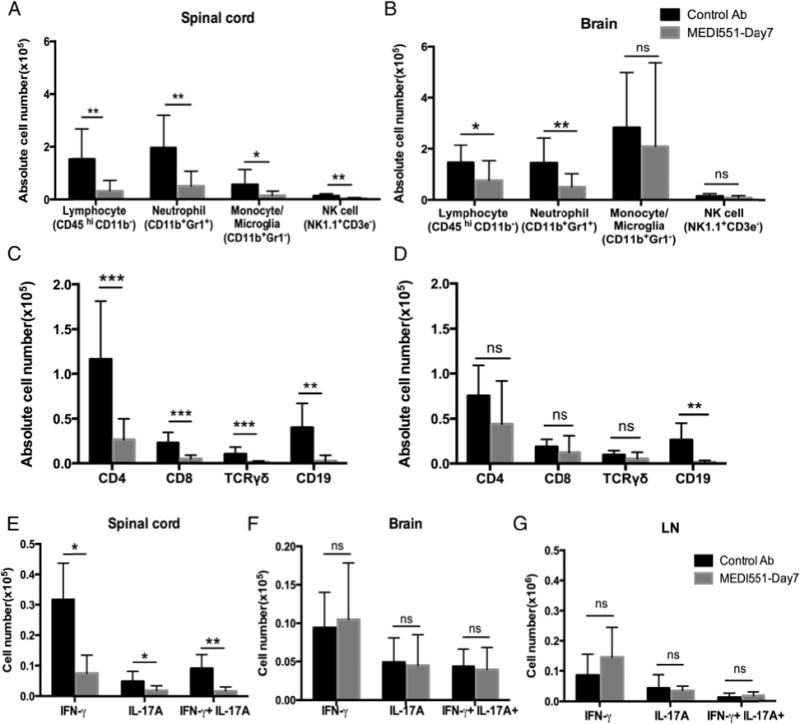
MEDI551 day 7 treatment suppresses immune cell infiltration and decreases cytokine-producing encephalitogenic T cell frequency in the spinal cord. At peak of the disease (days 14–16), brain and spinal cord mononuclear cells or bulk draining LNs from mice treated with MEDI551 or control Ab on day 7 were isolated for FACS analysis to identify leukocyte subsets or cytokine-producing T cells. (A–D) CNS cells were analyzed using our 10-color survey panel. The frequency of each major leukocyte subset (A and B) and lymphocyte subset (C and D) was presented for spinal cord (A and C) and brain (B and D). (E–G) CNS or LN cells were stimulated with PMA/ionomycin mixture before intracellular staining of cytokines followed by flow cytometry analysis. Data shown are the absolute cell count of each cytokine-producing T subset. Error bars indicate SEM. Data from eight mice per group are shown. Data are representative of three independent experiments. *p, 0.05, **p, 0.01, ***p, 0.001.
MEDI551 treatment at day 7 led to a significant reduction in the numbers of all three major CD45+ leukocyte populations, as well as NK cells in the spinal cord (Fig. 3A). Lymphocytes and neutrophils were also reduced in the brain, but not monocytes/microglia or NK cells (Fig. 3B). Specific to CD45hi CD11b2 lymphocytes in the spinal cord, a significant decrease in CD4, CD8, gd T cells, and CD19+ B cells was observed in MEDI551-treated mice (Fig. 3C). However, infiltration of T cell subsets in the brain was not significantly affected by the treatment (Fig. 3D).
Previous studies suggest that infiltration of cytokine-producing encephalitogenic T cells in the CNS contributes to CNS tissue damage in EAE (19). Thus, we examined the influence of MEDI551 treatment on proinflammatory T cell responses in the CNS. Despite our observation that frequencies of IFN-g–producing (Th1), IL-17A–producing (Th17), and IFN-g/IL-17A double-positive T cells (20) in the CD4+ T cell pool did not change in either the peripheral or CNS compartments (Supplemental Fig. 4A–C), absolute numbers of all three cytokine producing T cell subsets were reduced in spinal cords of postinduction treated animals (Fig. 3E). In the brain and LN, the number of each cytokine-producing T cell subset was comparable in MEDI551-treated mice and control mice (Fig. 3F, 3G). Overall, MEDI551 day 7 treatment preferentially inhibits leukocyte infiltration into the spinal cord rather than into the brain.
MEDI551 d7 treatment eliminates long-lived autoreactive plasma cells and reduces MOG-specific Abs in the CNS
A major benefit of anti-CD19 immunotherapy in general for treating B cell–dependent diseases is targeting Ab-secreting plasma cells. We examined the presence of CD138+ plasma cells at peak of the disease and found that the frequency and number of CD138+ short-lived plasma cells in the spleen and long-lived plasma cells in the bone marrow were significantly reduced after MEDI551 treatment (Fig. 4A). ELISPOT assays further confirmed that the frequency of MOG-specific IgM and IgG ASCs were significantly reduced with MEDI551 treatment in the spleen and the bone marrow (Fig. 4B). Furthermore, MEDI551-treated mice showed a significant reduction in serum and CNS tissue levels of total IgG and MOG-specific IgG, just 7–9 d after MEDI551 treatment (Fig. 4C, 4D). In addition, serum Ab levels were significantly reduced as early as 7 d with MEDI551 administration in naive hCD19Tg mice (Fig. 4E). Overall, these results suggest that both short-lived splenic and long-lived bone marrow ASCs were effectively targeted by MEDI551 in mice with ongoing EAE.
FIGURE 4.
