

Abstract
Mitochondrial DNA (mtDNA) copy number plays a key role in the pathophysiology of metabolic syndrome-related phenotypes, but its role in non-alcoholic fatty liver disease (NAFLD) is not well understood. We evaluated the molecular mechanisms that may be involved in the regulation of liver mtDNA content in a high-fat-induced rat model of NAFLD. In particular, we tested the hypothesis that liver mtDNA copy number is associated with liver expression of HIF-1α. Rats were given either standard chow diet (SCD, n= 10) or high-fat diet (HFD, n= 15) for 20 weeks. Subsequently, mtDNA quantification using nuclear DNA (nDNA) as a reference was carried out using real time quantitative PCR. HFD induced a significant increase in liver mtDNA/nDNA ratio, which significantly correlated with the liver triglyceride content (R: 0.29, P < 0.05). The liver mtDNA/nDNA ratio significantly correlated with the hepatic expression of HIF-1α mRNA (R: 0.37, P < 0.001); liver HIF-1α mRNA was significantly higher in the HFD group. In addition, liver cytochrome c oxidase subunit IV isoform 1 (COX4I1) mRNA expression was also positively correlated with liver mtDNA content. The hepatic expression of mRNA of transcriptional factors that regulate mitochondrial biogenesis, including peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α) and PGC-1β, nuclear respiratory factor-1 (NRF-1), peroxisome proliferator-activated receptor δ and Tfam, was not associated with the liver mtDNA content. Neither hepatocyte apoptosis nor oxidative stress was involved in the HIF-1α-mediated increase in mtDNA copy number. In conclusion, we found that HFD promotes an increase in liver mitochondrial biogenesis in response to hypoxia via HIF-1α, probably to enhance the mitochondrial function as well as to accommodate the metabolic load.
Keywords: liver, mitochondria, fatty liver, NAFLD, high-fat diet, HIF-1α, hypoxia, gene expression, 8-isoprostane, mitochondrial DNA content, type 1 angiotensin II receptor antagonist, COX4I1, PGC-1α, NRF-1, NTF-α, PPARδ
Introduction
Non-alcoholic fatty liver disease (NAFLD) is a highly prevalent disease that results from excessive fat accumulation in the liver. NAFLD not only affects the adult population (about 20–40% of adults in Western countries have fatty liver) [1], but is also the most common cause of paediatric liver disease [2].
NAFLD is now recognized as the hepatic manifestation of the metabolic syndrome (MS). In fact, there is a nearly universal association between MS comorbidities and NAFLD, because obesity, type 2 diabetes and hyperlipidaemia are known to co-exist in patients with fatty liver. NAFLD is also related to increased risk of cardiovascular diseases, such as carotid atherosclerosis [3]. Although many risk factors of NAFLD are well defined, its pathogenesis remains poorly understood. Recent evidence has implicated insulin resistance as a major contributor to the pathogenesis and disease progression of NAFLD [4]. In addition, NAFLD is associated with increased fatty acid β-oxidation, hepatic oxidative stress and mitochondrial structural defects [5]. For instance, crystalline inclusions are observed in hepatic mitochondria in non-alcoholic steatohepatitis (NASH), and their presence has been correlated either with an adaptive process or mitochondrial injury [6].
Mitochondrial function is critical in the physiopathology of NAFLD because liver mitochondria is a primary site for the oxidation of fatty acids and oxidative phosphorylation – a process that requires multiple enzymes that are encoded by both nuclear DNA (nDNA) and mitochondrial DNA (mtDNA).
Previous studies have revealed that qualitative and quantitative changes in skeletal muscle mtDNA are involved in the pathogenesis of type 2 diabetes [7]. Moreover, mtDNA content in peripheral blood leucocytes has been associated with insulin resistance in children and adult [8, 9]. These findings suggest that mitochondrial density and mtDNA copy number play a role in the mitochondrial dysfunction associated with MS-related phenotypes. However, its role in NAFLD is not well understood.
An interesting mechanism recently related to the pathogenesis of NAFLD is the association between mitochondrial dysfunction and hepatocyte sensitivity to hypoxic stress [10]. Although imbalance in oxygen homeostasis is a well-known mechanism in many pathophysiological processes, including cardiovascular and cerebrovascular disease, cancer, etc., its role in NAFLD is scarcely explored.
Hypoxia-inducible factor-1 (HIF-1) is a key transcription factor involved in cellular and systemic homeostatic response to hypoxia; HIF-1 is a heterodimeric protein complex comprising two subunits, among which, the redox-sensitive HIF-1α is considered to be the major regulator of O2-tension-sensitive genes in cells [11]. Interestingly, HIF-1α has been described as an important regulator of mitochondrial biogenesis in muscle [12], whereas sustained hypoxia in brain has been associated with an increase in neuronal mtDNA content [13].
In view of the evidence described earlier, we evaluated the molecular mechanisms that may be involved in the regulation of liver mtDNA content in a high-fat-induced rat model of NAFLD. In particular, we tested the hypothesis that liver mtDNA copy number is associated with liver expression of HIF-1α. We also examined liver mRNA abundance of master nuclear genes involved in the regulation of mitochondrial biogenesis and mtDNA transcription and replication. Furthermore, the occurrence of oxidative stress in the liver of high-fat solid diet (HFD)-induced NAFLD was also assessed. Finally, the effect of pharmacological reversion of hepatic steatosis on liver mtDNA content was evaluated. For the intervention experiment we chose telmisartan, an angiotensin II receptor blocker considered also to act as a partial agonist of PPARγ, as we and others previously demonstrated its efficacy in reducing hepatic lipid accumulation and reverting liver steatosis in a rat model of NAFLD [14, 15].
Materials and methods
Animals
Twelve-week-old male Sprague-Dawley rats weighing 280 ± 20 g were purchased from the Research Animal Facility of School of Veterinary Medicine, University of Buenos Aires. All the animals were housed individually with food and water freely available and were maintained at room temperature (23 + 1°C) under a 12 hr light/dark cycle.
All the animals received humane care, and the studies were conducted according to the regulations for the use and care of experimental animals.
After acclimatization for 1 week, the rats were randomly divided into two experimental groups. One group included 10 rats that received standard chow diet (SCD) for 20 weeks, and the quantity of the diet was restricted to that spontaneously consumed at the beginning of the experiment (control group, SCD). The other group, including 15 animals, was allowed ad libitum access to HFD (40% (w/w) bovine and porcine fat added to the standard chow, as previously described [16] for the same period of time. In all the animals, food intake and body weight were monitored weekly for the 20 week period.
At the completion of the study, the animals were anesthetized with pentobarbital and killed. Blood from individual rats was collected by cardiac puncture to determine the plasma and serum levels of different biochemical parameters. Food was withdrawn from 8:00 a.m. to 4:00 p.m., before the rats were killed, and the intraperitoneal and retroperitoneal fats were measured by weighting them directly.
Liver was quickly snap-frozen and stored at −76°C until gene expression analysis. A portion of each liver was fixed in 10% formalin for histological analysis. Two additional samples of liver tissue (150 mg) were stored at −80°C for quantifying liver lipids.
Biochemical measurements
Serum and sodium ethylenediaminetetraacetic acid plasma were obtained by centrifugation and stored at −80°C until needed. Fasting glucose and serum alanine aminotransferase (ALT) were measured by an automatic biochemical analytical system (Architect, Abbott, Buenos Aires, Argentina). The plasma insulin levels were determined using a commercial quantitative ultrasensitive ELISA rat kit according to the manufacturer’s instructions (CRYSTAL CHEM, INC., Downers Grove, IL, USA). Insulin resistance was calculated using the homeostasis model of assessment (HOMA) index (fasting plasma insulin [μU/ml]× fasting plasma glucose [mmol/l]/22.5).
Histological analysis of liver tissue
Formalin-fixed liver tissue was processed, and 5-μm-thick paraffin sections were stained with haematoxylin and eosin and Masson’s trichrome (for selective demonstration of collagen fibres and fibrin) and Gomori’s reticulin silver impregnation (for the evaluation of reticular fibres and collagen type III/IV) for histological analysis. In both experimental groups, osmium tetroxide staining was also carried out to estimate the degree of hepatic steatosis. The degree of steatosis was assessed irrespective of the experimental groups as previously described [17], based on the percentage of hepatocytes containing macrovesicular fat droplets: grade 0, no steatosis or steatosis <5%; grade 1, >5% to <33% of hepatocytes-containing macrovesicular fat droplets; grade 2, >33–66% of hepatocytes-containing macrovesicular fat droplets and grade 3, >66% of hepatocytes containing macrovesicular fat droplets.
Electron microscopy
Electron microscopy studies were performed on liver specimens by immersion in 3% glutaraldehyde buffered by cacodile buffer of pH 7.4, and then post-fixed in 1% osmium tetroxide and embedded in epoxy resin (Polibed®, Poly-Sciences, Warrington, PA, USA).
The samples were examined and photographed in an electron microscope (Zeiss EM 109, Zeiss, Oberkochen, Germany), and the sections were evaluated by an electron microscopist (NL).
Measurement of liver triglyceride content
The liver triglyceride content was determined using an automatic biochemical analytical system (Architect, Abbott), and the results were expressed as micrograms of triglyceride per milligram of liver tissue (μg/mg liver).
RNA preparation and Real-Time RT-PCR for quantitative assessment of mRNA expression
Total RNA was prepared from rat livers using phenol extraction step method, with an additional DNAse digestion. For RT-PCR, 3 μg of total RNA was reverse-transcribed using random hexamers and Moloney murine leukaemia virus (MMLV) reverse transcriptase (Promega, Madison, WI, USA). Real-time PCR was performed for quantitative assessment of mRNA expression in an iCycler thermocycler (BioRad, Hercules, CA, USA) using the fluorescent dye, SYBR-Green (Invitrogen, Buenos Aires, Argentina). All the real-time PCR reactions were run in triplicate and all the samples of the experimental groups were tested. The relative expression of target genes’ mRNA was normalized to the amount of a housekeeping gene (peptidylprolyl isomerase A, PPIA, also termed as cyclophilin A) to carry out comparisons between the groups. Cyclophilin was found to be the most stable reference gene for testing liver mRNA expression among other housekeeping genes tested before starting the experiment [β-actin, TATA box binding protein, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH)]. The levels of mRNA were expressed as the ratio of the estimated amount of the target gene relative to the PPIA mRNA levels using fluorescence threshold cycle values (Ct) calculated for each sample, and the estimated efficiency of the PCR for each product was expressed as the average of each sample efficiency value obtained [18].
The specificity of amplification and absence of primer dimers were confirmed using melting curve analysis at the end of each run. The primer sequences and the resulting PCR product lengths are shown Table 1.
Table 1.
Primers Used for the mRNA gene expression–PCR reaction and quantification of mtDNA
| Gene name | Primer sequence | Amplicon size (bp) |
|---|---|---|
| HIF1-α | Forward 5′-TGT GTG TGT GAA TTA TGT TG-3′ Reverse 5′-GTC CTC AGA TTC CAC TTT AG-3′ | 142 |
| PGC1α | Forward 5′-AAA AGC TTG ACT GGC GTC AT-3′ Reverse 5′-TCA GGA AGA TCT GGG CAA AG-3′ | 129 |
| Tfam | Forward 5′-GGC AGA AAC GCC TAA AGA AG-3′ Reverse 5′-TCA TCC TTA GCC TCC TGG AA-3′ | 127 |
| NRF-1 | Forward 5′-CGC AGT GAC GTC CGC ACA GA-3′ Reverse 5′-AAG GTC CTC CCG CCC ATG CT-3′ | |
| PPARδ | Forward 5′-AGG CCT CAG GCT TCC ACT AC-3′ Reverse 5′-TTG CGG TTC TTC TTC TGG AT-3′ | 130 |
| COX4I1 | Forward 5′-CTT GTC CTG ATC TGG GAG AA-3′ Reverse 5′-GAC CTT CAT GTC CAG CAT CC-3′ | 105 |
| PGC1b | Forward 5′-TTG ACA GTG GAG CTT TGT GG-3′ Reverse 5′-CTG TGC TTG GTG TCC TGC T-3′ | 101 |
| Bax | Forward 5′-CTG CAG AGG ATG ATT GCT GA-3′ Reverse 5′-GGG CAC TTT AGT GCA CAG G-3′ | 165 |
| Bcl2 | Forward 5′-GAT AAC GGA GGC TGG GAT G-3′ Reverse 5′-CTC ACT TGT GGC CCA GGT AT-3′ | 151 |