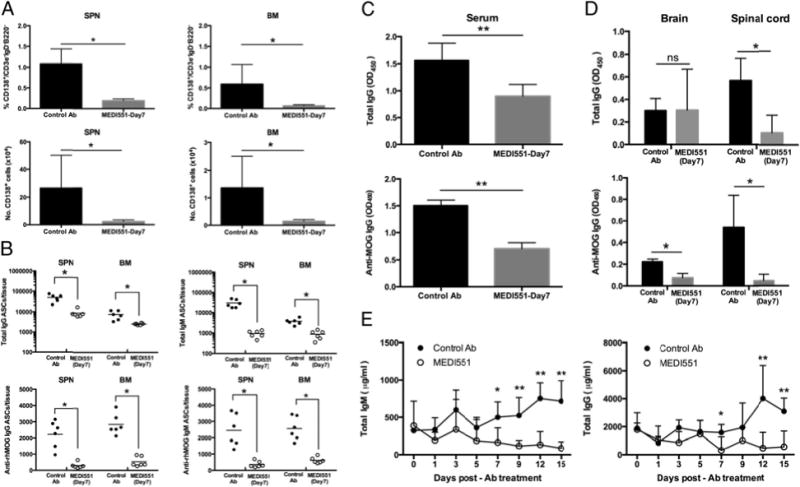
MEDI551 day 7 treatment depletes plasma cells and decreases total IgG and MOG-specific IgG levels in various tissues. hCD19Tg mice were immunized with rhMOG on day 0 and treated with 250 mg MEDI551 or control Ab on day 7. Tissues were harvested at peak of the disease and subjected to different assays. (A) FACS analysis was performed on isolated cells. Frequency and cell numbers of CD138+ plasma cells in the spleen and the bone marrow from MEDI551 day 7–treated and control Ab-treated mice were shown. After live and singlet gating, cells were gated on IgD and CD3e to identify IgD2CD3e2 populations. IgD2CD3e2 cells were then gated on B220 to exclude B220+ cells because some pre-B cells in the bone marrow are B220+CD138+. From the B2202 population, CD138+ plasma cells were identified as CD138hi CD19low. (B) Cells from the spleen and the bone marrow were collected, and the numbers of total IgG/IgM and MOG-specific IgG/M ASCs were determined by ELISPOT. Horizontal bar represents mean number of indicated ASCs per tissue. Serum (C), brain, and spinal cord supernatants (D) were harvested at peak of the disease (days 14–16), and total IgG and MOG-specific IgG levels were determined by ELISA. Ab titers were quantified as relative OD450 for total IgG or MOG-specific IgGs. Significant differences between the means of the two groups are indicated. Data from four to six mice per group were shown and are representative of two independent experiments. (E) MEDI551 reduces existing serum Ig levels in hCD19Tg mice. Naive hCD19Tg mice were i.p. injected with a single dose of 250 mg MEDI551 on day 0. Blood were collected every 2 d, and ELISAs detecting total IgM and total IgG were performed. Data from five mice per group were shown. *p, 0.05, **p, 0.01.
CD1dhigh CD5+ B cells are resistant to MEDI551-mediated B cell depletion
Although MEDI551 showed potent B cell depletion activity in various tissues including the CNS compartments (Supplemental Fig. 3), we observed that the percentage of CD1dhigh CD5+ Bregs was increased in the spleen (data not shown) and in the LNs of the MEDI551-treated group compared with the control Ab-treated group (7.80 6 2.64% to 1.62 6 0.18%; p = 0.0043; Fig. 5A, 5B). The number of Bregs was significantly reduced after MEDI551 treatment, as were all B cell subsets, but the reduction rate for the Breg subset was lower (72% of reduction) than the non-Bregs (98% of reduction; Fig. 5C). In addition, the Breg/non-Breg ratio in this compartment was significantly increased in the MEDI551 cohort (compare control Ab ratio 1:61 with MEDI551 ratio 1:12; p = 0.0043; Fig. 5D). Taken together, these results suggested that CD1dhigh CD5+ B cells are more resistant to MEDI551-mediated B cell depletion.
FIGURE 5.
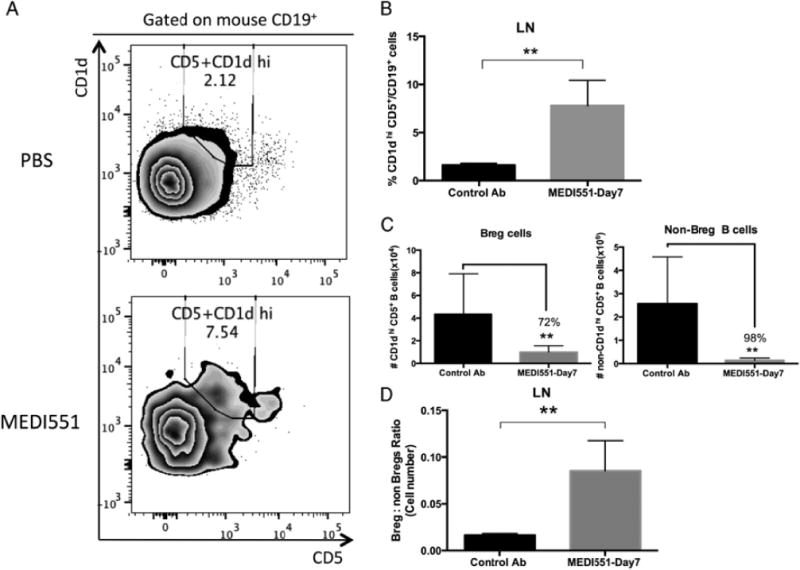
CD1dhigh CD5+ Bregs are resistant to MEDI551 depletion. LN cells were isolated at peak of the disease (days 14–16) from MEDI551 or control Ab (250 mg)-treated mice and assessed by immunofluorescence staining followed by flow cytometry analysis. (A) Representative plot of FACS analysis of the CD1dhigh CD5+ Bregs in the LN. Numbers represent the relative frequencies of CD1dhigh CD5+ Bregs within mouse CD19+ gates. (B) Bar graphs indicate mean (6 SEM) percentages of CD1dhigh CD5+ cells in mouse CD19+ gates as shown in (A). (C) Bar graphs indicate mean (6 SEM) absolute cell number of CD1dhigh CD5+ cells in mouse CD19+ gates as shown in (A) and (B). (D) Ratio of cell numbers of Bregs and non-Breg B cells (other CD19+ B cells) in the LNs were bar-graphed in the MEDI551-treated compared with control Ab-treated mice. Data from five to six mice per group are shown and are representative of two independent experiments. **p, 0.01.
MEDI551 d7 treatment inhibits Ag-specific Th17 responses but induces Ag-specific Treg expansion in the periphery
To investigate whether the deficiency of CNS-infiltrating leukocytes in MEDI551-treated mice was due to a T cell priming defect in the periphery, we analyzed functional responses of MOG-pecific CD4+ and CD8+ T cells from draining LNs (gating strategy provided in Fig. 6A, 6E). MEDI551 day 7 treatment suppressed the proliferation of MOG-specific CD4+ and CD8+ T cells upon in vitro stimulation (Fig. 6A, 6B). IL-17A was significantly suppressed in the culture supernatants of rhMOG-responsive lymphocytes from draining LNs of MEDI551-treated mice compared with controls (Fig. 6D), whereas IFN-g in the culture supernatants was not significantly affected by MEDI551 treatment (Fig. 6C), despite the lower frequency of MOG-specific Th1 cells (Fig. 6E, 6F). IL-6, IL-10, TNF-a, and IL-1b in the culture supernatant were also detected, but at much lower levels compared with IFN-g and IL-17A (Supplemental Fig. 4G). We also found MOG-specific proliferation by both Th1 and Th17 cells identified by intracellular staining of IFN-g or IL-17A was significantly suppressed in the MEDI551-treated group compared with control Ab-treated mice (Fig. 6E, 6F), but the frequency of rhMOG-responsive Foxp3+ Tregs was significantly increased from the control group (control Ab 9.78 6 2.32% versus MEDI551 17.88 6 7.06, p = 0.0142).
FIGURE 6.
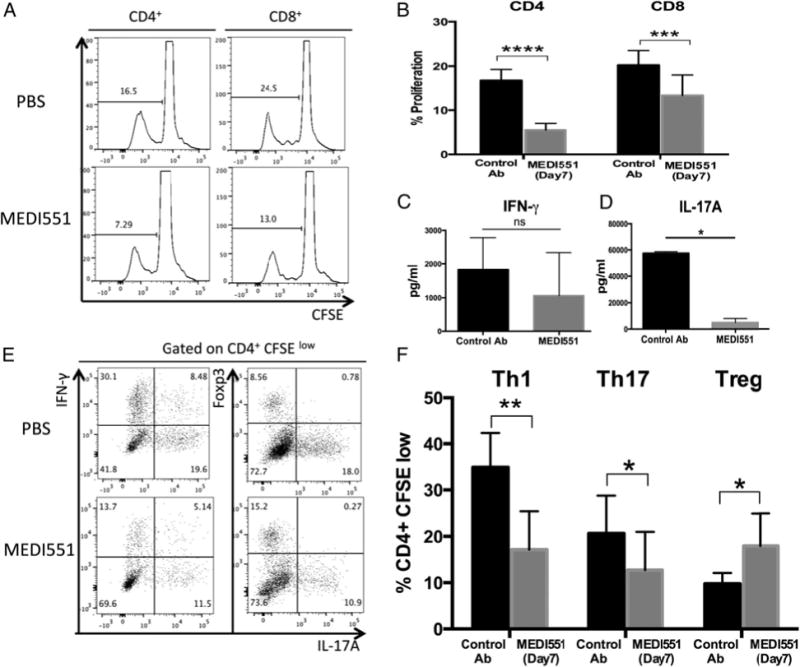
MEDI551 day 7 treatment inhibits the induction of MOG-specific Th17 and Th1 cells but promotes expansion of MOG-specific Foxp3+ Tregs. Bulk draining LN cells harvested at peak of the disease (days 14–16) were CFSE labeled and stimulated with 30 mg/ml rhMOG in culture. Ninety-six hours later, MOG-specific T cells were analyzed by intracellular staining and flow cytometry analysis, and supernatants were harvested for cytokine measurements. (A) Representative histograms and (B) bar graphs show the proliferation of rhMOG-responsive CD4 and CD8 T cells determined by CFSE dilution. (C and D) Quantification of IFN-g and IL-17A secretion in the culture supernatants by Bioplex. (E) CD4+CFSElow populations were further gated based on the expression of Foxp3, IFN-g, and IL-17A. (F) Bar graphs show the summarized data of frequencies of MOG-specific Th1 (IFN-g+) cells, Th17 (IL-17A+) cells, and Tregs (Foxp3+) in the CD4+CFSElow populations. Error bars indicate SEM. Data from six mice per group are shown and are representative of three independent experiments. *p, 0.05, **p, 0.01, ***p, 0.001, ****p, 0.0001.
Discussion
Targeting plasma cells and the autoreactive Abs they produce is of critical importance in MS because dysregulated humoral immunity likely contributes to the pathology of the disease. MEDI551 is designed to target plasmablasts and plasma cells, and thus reduce autoantibody levels. However, it was unclear whether this biologic would provide a favorable risk/benefit ratio considering more recent findings that not all B cell targeting therapies demonstrate efficacy (9, 11). This study evaluates the efficacy of MEDI551 in a B cell–dependent EAE model.