| Casp3 | Forward 5′-GGA CCT GTG GAC CTG AAA AA-3′ Reverse 5′-ATA CCG CAG TCC AGC TCT GT-3′ | 122 |
| Col1a1 | Forward 5′-GCG TGC TAT GCA AAG AAG ACT-3′ Reverse 5′-TGA CTT CTG CGT CTG GTG AT-3′ | 105 |
| Acta2 | Forward 5′-TCG GGA CCT CAC TGA CTA CC-3′ Reverse 5′-AAT CCA GGG CGA CAT AAC AC-3′ | 122 |
| PPIA | Forward 5′-AGC ACT GGG GAG AAA GGA TT-3′ Reverse 5′-CTT GCC ACC AGT GCC ATT AT-3′ | 111 |
| β-actin | Forward 5′-TTC CTG GGT ATG GAA TCC TG-3′ Reverse 5′-CAG CAA TGC CTG GGT ACA T-3′ | 136 |
| GAPDH | Forward 5′-CTG ACA TGC CGC CTG GAG AAA C-3′ Reverse 5′-CCA GCA TCA AAG GTG GAA GAA T-3′ | 161 |
| TBP | Forward 5′-TGG GAT TGT ACC ACA GCT CCA-3′ Reverse 5′-CTC ATG ATG ACT GCA GCA AAC C-3′ | 132 |
| The primer sequences for mtDNA and nDNA amplification | ||
| Rnr2 | Forward 5′-AGC TAT TAA TGG TTC GTT TGT-3′ Reverse 5-AGG AGG CTC CAT TTC TCT TGT-3′ | 132 bp |
| GAPDH | Forward 5′-GGA AAG ACA GGT GTT TTG CA-3′ Reverse 5-AGG TCA GAG TGA GCA GGA CA-3′ | 129 bp |
HIF-1α: hypoxia-inducible factor 1, α subunit (basic helix-loop-helix transcription factor); PGC1α: peroxisome proliferator-activated receptor gamma, coactivator 1α; Tfam: mitochondrial transcription factor A; COX4I1: cytochrome c oxidase subunit IV isoform 1; NRF-1: Nuclear respiratory factor 1; PPARδ: Peroxisome proliferator-activated receptor γ; PGC1β: peroxisome proliferator-activated receptor gamma, coactivator 1β; Col1a1: collagen, type I, α1; Acta2: smooth muscle α-actin; Bax: Bcl2-associated X protein; Bcl2: B-cellCLL/lymphoma 2; Casp3: caspase 3; PPIA: peptidylprolyl isomerase A (cyclophilin A) (relative quantification of gene expression); GAPDH: glyceraldehyde 3-phosphate dehydrogenase; TBP: TATA box binding protein; Rnr2: mitochondrially encoded 16S RNA.
Quantification of mtDNA
An assay based on real-time quantitative PCR was used for both nDNA and mtDNA quantification using SYBR-Green as the fluorescent dye (Invitrogen). For the detection of liver nDNA, we selected GAPDH between the nucleotides 1443 and 1571, and for the detection of mtDNA, we selected mitochondrially encoded 16S RNA (Rnr2) between the nucleotides 2451 and 2583 [19]. The results were presented as the mtDNA/nDNA ratio. The primer sequences for both mtDNA and nDNA for loading normalization are given in Table 1.
Triplicate amplifications of mitochondrial and nuclear products were performed separately.
Real-time quantitative PCR was carried out in a BioRad iCycler (Bio-Rad Laboratories, Hercules, CA, USA). The calculation of DNA copy number involved extrapolation from the fluorescence readings in the mode of background subtracted from the readings of BioRad iCycler according to the above-described procedure for gene expression. The specificity of amplification and absence of primer dimers were confirmed using the melting curve analysis at the end of each run.
The two target amplicon sequences (mtDNA and nDNA) were visualized in 2% agarose and purified using Qiagen Qiaex II, Gel extraction Kit (Tecnolab, Buenos Aires, Argentina). The dilutions of the purified amplicons were used as the standard curve to verify the PCR-efficiency values estimated by using the procedure described earlier.
Western blot analysis of liver HIF-1α protein
Nuclear proteins from liver tissue were denatured in SDS sample buffer, separated with 8% SDS-acrylamide gel and electrotransferred to Hybond-hydrophobic polyvinylidene difluoride (PVDF) membranes (GE Healthcare UK Limited, Buckinghamshire, UK). After blocking with 5% non-fat dry milk in tris-buffered saline tween (TBST) buffer (20 mmol/l of Tris-HCl [pH 7.6], 137 mmol/l of NaCl, 0.25% of Tween-20), the membranes were probed with rabbit polyclonal anti-HIF-1a (1:1.000) (GeneTex, Inc., GTX30603, Irvine, CA, USA), followed by incubation with horseradish peroxidase (HRP)-conjugated anti-rabbit polyclonal immunoglobulin G secondary antibody (1:15.000) (GeneTex, Inc., GTX26795). Equal protein loading was confirmed by reblotting of the membranes with a goat polyclonal antibody against rabbit polyclonal anti-β-actin (1:500) (GeneTex, Inc., GTX16039). Binding of the antibody was subsequently visualized with enhanced chemiluminescence reagent (GE Healthcare UK Limited), and the band images were detected and analysed using the Lab Works Analysis Software (Ultra-Violet Products Ltd., Cambridge, UK).
Measurement of liver 8-isoprostane
Measurement of 8-isoprostane (also known as 15-isoprostane F2t, 8-epi-PGF2α, or 8-iso-PGF2α) in liver tissue was carried out using ELISA by employing ethyl acetate extraction method, according to the manufacturer’s instruction (Detroit R&D, Inc., Detroit, MI, USA).
Statistical analysis
Quantitative data were expressed as mean ± S.E. Pairwise mean differences were evaluated using nonparametric Mann-Whitney test, as most of the variables were ratios and not normally distributed, and non-homogeneous variances between the groups were evident. For the comparison of more than two groups, we used the Kruskal–Wallis test. For testing the differences in steatosis gradation (as a categorical response variable), we used a model with ordinal multinomial distribution and probit as a link function, with animal length and adipose tissue as continuous predictor variables.
The correlation between two variables was determined using Spearman rank correlation test. Logistic and multiple regressions were used on log-transformed variables for testing multivariate association between variables or for adjusting the cofactors.
The data were also adjusted for body weight whenever applicable. A value of P < 0.05 was considered to be statistically significant. We used the Statistica program package, StatSoft (Tulsa, OK, USA), to perform all the analyses.