In our model, rhMOG immunization induced a classical EAE with ascending paralysis and immune cell infiltration in the CNS (21). Th1 (IFN-g–producing) and Th17 (IL-17–producing) cells are involved in the development of EAE (22, 23), and indeed, the MEDI551-treated EAE mice had a decrease of infiltrated Th1, Th17, and IFN-g/IL-17A double-positive CD4+ T cells (20) in the spinal cord (Fig. 3E). Because pathogenic T cells in the CNS are the driving force for recruitment of other inflammatory cells, we predicted that MEDI551 treatment would thus also result in diminished recruitment of other inflammatory cells, including activated macrophages into the CNS (24). Indeed, besides a paucity of T lymphocytes, we observed an overall decrease of myeloid cells, including neutrophils and macrophages in the spinal cord of MEDI551-treated mice, which otherwise would mediate demyelination and axonal damage (Fig. 3A, 3C) (1). In contrast with the spinal cord, where infiltration of all major leukocyte subsets including pathogenic T cells was inhibited by MEDI551 treatment, the brain showed only a significant decrease of neutrophils and total lymphocytes, with little effect on specific lymphocyte subsets or cytokine-producing T cells (Fig. 3B, 3D, 3F). These data suggest that the brain and spinal cord are differentially affected by MEDI551-mediated B cell depletion, and inhibition of inflammation in the spinal cord, but not the brain, is sufficient to prevent disease.
We hypothesized that the reduced infiltration of inflammatory cytokine-producing T cells in the spinal cord after MEDI551 day 7 treatment is due to a defect in the activation and expansion of pathogenic T cells in the periphery. In recall assays, the proliferation of MOG-specific CD4+ T cells was inhibited, and significantly lower frequencies of Th1 and Th17 cells were identified in the rhMOG-responding CD4+ T cell pool from mice treated with MEDI551 (Fig. 6). A possible mechanism for the inhibitory effect of MEDI551 on Ag-specific Th1 and Th17 responses may be related to the induction of MOG-specific Foxp3+ Tregs (25) found in this study (Fig. 6E, 6F). Previous studies demonstrated that stimulation of Ag-specific Tregs reverses IL-17 action via the production of IL-10 and other regulatory cytokines, and enhances resolution of autoimmune diseases including EAE (19, 26). In the recall supernatants, we detected more than a 10-fold decrease of IL-17A from MEDI551-treated mice, which corresponds with the detection of lower frequency of Th17 cells (Fig. 6D, 6E). However, other Th17-related cytokines, including IL-10, showed only a slightly lower level in MEDI551-treated mice compared with the control Ab-treated mice (Supplemental Fig. 4G). In addition, the Th17-related cytokines in the serum and the CNS supernatants showed a similar level in MEDI551-treated mice compared with the control Ab-treated mice (Supplemental Fig. 4D–F).
T cell differentiation into T effector and inhibitory subsets is regulated by multiple mechanisms, including the cytokine environment present at the time of Ag recognition (24, 27). Recent studies show that IL-6 promotes Th17 immunity both in vitro and in vivo, by inhibiting the conversion of conventional T cells into Foxp3+ Tregs (28–30). B cells are a major source of IL-6, and ablation of IL-6–producing B cells suppresses EAE (30, 31). Thus, it is possible that MEDI551 promotes MOG-specific Treg responses via depleting IL-6–secreting pathogenic B cells. The expanded MOG-specific Tregs in MEDI551-treated mice could inhibit Th1 and Th17 differentiation, as well as MOG-specific CD4 and CD8 T cell proliferation in the periphery (Fig. 6) (32–34). Studies are under way to determine whether the potent inhibitory effect of MEDI551 on Th17 responses in the periphery in our B cell–dependent EAE model is mediated by depletion of IL-6– secreting B cells, which allows for induction of Ag-specific Tregs. In addition, previous studies showed that B cell deficiency achieved either genetically (mMT) or by depletion with anti-CD20 resulted in a reduction of Treg frequency in the CNS (35, 36). In our study, we confirmed that this anti-CD19 Ab also depletes this population in the CNS (data not shown). However, the contribution of this undesired effect on CNS-residing Tregs to the overall recovery of MEDI551-treated mice still needs further investigation.
We were surprised to observe that CD1dhi CD5+ B cells, which contain mostly Bregs (16, 37), were resistant to depletion by MEDI551 in the periphery (Fig. 5). CD1dhi CD5+ B cells in the spinal cord also showed the trend of increased resistance to MEDI551 depletion as well, but this observation is complicated by the very low frequency of Bregs in this compartment even without B cell depletion. Decreased susceptibility of CD1dhi CD5+ B cells to MEDI551 depletion is not due to decreased CD19 expression because these Bregs express similar levels of CD19 on the surface as marginal zone B cells and even higher levels of CD19 than follicular and transitional B cells (data not shown) (16). The increased resistance of Bregs to depletion could be because of their increased activation state, as has been suggested by others using anti-CD20 depletion (38, 39). Other factors like physical niche or an increase in circulating survival factors after B cell depletion could also contribute to the depletion resistance of Bregs in our model (38) and others (40). Furthermore, reconstituted B cell populations postrituximab treatment in a cohort of patients with rheumatoid arthritis exhibited a specific expansion of the Breg population, suggesting a possibly inefficient depletion of this population in the first place (41). In addition, it is possible that the residual Bregs resistant to the depletion may also contribute to the shift of Th17 to T regulatory responses (32, 33). Given the im portance of Bregs in suppressing multiple autoimmune diseases, as well as the potential role in B cell depletion therapy indicated by this study, further characterization of this population of B cells in the context of B cell depletion and disease suppression is under way.
The impact of MEDI551 on Tregs and Bregs is of potential interest, but another important advantage of MEDI551 compared with anti-CD20 therapies is depletion of autoreactive Ab-producing plasma cells (42, 43). CD20+ B cell depletion reduces splenic ASCs but has no effect on bone marrow ASC numbers or established Ag-specific Ab responses unless multiple treatments of anti-CD20 are given (44). In contrast with anti-CD20 therapies, our data indicate that a single dose of MEDI551 given at the early stage of EAE significantly depletes long-lived plasma cells including the Ag-specific ones in the bone marrow, which are the major source of persistent Ag-specific Ab titers (Fig. 4A, 4B). The observation of significantly reduced serum Ig levels shortly after MEDI551 treatment in naive hCD19Tg mice further supports the notion that long-lived plasma cells are directly targeted by MEDI551 (Fig. 4E). Thus, MEDI551 has access to the bone marrow and effectively targets autoreactive plasma cells despite their lower level of CD19 expression in comparison with other stages of B cell development resulting in lower serum and CNS Ab levels (Fig. 4C, 4D). Higher frequency of plasma cells expressing CD19 than CD20 in our EAE model (CD191CD201, 31.5%; CD191CD202, 41.7%; CD192CD201, 1.79%) further indicates that anti-CD19 Ab likely more efficiently targets plasma cells than anti-CD20 Ab (Supplemental Fig. 1D). In addition, depletion of marginal zone, germinal center, and memory B cells may also contribute to the decreased CD138+ population by preventing the formation of new plasmablasts and autoreactive Ab levels (44). Despite a low frequency of B cells in the brain and spinal cord, we detected relatively high levels of total and MOG-specific IgG in the CNS compartments after rhMOG immunization (Fig. 4D). In EAE models such as ours, which are induced by recombinant myelin proteins, Ag-specific Abs are essential for EAE pathogenesis (5, 45, 46). MEDI551 treatment led to a significant reduction of MOG-specific IgGs in the spinal cord and, to a lesser degree, in the brain (Fig. 4D). In fact, a small fraction of mice that had high frequencies of ASCs after MEDI551 treatment did not recover as did their counterparts whose ASCs were almost completely depleted (data not shown). Thus, our data support the notion that one of the primary mechanisms by which MEDI551 treatment suppresses EAE is by depleting humoral immune components involved in EAE pathogenesis in the bone marrow and spinal cord.
In summary, our studies demonstrate that MEDI551, a CD19-directed B cell depletion therapy, effectively disrupts EAE pathogenesis in a B cell–dependent EAE model. The protective effect of MEDI551 treatment in EAE is likely mediated by inhibiting pathogenic properties while sparing regulatory mechanisms of B cells including: 1) depletion of plasma cells in the bone marrow; 2) reduction of autoantibodies in the spinal cord; 3) inhibition of Ag-specific Th17 responses; 4) preservation of Bregs; and 5) promotion of Ag-specific Treg responses. Together, these findings support further development of MEDI551 in MS treatment as a means to extend the efficacy of B cell–depleting biologics to plasma cell depletion and reduction of autoantibodies.
Supplementary Material
Supplemental Figure 1. Gating strategies used in this study. A–C) 10-color survey panel to identify leukocyte subsets in the CNS. At peak of the disease, brain and spinal cord mononuclear cells were isolated from PBS perfused animals and subjected to FACS analysis. Data shown is the FACS analysis of MNC isolated from the spinal cord of control Ab-treated mice. A) Cells were first gated on FSC-A and SSC-A to identify mononuclear cells (MNC). After singlet gating, cells were gated on yellow viability dye to define viable cells. B) Neutrophils (CD11b+Gr1+), activated monocyte/microglia (CD11b+Gr1−), NK cell (NK1.1+CD3e−) and lymphocytes (CD45hiCD11b−) were identified in the viable cell population. C) Lymphocytes were further subdivided based on the expression of CD3e, TCRβ, CD4, B220 and mouse CD19. T cells were identified as CD3e+ and further divided to αβT cells (TCRβ+) and γδ T cells (TCRβ−). CD4 and CD8 T cells were identified in αβT cell population based on the presence or absence of CD4 expression. CD19+ B cells were identified in the CD3e− fraction. Of note is that the population negative for CD19 but positive for B220 which may represent plasmacytoid dendritic cells (pDC) was excluded from the B cell gate. D) Analysis of CD19 and CD20 expression on CD138+ plasma cells. Splenocytes from EAE mice at the peak of disease were stained with antibodies to detect human CD19 and mouse CD20 expression on CD138+ plasma cells (IgD−CD3−CD138+B220−) and IgD+ B cells (IgD+mCD19+B220+). Data are representative of two independent experiments.
Supplemental Figure 3. B cell depletion by MEDI551 in vivo. hCD19Tg mice immunized with rhMOG1–125 were i.p. injected with 250μg MEDI551 or Control Ab on day 7 post-immunization. A) Blood were taken at indicated time points and subjected to FACS analysis for monitoring circulating CD19+ B cells (n=8). B) At peak of the disease (day 14–16), cells from bone marrow, spleen, lymph node, peritoneal cavity, brain and spinal cord were harvested for FACS analysis. Cells were stained with anti-CD45, anti-B220 and anti-mouse CD19, and then examined by flow cytometry. Upper panel: Numbers indicate the percentage of CD19+ B cells in CD45+ lymphocyte gates. Bottom panel: Bar graphs indicate the mean (±SEM) percentage of CD19+ B cells within CD45+ lymphocyte gates (n=4). Data are representative of three independent experiments.
Supplemental Figure 4. Detection of cytokine-secreting T cells and Th17-related cytokines in MEDI551 day7 treated mice. Mice were sacrificed at peak of the disease and subjected to various assays. A–C) Frequency of each cytokine-producing T cell subset in CD4+ T populations from the spinal cord (A), brain (B) and LN (C). (D–G) Cytokines including IFN-γ, IL-17A, IL-1β, TNF-α, IL-10 and IL-6 were quantified by Bioplex. D) Spinal cord supernatants; E) brain supernatants; F) serum; G) Culture supernatants of LN cells in the recall assay shown in Figure 6. Data from five to six mice per group are shown. Data are representative of three independent experiments.
Acknowledgments
We thank Dr. Erik Plautz and Sherry Rovinsky in the Animal Facility of the Department of Neurology and Neurotherapeutics (University of Texas Southwestern Medical Center) for technical assistance. We also thank Dr. Sean Morrison and his team in the Moody Foundation Flow Cytometry Facility at Children’s Research Institute for use of instruments (University of Texas Southwestern Medical Center).