Experimental results
Effect of HFD on liver histology and organ fat accumulation
HFD-fed rats developed severe hepatic microvesicular and macrovesicular steatosis (Fig. 1). As shown in Figure 1G, quantitative evaluation of steatosis score from haematoxylin and eosin and osmium tetroxide staining of liver sections at the end of the experiment demonstrated significant differences between the groups (ancova with steatosis gradation as a categorical response variable with ordinal multinomial distribution and probit as a link function, with animal length and adipose tissue as continuous predictor variables). This finding was confirmed by biochemical analysis of hepatic triglyceride content (Fig. 1H).
Fig 1.
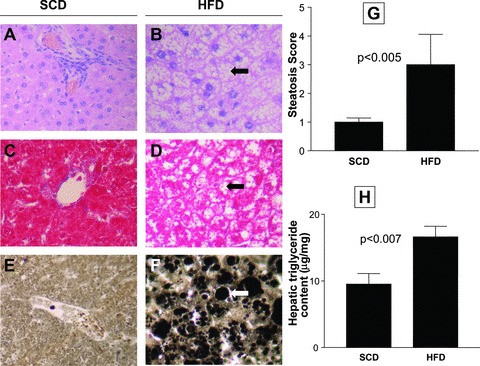
Left panel: Liver histology of a representative animal from each experimental group. Haematoxylin and eosin (A and B), Masson’s trichrome (C and D), and osmium tetroxide (E and F) staining of liver sections at the end of the experiment. The livers of rats fed with SCD show normal histology and absence of fat accumulation. The livers of rats fed with HFD show severe panlobular micro and macrovesicular steatosis. Arrows indicate large or small droplet steatosis, and lipid globules appear black after osmium tetroxide staining (F). Original magnification: 400×. (G) Quantitative evaluation of steatosis score. Steatosis was given a score from 0 to 3 as described in the ‘Materials and methods’ section. The data are presented as mean ± S.E. (H) Biochemical analysis of hepatic triglyceride content. The results (μg/mg liver) are expressed as mean ± S.E.
Furthermore, plasma glucose in the HFD group was not significantly different from that in the SCD group (199 ± 6 mg/dl and 186 ± 7 mg/dl, respectively). Similarly, the plasma insulin levels and HOMA index, despite being lower in the SCD group (96 ± 35 and 45 ± 19 μU/l, respectively), were not statistically different when compared with those observed in the HFD group (187 ± 30 and 96 ± 16 μU/l, respectively). Furthermore, ALT levels (UI/l) were similar in both groups (33 ± 3.2 in SCD rats and 39 ± 2.8 in HFD rats), and HFD rats showed higher visceral fat content at the end of the experiment (26.9 ± 4.5 g) when compared with that in the controls (12.5±5.2 g), P < 0.05.
Effect of HFD on liver mtDNA content and liver mitochondrial density
As shown in Figure 2A, we observed that HFD induced a significant increase in liver mtDNA content when compared with that in the SCD rats. In addition, the liver mtDNA/nDNA ratio was significantly correlated with the liver triglyceride content (R: 0.29, P < 0.05).
Fig 2.
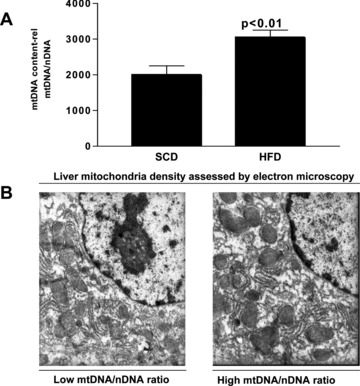
(A) Liver mtDNA/nDNA ratio assessed by quantitative real-time PCR in each experimental group. Each bar represents the mean ± S.E. values of 10 and 15 animals in SCD and HFD, respectively. mtDNA copy number was calculated as the ratio of GAPDH to 16S RNA gene. The results are shown as mtDNA/nDNA ratio. (B) Electron microscopic images depicting the difference in the abundance of the liver mitochondrial density with respect to the mtDNA content. The left-hand picture shows an exemplar of low mtDNA/nDNA ratio, and the right-hand picture shows an exemplar of high mtDNA/nDNA ratio. Original magnification: 12,000×.
Furthermore, electron microscopic evaluation of liver tissue showed that the observed increase in liver mtDNA copy number was accompanied by higher mitochondrial density (Fig. 2B).
Liver expression of HIF-1α mRNA was significantly associated with liver mtDNA content
To address the question of whether the increase in liver mtDNA copy number is associated with HIF-1α, we examined the liver expression of HIF-1α mRNA, and observed a significant positive correlation between liver mtDNA/nDNA ratio and hepatic levels of HIF-1α mRNA (R: 0.37, P < 0.001). We found that the level of HIF-1α mRNA was significantly higher in the HFD group (Fig. 3A). Western blot analysis showed that the level of protein, in parallel with the liver mRNA HIF-1α expression, was also significantly higher in the livers with increased mtDNA/nDNA ratio (Fig. 3B).
Fig 3.
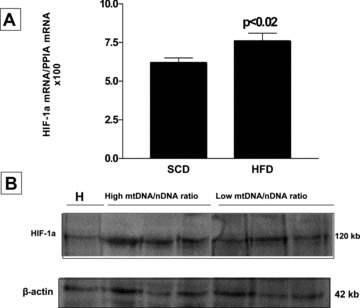
(A) Liver expression analysis of HIF-1α mRNA by quantitative real-time PCR in each experimental group. Each bar represents the mean ± S.E. values of 10 and 15 animals in SCD and HFD, respectively. In each sample, the gene expression was normalized to the expression of cyclophilin A and multiplied by 100. (B) Representative Western blot analysis of liver HIF-1α protein expression in each experimental group. Hypoxia-positive control (H) obtained from a rat ischemic skeletal muscle. β-actin was used as loading control.
Liver expression of cytochrome c oxidase subunit IV isoform 1 (COX4I1) mRNA was significantly associated with liver mtDNA content
Cytochrome c oxidase (COX), located in the inner mitochondrial membrane, is a dimer in which each monomer consists of 13 subunits; subunit IV (COX4) is regulated in an O2-dependent manner and is also a terminal enzyme in the mitochondrial respiratory chain, which catalyses the electron transfer from reduced cytochrome c to molecular oxygen. COX4 subunit determines the efficiency of mitochondrial respiration in response to different oxygen tensions. In addition, COX4I1 is a marker of mitochondrial mass. Thus, we evaluated liver COX4I1 mRNA expression and provided further evidence for an increase in liver mtDNA content, probably mediated by the effect of oxygen concentration in the liver, as the mtDNA/nDNA ratio was positively correlated with COX4I1 expression (R: 0.30, P < 0.02).