Abbreviations
- ADCC
antibody-dependent cellular cytotoxicity
- Breg cells
regulatory B cells
- EAE
experimental autoimmune encephalomyelitis
- hCD19Tg
human CD19 transgenic
- LN
lymph node
- MOG
myelin oligodendrocyte glycoprotein
- MS
Multiple Sclerosis
- rhMOG
recombinant human myelin oligodendrocyte glycoprotein
- Treg cells
regulatory T cells
- WT
wild-type
Footnotes
This work was supported by a sponsored research agreement with MedImmune Inc. (Gaithersburg, MD).
Disclosures
N.L.M. receives grant funding from the National Multiple Sclerosis Society, Diogenix, Inc., MedImmune LLC, and TEVA Neuroscience. The other authors have no financial conflicts of interest.
References
- 1.Frohman EM, Racke MK, Raine CS. Multiple sclerosis—the plaque and its pathogenesis. N Engl J Med. 2006;354:942–955. doi: 10.1056/NEJMra052130. [DOI] [PubMed] [Google Scholar]
- 2.Ireland S, Monson N. Potential impact of B cells on T cell function in multiple sclerosis. Mult Scler Int. 2011;2011:423971. doi: 10.1155/2011/423971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weber MS, Hemmer B, Cepok S. The role of antibodies in multiple sclerosis. Biochim Biophys Acta. 2011;1812:239–245. doi: 10.1016/j.bbadis.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 4.Krumbholz M, Derfuss T, Hohlfeld R, Meinl E. B cells and anti-bodies in multiple sclerosis pathogenesis and therapy. Nat Rev Neurol. 2012;8:613–623. doi: 10.1038/nrneurol.2012.203. [DOI] [PubMed] [Google Scholar]
- 5.Lyons JA, Ramsbottom MJ, Cross AH. Critical role of antigen-specific antibody in experimental autoimmune encephalomyelitis induced by recombinant myelin oligodendrocyte glycoprotein. Eur J Immunol. 2002;32:1905–1913. doi: 10.1002/1521-4141(200207)32:7<1905::AID-IMMU1905>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 6.Lyons JA, San M, Happ MP, Cross AH. B cells are critical to induction of experimental allergic encephalomyelitis by protein but not by a short encephalitogenic peptide. Eur J Immunol. 1999;29:3432–3439. doi: 10.1002/(SICI)1521-4141(199911)29:11<3432::AID-IMMU3432>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 7.Lassmann H, Brunner C, Bradl M, Linington C. Experimental allergic encephalomyelitis: the balance between encephalitogenic T lymphocytes and demyelinating antibodies determines size and structure of demyelinated lesions. Acta Neuropathol. 1988;75:566–576. doi: 10.1007/BF00686201. [DOI] [PubMed] [Google Scholar]
- 8.Linington C, Bradl M, Lassmann H, Brunner C, Vass K. Augmentation of demyelination in rat acute allergic encephalomyelitis by circulating mouse monoclonal antibodies directed against a myelin/oligodendrocyte glycoprotein. Am J Pathol. 1988;130:443–454. [PMC free article] [PubMed] [Google Scholar]
- 9.Lehmann-Horn K, Kronsbein HC, Weber MS. Targeting B cells in the treatment of multiple sclerosis: recent advances and remaining challenges. Ther Adv Neurol Disord. 2013;6:161–173. doi: 10.1177/1756285612474333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderton SM, Fillatreau S. Activated B cells in autoimmune diseases: the case for a regulatory role. Nat Clin Pract Rheumatol. 2008;4:657–666. doi: 10.1038/ncprheum0950. [DOI] [PubMed] [Google Scholar]
- 11.Hartung HP, Kieseier BC. Atacicept: targeting B cells in multiple sclerosis. Ther Adv Neurol Disord. 2010;3:205–216. doi: 10.1177/1756285610371146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kappos L, Hartung HP, Freedman MS, Boyko A, Rad€u EW, Mikol DD, Lamarine M, Hyvert Y, Freudensprung U, Plitz T, van Beek J, ATAMS Study Group Atacicept in multiple sclerosis (ATAMS): a randomised,placebo-controlled, double-blind, phase 2 trial. Lancet Neurol. 2014;13:353–363. 13. doi: 10.1016/S1474-4422(14)70028-6. [DOI] [PubMed] [Google Scholar]
- 13.Hoffmann F, Meinl E. B cells in multiple sclerosis: good or bad guys?: An article for 28 May 2014 – World MS Day 2014. Eur J Immunol. 2014;44:1247–1250. doi: 10.1002/eji.201470045. [DOI] [PubMed] [Google Scholar]
- 14.Yazawa N, Hamaguchi Y, Poe JC, Tedder TF. Immunotherapyusing unconjugated CD19 monoclonal antibodies in animal models for B lymphocyte malignancies and autoimmune disease. Proc Natl Acad Sci USA. 2005;102:15178–15183. doi: 10.1073/pnas.0505539102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herbst R, Wang Y, Gallagher S, Mittereder N, Kuta E, Damschroder M, Woods R, Rowe DC, Cheng L, Cook K, et al. B-cell depletion in vitro and in vivo with an afucosylated anti-CD19 antibody. J Pharmacol Exp Ther. 2010;335:213–222. doi: 10.1124/jpet.110.168062. [DOI] [PubMed] [Google Scholar]
- 16.Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest. 2008;118:3420–3430. doi: 10.1172/JCI36030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DiLillo DJ, Hamaguchi Y, Ueda Y, Yang K, Uchida J, Haas KM, Kelsoe G, Tedder TF. Maintenance of long-lived plasma cells and serological memory despite mature and memory B cell depletion during CD20 immunotherapy in mice. J Immunol. 2008;180:361–371. doi: 10.4049/jimmunol.180.1.361. [DOI] [PubMed] [Google Scholar]
- 18.Hao J, Liu R, Piao W, Zhou Q, Vollmer TL, Campagnolo DI, Xiang R, La Cava A, Van Kaer L, Shi FD. Central nervous system (CNS)- resident natural killer cells suppress Th17 responses and CNS autoimmune pathology. J Exp Med. 2010;207:1907–1921. doi: 10.1084/jem.20092749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maddur MS, Miossec P, Kaveri SV, Bayry J. Th17cells:biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. Am J Pathol. 2012;181:8–18. doi: 10.1016/j.ajpath.2012.03.044. [DOI] [PubMed] [Google Scholar]
- 20.Duhen R, Glatigny S, Arbelaez CA, Blair TC, Oukka M, Bettelli E. Cutting edge: the pathogenicity of IFN-g-producing Th17 cells is independent of T-bet. J Immunol. 2013;190:4478–4482. doi: 10.4049/jimmunol.1203172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rao P, Segal BM. Experimental autoimmune encephalomyelitis. Methods Mol Biol. 2012;900:363–380. doi: 10.1007/978-1-60761-720-4_18. [DOI] [PubMed] [Google Scholar]
- 22.Lovett-Racke AE, Yang Y, Racke MK. Th1 versus Th17: are T cell cytokines relevant in multiple sclerosis? Biochim Biophys Acta. 2011;1812:246–251. doi: 10.1016/j.bbadis.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM. Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat Med. 2008;14:337–342. doi: 10.1038/nm1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol. 2010;162:1–11. doi: 10.1111/j.1365-2249.2010.04143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. 2009;30:636–645. doi: 10.1016/j.immuni.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 26.Mills KH. Regulatory T cells: friend or foe in immunity to infection? Nat Rev Immunol. 2004;4:841–855. doi: 10.1038/nri1485. [DOI] [PubMed] [Google Scholar]
- 27.Jäger A, Kuchroo VK. Effector and regulatory T-cell subsets in autoimmunity and tissue inflammation. Scand J Immunol. 2010;72:173–184. doi: 10.1111/j.1365-3083.2010.02432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Serada S, Fujimoto M, Mihara M, Koike N, Ohsugi Y, Nomura S, Yoshida H, Nishikawa T, Terabe F, Ohkawara T, et al. IL-6 blockade inhibits the induction of myelin antigen-specific Th17 cells and Th1 cells in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2008;105:9041–9046. doi: 10.1073/pnas.0802218105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fujimoto M, Nakano M, Terabe F, Kawahata H, Ohkawara T, Han Y, Ripley B, Serada S, Nishikawa T, Kimura A, et al. The influence of excessive IL-6 production in vivo on the development and function of Foxp3+ regulatory T cells. J Immunol. 2011;186:32–40. doi: 10.4049/jimmunol.0903314. [DOI] [PubMed] [Google Scholar]
- 30.Barr TA, Shen P, Brown S, Lampropoulou V, Roch T, Lawrie S, Fan B, O’Connor RA, Anderton SM, Bar-Or A, et al. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. J Exp Med. 2012;209:1001–1010. doi: 10.1084/jem.20111675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Molnarfi N, Schulze-Topphoff U, Weber MS, Patarroyo JC, Prod’homme T, Varrin-Doyer M, Shetty A, Linington C, Slavin AJ, Hidalgo J, et al. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J Exp Med. 2013;210:2921–2937. doi: 10.1084/jem.20130699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Flores-Borja F, Bosma A, Ng D, Reddy V, Ehrenstein MR, Isenberg DA, Mauri C. CD19+CD24hiCD38hi B cells maintain regulatory T cells while limiting TH1 and TH17 differentiation. Sci Transl Med. 2013;5:173ra123. doi: 10.1126/scitranslmed.3005407. [DOI] [PubMed] [Google Scholar]
- 33.Carter NA, Vasconcellos R, Rosser EC, Tulone C, Muñoz-Suano A, Kamanaka M, Ehrenstein MR, Flavell RA, Mauri C. Mice lacking endogenous IL-10-producing regulatory B cells develop exacerbated disease and present with an increased frequency of Th1/Th17 but a decrease in regulatory T cells. J Immunol. 2011;186:5569–5579. doi: 10.4049/jimmunol.1100284. [DOI] [PubMed] [Google Scholar]
- 34.McNally A, Hill GR, Sparwasser T, Thomas R, Steptoe RJ. CD4+ CD25+ regulatory T cells control CD8+ T-cell effector differentiation by modulating IL-2 homeostasis. Proc Natl Acad Sci USA. 2011;108:7529–7534. doi: 10.1073/pnas.1103782108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ray A, Basu S, Williams CB, Salzman NH, Dittel BN. A novel IL-10-independent regulatory role for B cells in suppressing auto-immunity by maintenance of regulatory T cells via GITR ligand. J Immunol. 2012;188:3188–3198. doi: 10.4049/jimmunol.1103354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoehlig K, Shen P, Lampropoulou V, Roch T, Malissen B, O’Connor R, Ries S, Hilgenberg E, Anderton SM, Fillatreau S. Activation of CD4+ Foxp3+ regulatory T cells proceeds normally in the absence of B cells during EAE. Eur J Immunol. 2012;42:1164–1173. doi: 10.1002/eji.201142242. [DOI] [PubMed] [Google Scholar]
- 37.Watanabe R, Ishiura N, Nakashima H, Kuwano Y, Okochi H, Tamaki K, Sato S, Tedder TF, Fujimoto M. Regulatory B cells (B10 cells) have a suppressive role in murine lupus: CD19 and B10 cell deficiency exacerbatessystemic autoimmunity. J Immunol. 2010;184:4801–4809. doi: 10.4049/jimmunol.0902385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gong Q, Ou Q, Ye S, Lee WP, Cornelius J, Diehl L, Lin WY, Hu Z, Lu Y, Chen Y, et al. Importance of cellular microenvironment and circulatory dynamics in B cell immunotherapy. J Immunol. 2005;174:817–826. doi: 10.4049/jimmunol.174.2.817. [DOI] [PubMed] [Google Scholar]