Multiple regression analysis showed that liver mtDNA/nDNA ratio was strongly correlated with the expression of HIF-1α (β: 0.32, S.E.: ±0.12, P < 0.009) and COX4I1 (β 0.37, S.E. ± 0.14, P < 0.01) mRNA in the liver. However, backward stepwise regression showed that the expression of HIF-1α mRNA in the liver was the only variable that was retained in the model and was strongly correlated with liver mtDNA/nDNA ratio (β: 0.40, S.E.: 0.11, P < 0.001).
Measurement of liver 8-isoprostane as a biomarker of local oxidative stress
Isoprostanes are prostaglandin-like compounds produced by free-radical-mediated peroxidation of lipoproteins. Numerous studies have shown that the quantification of isoprostanes is an accurate measure of oxidative stress [20, 21]. Evidence from both human and animal studies showed that liver steatosis increases oxidative stress [22]; intracellular levels of reactive oxygen species (ROS) have been found to be associated with changes in mtDNA copy number [23]. To determine whether oxidative stress is related to both hepatic steatosis and mtDNA content, we measured the liver 8-isoprostane levels in both experimental groups, and observed that they were significantly correlated with liver fat content (R: 0.42, P < 0.004). Nevertheless, no significant association was observed either with liver mtDNA content or liver HIF-1α mRNA expression.
The liver abundance of mRNA of master nuclear genes that regulate normal mitochondrial biogenesis and replication was not associated with the liver mtDNA content
To evaluate whether the hepatic expression of other transcription factors was associated with the observed increase in liver mtDNA content, we measured the following: mitochondrial transcription factor (Tfam) mRNA (a key regulator of mtDNA copy number required for the replication and maintenance of mtDNA [24]), peroxisome proliferator-activated receptor gamma coactivator-1-α (PGC-1α) and -β (PGC-1β) mRNA (physiological transcriptional regulators of oxidative metabolism and mitochondrial biogenesis [24]); PGC-1β is also considered to preferentially induce the expression of genes involved in the removal of ROS, nuclear respiratory factor-1 (NRF-1) mRNA (a gene involved in oxidative stress induced mitochondrial biogenesis [23]), and peroxisome proliferator-activated receptor δ (PPARδ) mRNA (a nuclear receptor capable of increasing the skeletal muscle mitochondria even in the absence of an increase in PGC-1α mRNA).
On the whole, we observed that the mRNA expression of the above-mentioned genes in the liver was not significantly associated with the liver mtDNA/nDNA ratio. In addition, we found no differences between the groups (HFD versus SCD) with regard to the mRNA expression of either of these genes (Fig. 4).
Fig 4.
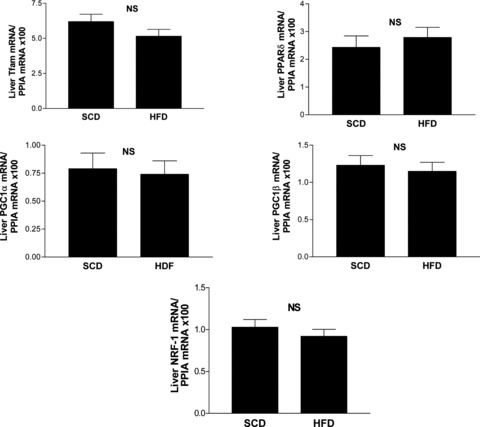
Liver mRNA expression of mitochondrial biogenesis genes assessed by quantitative real-time PCR in each experimental group. Each bar represents the mean ± S.E. values of 10 and 15 animals in SCD and HFD, respectively. In each sample, the gene expression was normalized to the expression of cyclophilin A and multiplied by 100. Tfam: mitochondrial transcription factor A; PPARδ: peroxisome proliferator-activated receptor γ; PGC-1α: peroxisome proliferator-activated receptor gamma coactivator-1α; PGC-1β: peroxisome proliferator-activated receptor gamma coactivator-1β; NRF-1: nuclear respiratory factor 1; PPIA: peptidylprolyl isomerase A (cyclophilin A); NS: not significant.
Evaluation of hepatic mRNA expression of apoptosis-related genes (Bcl-2, Bax and caspase-3), and fibrosis-related genes (collagen type Iα1 and smooth muscle α-actin)
Hepatocyte apoptosis and caspase activation are prominent features of human NALFD [25, 26]. In our experimental model, the liver tissue of HFD rats did not show histological signs of liver injury, inflammation or liver fibrosis, and this observation is in agreement with other reports [27]. Even so, to understand the potential role that early events associated with hepatocyte apoptosis might have in the HIF-1α-induced increase of the liver mtDNA content, we measured the hepatic mRNA expression of three apoptotic genes, such as: caspase 3 – a marker of early apoptosis and a reliable indicator of apoptotic rate – and two members of the Bcl-2 superfamily of proteins, the pro-apoptotic Bax and the anti-apoptotic Bcl-2, which act regulating the permeability of the mitochondrial membrane. Interestingly, the liver mRNA expression of none of the above-mentioned genes was significantly associated with the liver mtDNA/nDNA ratio. In addition, there was no significant difference in the liver Bax/Bcl-2 expression ratio between the groups (HFD versus SCD), Figure 5A. Nevertheless, the levels of hepatic mRNA expression of caspase 3 were significantly higher in the HFD group (Fig. 5A).
Fig 5.