- 39.Hamel K, Doodes P, Cao Y, Wang Y, Martinson J, Dunn R, Kehry MR, Farkas B, Finnegan A. Suppression of proteoglycan-induced arthritis by anti-CD20 B Cell depletion therapy is mediated by reduction in autoantibodies and CD4+ T cell reactivity. J Immunol. 2008;180:4994–5003. doi: 10.4049/jimmunol.180.7.4994. [DOI] [PubMed] [Google Scholar]
- 40.Yang M, Rui K, Wang S, Lu L. Regulatory B cells in autoimmune diseases. Cell Mol Immunol. 2013;10:122–132. doi: 10.1038/cmi.2012.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heidt S, Hester J, Shankar S, Friend PJ, Wood KJ. B cell repopulation after alemtuzumab induction-transient increase in transitional B cells and long-term dominance of naive B cells. Am J Transplant. 2012;12:1784–1792. doi: 10.1111/j.1600-6143.2012.04012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tedder TF. CD19: a promising B cell target for rheumatoid arthritis. Nat Rev Rheumatol. 2009;5:572–577. doi: 10.1038/nrrheum.2009.184. [DOI] [PubMed] [Google Scholar]
- 43.Mei HE, Schmidt S, Dörner T. Rationale of anti-CD19 immunotherapy: an option to target autoreactive plasma cells in autoimmunity. Arthritis Res Ther. 2012;14(Suppl. 5):S1. doi: 10.1186/ar3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang W, Rangel-Moreno J, Owen T, Barnard J, Nevarez S, Ichikawa HT, Anolik JH. Long-term B cell depletion in murine lupus eliminates autoantibody-secreting cells and is associated with alterations in the kidney plasma cell niche. J Immunol. 2014;192:3011–3020. doi: 10.4049/jimmunol.1302003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bansal P, Khan T, Bussmeyer U, Challa DK, Swiercz R, Velmurugan R, Ober RJ, Ward ES. The encephalitogenic, human myelin oligodendrocyte glycoprotein-induced antibody repertoire is directed toward multiple epitopes in C57BL/6-immunized mice. J Immunol. 2013;191:1091–1101. doi: 10.4049/jimmunol.1300019. [DOI] [PubMed] [Google Scholar]
- 46.Challa DK, Bussmeyer U, Khan T, Montoyo HP, Bansal P, Ober RJ, Ward ES. Autoantibody depletion ameliorates disease in murine experimental autoimmune encephalomyelitis. MAbs. 2013;5:655–659. doi: 10.4161/mabs.25439. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Gating strategies used in this study. A–C) 10-color survey panel to identify leukocyte subsets in the CNS. At peak of the disease, brain and spinal cord mononuclear cells were isolated from PBS perfused animals and subjected to FACS analysis. Data shown is the FACS analysis of MNC isolated from the spinal cord of control Ab-treated mice. A) Cells were first gated on FSC-A and SSC-A to identify mononuclear cells (MNC). After singlet gating, cells were gated on yellow viability dye to define viable cells. B) Neutrophils (CD11b+Gr1+), activated monocyte/microglia (CD11b+Gr1−), NK cell (NK1.1+CD3e−) and lymphocytes (CD45hiCD11b−) were identified in the viable cell population. C) Lymphocytes were further subdivided based on the expression of CD3e, TCRβ, CD4, B220 and mouse CD19. T cells were identified as CD3e+ and further divided to αβT cells (TCRβ+) and γδ T cells (TCRβ−). CD4 and CD8 T cells were identified in αβT cell population based on the presence or absence of CD4 expression. CD19+ B cells were identified in the CD3e− fraction. Of note is that the population negative for CD19 but positive for B220 which may represent plasmacytoid dendritic cells (pDC) was excluded from the B cell gate. D) Analysis of CD19 and CD20 expression on CD138+ plasma cells. Splenocytes from EAE mice at the peak of disease were stained with antibodies to detect human CD19 and mouse CD20 expression on CD138+ plasma cells (IgD−CD3−CD138+B220−) and IgD+ B cells (IgD+mCD19+B220+). Data are representative of two independent experiments.
Supplemental Figure 3. B cell depletion by MEDI551 in vivo. hCD19Tg mice immunized with rhMOG1–125 were i.p. injected with 250μg MEDI551 or Control Ab on day 7 post-immunization. A) Blood were taken at indicated time points and subjected to FACS analysis for monitoring circulating CD19+ B cells (n=8). B) At peak of the disease (day 14–16), cells from bone marrow, spleen, lymph node, peritoneal cavity, brain and spinal cord were harvested for FACS analysis. Cells were stained with anti-CD45, anti-B220 and anti-mouse CD19, and then examined by flow cytometry. Upper panel: Numbers indicate the percentage of CD19+ B cells in CD45+ lymphocyte gates. Bottom panel: Bar graphs indicate the mean (±SEM) percentage of CD19+ B cells within CD45+ lymphocyte gates (n=4). Data are representative of three independent experiments.
Supplemental Figure 4. Detection of cytokine-secreting T cells and Th17-related cytokines in MEDI551 day7 treated mice. Mice were sacrificed at peak of the disease and subjected to various assays. A–C) Frequency of each cytokine-producing T cell subset in CD4+ T populations from the spinal cord (A), brain (B) and LN (C). (D–G) Cytokines including IFN-γ, IL-17A, IL-1β, TNF-α, IL-10 and IL-6 were quantified by Bioplex. D) Spinal cord supernatants; E) brain supernatants; F) serum; G) Culture supernatants of LN cells in the recall assay shown in Figure 6. Data from five to six mice per group are shown. Data are representative of three independent experiments.