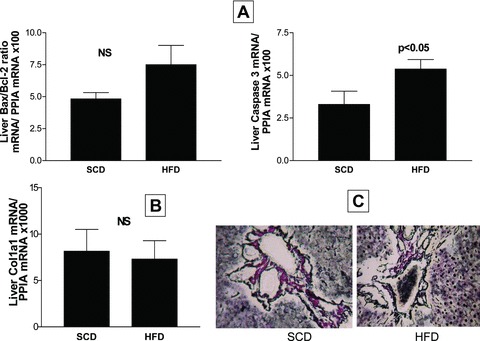
Liver mRNA expression of apoptosis-related genes (Bax, Bcl2 and caspase 3) (A) and a fibrosis-related gene (Col1a1) (B), assessed by quantitative real-time PCR in each experimental group. Each bar represents the mean ± S.E. values of 10 and 15 animals in SCD and HFD, respectively. In each sample, the gene expression was normalized to the expression of cyclophilin A and multiplied by 100/1000 as shown. Bax: Bcl2-associated X protein; Bcl2: B-cell CLL/lymphoma 2; Casp3: caspase 3; Col1a1: collagen type I α1. (C) Gomori’s reticulin silver impregnation of liver sections at the end of the experiment using a representative rat from each experimental group. Figure shows comparisons of the reticulin pattern of the portal tract and hepatic parenchyma across groups.
We further evaluated the liver mRNA expression of collagen type I α1– an extracellular matrix component, and smooth muscle α-actin, a marker of activation of resident stellate cells to myofibroblast-like cells, to gauge potential progression along the fibrosis development. There were no significant differences between the groups about liver collagen type I α1 mRNA expression (Fig. 5B); the abundance of smooth muscle α-actin mRNA was completely undetectable in both experimental groups, by our real-time PCR-based method. Moreover, neither the trichrome (Fig. 1C and D) nor the reticulin stains showed alterations of the collagenous stroma (Fig. 5C).
Effect of reversion of hepatic steatosis on liver mtDNA content and liver HIF-1α mRNA expression
To further understand the role of liver steatosis in HIF-1α-mediated increase in the liver mtDNA content, we evaluated the effect of telmisartan – a type 1 angiotensin II receptor blocker also considered to act as a partial agonist of PPARγ– on both liver HIF-1α mRNA expression and liver mtDNA/nDNA ratio. We chose this pharmacological agent because we previously demonstrated that telmisartan could revert fatty liver in an HFD-induced rat model of NAFLD [14]. In addition, previous evidence also showed that telmisartan could inhibit the generation of ROS and lipid peroxidation products in kidney [28].
The animals were given HFD for 8 weeks and after this period, for 12 weeks, they received vehicle (HFD) or telmisartan (Bago, Buenos Aires, Argentina) at 10 mg/kg intraperitoneally for every 24 hrs, along with the same access to HFD (T + HFD). The liver tissue was then collected to determine the mtDNA content, HIF-1α, mitochondria-related mRNA gene expression levels, and liver 8-isoprostane, as described earlier.
Telmisartan was associated with a significant decrease in the liver mtDNA/nDNA ratio (Fig. 6A) and liver HIF-1α mRNA expression (Fig. 6B). The liver HIF-1α protein level was also decreased in the T + HFD group when compared with the HFD group and SCD group (Fig. 6D). As shown in Figure 6C, fatty liver was efficiently reverted by telmisartan. Interestingly, rats treated with telmisartan showed a significant decrease in liver 8-isoprostane levels when compared with the HFD group (Fig. 6E).
Fig 6.
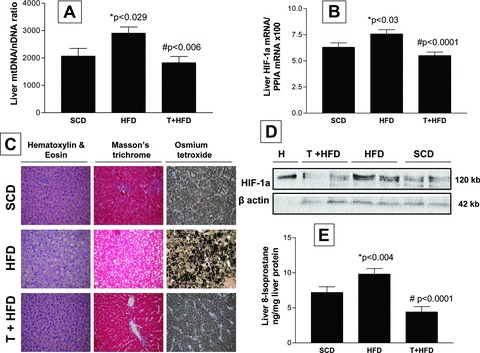
Effect of telmisartan on the reversion of hepatic steatosis, liver mtDNA content, HIF-1α mRNA and protein abundance, and liver 8-isoprostane levels. (A) Liver mtDNA/nDNA ratio assessed by quantitative real-time PCR in each experimental group. *P < 0.029, HFD versus SCD; #P < 0.006, T + HFD versus HFD. (B) HIF-1α mRNA analysed using quantitative real-time PCR in each experimental group. *P < 0.03, HFD versus SCD; #P < 0.0001, T + HFD versus HFD. (C) Liver histology of a representative animal from each experimental group. Haematoxylin and eosin, Masson’s trichrome and osmium tetroxide staining of liver sections at the end of the experiment using a representative rat from each experimental group as described in the ‘Materials and methods’ section. (D) Representative Western blot analysis of liver HIF-1α protein levels in each experimental group. Hypoxia-positive control (H) obtained from a rat ischemic skeletal muscle. β-actin was used as loading control. (E) Liver 8-isoprostane levels as a biomarker of local oxidative stress, assessed by ELISA in each experimental group. The results expressed as ng/mg liver protein. *P < 0.004, HFD versus SCD; #P < 0.0001, T + HFD versus HFD, adjusted by liver triglyceride content using ANCOVA. SCD: standard chow diet, HFD: high-fat diet; T + HFD: telmisartan plus high-fat diet.
Discussion
Progress in the understanding of the pathophysiology of NAFLD has been partly made through the evaluation of the molecular events that occur in the liver of experimental models. Using an HFD-induced rat model of NAFLD, we reproduced the features of human liver steatosis to evaluate the liver mtDNA content. We observed that feeding of rats with HFD resulted in an increase in the liver mtDNA/nDNA ratio, and this increase, was modestly but significantly correlated with the liver triglyceride content. We also found that livers of rats with increased mtDNA copy number also appear to have higher mitochondrial density, as assessed by electron microscopy. We further suggested that this increase in mtDNA copy number might be regulated by liver HIF-1α. Thus, this study, for the first time, demonstrated that the observed increase in the liver mtDNA/nDNA ratio was associated with the expression of HIF-1α mRNA in the liver. In addition, we provided further evidence to the hypoxic-mediated increase in liver mitochondrial biogenesis by demonstrating that liver COX4I1 mRNA expression was positively correlated with liver mtDNA content. We found that an increase in the mitochondrial mass was also supported by the increased expression of COX4I1 mRNA.
Furthermore, we also evaluated the mRNA expression in the liver of several key transcriptional factors, which are known to regulate normal mitochondrial biogenesis, including, PGC-1α, PGC-1β, NRF-1, PPARδ and Tfam, and observed that the increase in liver mitochondrial biogenesis was not related to the expression of the genes that commonly regulate mtDNA content. Additionally, we also observed that oxidative stress, although increased by HFD, was not directly involved in the HIF-1α-mediated increase in mtDNA copy number. Similarly, while liver caspase 3 mRNA levels were higher in the HFD group suggesting the presence of early apoptotic events, they were not associated with the changes observed in the liver mtDNA content.
Finally, to explore whether the observed events were related to the presence of liver steatosis, we evaluated the liver mtDNA content and HIF-1α mRNA expression in rats treated with telmisartan (a pharmacological agent that was found to efficiently revert back fatty liver [14]). Treatment of rats with telmisartan resulted in a significant decrease in liver mtDNA/nDNA ratio as well as liver HIF-1α mRNA and protein level. Otherwise, telmisartan may be involved directly in the modulation of HIF-1α. This observation is supported by previous evidence that showed that chronic hypoxia is associated with activation of the renin–angiotensin system [29, 30]. Thus, it is reasonable to hypothesize that the blockade of the renin–angiotensin system may directly prevent the effects of the induction of HIF-1α.
Although these results are novel, they are in agreement with earlier studies that showed that HFD could induce increase in mitochondrial enzymes and mitochondrial biogenesis in muscle [31, 32].
Furthermore, the central finding of our study, i.e. HIF-1α-associated increase in liver mtDNA content, is also supported by previous experimental observations on other tissues. For instance, neuronal mitochondrial biogenesis was highly increased after hypoxia or hypoxic–ischemic injury [13, 33].
Interestingly, although hypoxia has been well defined as an important mediator of inflammation and insulin resistance [34], its role in fatty liver disease, particularly in modulating liver mtDNA content, was never described previously.
The most important finding of our study is the potential role that hypoxia seems to have in the molecular events related to the increased accumulation of fat in the liver, and its impact on mitochondrial function. This concept was initially described in ethanol-induced steatohepatitis [35]. Moreover, Mantena et al. showed that chronic exposure of rats to HFD could affect liver mitochondrial function and cause hepatocyte hypoxia [10]. These findings were also replicated in human studies. Fisher et al. recently evaluated the microsomes isolated from human liver samples, and observed that NAFLD progression was correlated with elevated HIF-1α expression [36]. Finally, the increased hepatocyte lipid content was shown as a key determinant of hepatocyte sensitivity to hypoxia, an observation that has a direct clinical impact on, for instance, decreased viability of donor steatotic liver after liver transplantation [37].
In summary, accumulating evidence suggests a major role of mitochondrial dysfunction in the physiopathology of NAFLD. Nevertheless, only a few studies have explained how lipid accumulation in the hepatocytes affects liver mtDNA content. We consider that the main objective of our study was to provide evidence on the notion that hypoxia may play a role in the mitochondrial changes associated with NAFLD.
In addition, the biological and prognostic implications of our findings are as follows. Based on our experimental results, it seems reasonable to speculate that the observed increase in mtDNA content is an adaptive mechanism either to enhance the liver mitochondrial function to manage the increased mitochondrial β-oxidation as well as protect the hepatocytes against lipid peroxidation, or to compensate the hypoxic condition resulting from microcirculatory disturbances associated with fatty liver [38]. However, this aspect deserves further investigation.
It is worth mentioning that we did not observe either inflammatory changes or fibrosis in the liver, and that the histological lesions were limited to the accumulation of lipids in the hepatocytes – a morphological change also known as simple steatosis. This seems to be a hallmark as well as a limitation of this particular rodent model. Hence, we can hypothesize that changes in mtDNA precede liver lipotoxicity damage with the subsequent progression to more severe disease, which is in agreement with previous works that have demonstrated that either intermittent or sustained exposure to hypoxia results in steatohepatitis [39, 40]. This observation has clinical relevance because the observed changes in mtDNA can be reverted using any intervention that can improve liver steatosis. Furthermore, our observations on the effects of telmisartan on improving fatty liver infiltration as well as decreasing liver mtDNA content and expression of HIF-1α in the liver are in agreement with this finding.
Finally, the presence of increased mtDNA content in the steatotic liver, either as an outcome of chronic effects of hepatocyte hypoxia or an adaptive mechanism to enhance mitochondrial function, needs to be assessed further in the context of advanced lesions of NAFLD, including necroinflammatory response and fibrosis, to evaluate whether the increase in liver mtDNA confers a physiological advantage during the initial stages of NAFLD.
Acknowledgments
This study was partially supported by the grants PICT 2006–124 (Agencia Nacional de Promoción CientÌfica y Tecnológica) and UBACYT M055 (Universidad de Buenos Aires). A.B., M.S.R., T.F.G., C.J.P. and S.S. are members of Consejo Nacional de Investigaciones CientÌficas.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Browning JD, Szczepaniak LS, Dobbins R, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40:1387–95. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 2.Barshop NJ, Sirlin CB, Schwimmer JB, et al. Review article: epidemiology, pathogenesis and potential treatments of paediatric non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2008;28:13–24. doi: 10.1111/j.1365-2036.2008.03703.x. [DOI] [PubMed] [Google Scholar]
- 3.Sookoian S, Pirola CJ. Non-alcoholic fatty liver disease is strongly associated with carotid atherosclerosis: a systematic review. J Hepatol. 2008;49:600–7. doi: 10.1016/j.jhep.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 4.Kotronen A, Yki-Jarvinen H. Fatty liver: a novel component of the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2008;28:27–38. doi: 10.1161/ATVBAHA.107.147538. [DOI] [PubMed] [Google Scholar]
- 5.Sanyal AJ, Campbell-Sargent C, Mirshahi F, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183–92. doi: 10.1053/gast.2001.23256. [DOI] [PubMed] [Google Scholar]
- 6.Caldwell SH, Swerdlow RH, Khan EM, et al. Mitochondrial abnormalities in non-alcoholic steatohepatitis. J Hepatol. 1999;31:430–4. doi: 10.1016/s0168-8278(99)80033-6. [DOI] [PubMed] [Google Scholar]
- 7.Kelley DE, He J, Menshikova EV, et al. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–50. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- 8.Gianotti TF, Sookoian S, Dieuzeide G, et al. A decreased mitochondrial DNA content is related to insulin resistance in adolescents. Obesity. 2008;16:1591–5. doi: 10.1038/oby.2008.253. [DOI] [PubMed] [Google Scholar]
- 9.Song J, Oh JY, Sung YA, et al. Peripheral blood mitochondrial DNA content is related to insulin sensitivity in offspring of type 2 diabetic patients. Diabetes Care. 2001;24:865–9. doi: 10.2337/diacare.24.5.865. [DOI] [PubMed] [Google Scholar]
- 10.Mantena SK, Vaughn DP, Andringa KK, et al. High fat diet induces dysregulation of hepatic oxygen gradients and mitochondrial function in vivo. Biochem J. 2009;417:183–93. doi: 10.1042/BJ20080868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Semenza GL. Hypoxia-inducible factor 1: master regulator of O2 homeostasis. Curr Opin Genet Dev. 1998;8:588–94. doi: 10.1016/s0959-437x(98)80016-6. [DOI] [PubMed] [Google Scholar]
- 12.Mason SD, Rundqvist H, Papandreou I, et al. HIF-1alpha in endurance training: suppression of oxidative metabolism. Am J Physiol Regul Integr Comp Physiol. 2007;293:R2059–69. doi: 10.1152/ajpregu.00335.2007. [DOI] [PubMed] [Google Scholar]
- 13.Lee HM, Greeley GH, Jr, Englander EW. Sustained hypoxia modulates mitochondrial DNA content in the neonatal rat brain. Free Radic Biol Med. 2008;44:807–14. doi: 10.1016/j.freeradbiomed.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosselli MS, Burgueno AL, Carabelli J, et al. Losartan reduces liver expression of plasminogen activator inhibitor-1 (PAI-1) in a high fat-induced rat nonalcoholic fatty liver disease model. Atherosclerosis. 2009;206:119–26. doi: 10.1016/j.atherosclerosis.2009.01.026. [DOI] [PubMed] [Google Scholar]
- 15.Sugimoto K, Qi NR, Kazdova L, et al. Telmisartan but not valsartan increases caloric expenditure and protects against weight gain and hepatic steatosis. Hypertension. 2006;47:1003–9. doi: 10.1161/01.HYP.0000215181.60228.f7. [DOI] [PubMed] [Google Scholar]
- 16.Landa MS, Garcia SI, Schuman ML, et al. Knocking down the diencephalic thyrotropin-releasing hormone precursor gene normalizes obesity-induced hypertension in the rat. Am J Physiol Endocrinol Metab. 2007;292:E1388–94. doi: 10.1152/ajpendo.00234.2006. [DOI] [PubMed] [Google Scholar]
- 17.Brunt EM, Janney CG, Di Bisceglie AM, et al. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol. 1999;94:2467–74. doi: 10.1111/j.1572-0241.1999.01377.x. [DOI] [PubMed] [Google Scholar]
- 18.Ruijter JM, Ramakers C, Hoogaars WM, et al. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009;37:e45–57. doi: 10.1093/nar/gkp045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lebrecht D, Geist A, Ketelsen UP, et al. Dexrazoxane prevents doxorubicin-induced long-term cardiotoxicity and protects myocardial mitochondria from genetic and functional lesions in rats. Br J Pharmacol. 2007;151:771–8. doi: 10.1038/sj.bjp.0707294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roberts LJ, Morrow JD. Measurement of F (2)-isoprostanes as an index of oxidative stress in vivo. Free Radic Biol Med. 2000;28:505–13. doi: 10.1016/s0891-5849(99)00264-6. [DOI] [PubMed] [Google Scholar]
- 21.Morrow JD, Hill KE, Burk RF, et al. A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc Natl Acad Sci USA. 1990;87:9383–7. doi: 10.1073/pnas.87.23.9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147–52. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee HC, Wei YH. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int J Biochem Cell Biol. 2005;37:822–34. doi: 10.1016/j.biocel.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 24.Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004;18:357–68. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- 25.Feldstein AE, Canbay A, Angulo P, et al. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology. 2003;125:437–43. doi: 10.1016/s0016-5085(03)00907-7. [DOI] [PubMed] [Google Scholar]
- 26.Feldstein AE, Wieckowska A, Lopez AR, et al. Cytokeratin-18 fragment levels as noninvasive biomarkers for nonalcoholic steatohepatitis: a multicenter validation study. Hepatology. 2009;50:1072–8. doi: 10.1002/hep.23050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buettner R, Parhofer KG, Woenckhaus M, et al. Defining high-fat-diet rat models: metabolic and molecular effects of different fat types. J Mol Endocrinol. 2006;36:485–501. doi: 10.1677/jme.1.01909. [DOI] [PubMed] [Google Scholar]
- 28.Sugiyama H, Kobayashi M, Wang DH, et al. Telmisartan inhibits both oxidative stress and renal fibrosis after unilateral ureteral obstruction in acatalasemic mice. Nephrol Dial Transplant. 2005;20:2670–80. doi: 10.1093/ndt/gfi045. [DOI] [PubMed] [Google Scholar]
- 29.Nangaku M, Fujita T. Activation of the renin-angiotensin system and chronic hypoxia of the kidney. Hypertens Res. 2008;31:175–84. doi: 10.1291/hypres.31.175. [DOI] [PubMed] [Google Scholar]
- 30.Chan WP, Fung ML, Nobiling R, et al. Activation of local renin-angiotensin system by chronic hypoxia in rat pancreas. Mol Cell Endocrinol. 2000;160:107–14. doi: 10.1016/s0303-7207(99)00258-0. [DOI] [PubMed] [Google Scholar]
- 31.Hancock CR, Han DH, Chen M, et al. High-fat diets cause insulin resistance despite an increase in muscle mitochondria. Proc Natl Acad Sci USA. 2008;105:7815–20. doi: 10.1073/pnas.0802057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nemeth PM, Rosser BW, Choksi RM, et al. Metabolic response to a high-fat diet in neonatal and adult rat muscle. Am J Physiol. 1992;262:C282–6. doi: 10.1152/ajpcell.1992.262.2.C282. [DOI] [PubMed] [Google Scholar]
- 33.Yin W, Signore AP, Iwai M, et al. Rapidly increased neuronal mitochondrial biogenesis after hypoxic-ischemic brain injury. Stroke. 2008;39:3057–63. doi: 10.1161/STROKEAHA.108.520114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ye J. Emerging role of adipose tissue hypoxia in obesity and insulin resistance. Int J Obes. 2009;33:54–66. doi: 10.1038/ijo.2008.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li L, Chen SH, Zhang Y, et al. Is the hypoxia-inducible factor-1 alpha mRNA expression activated by ethanol-induced injury, the mechanism underlying alcoholic liver disease? Hepatobiliary Pancreat Dis Int. 2006;5:560–3. [PubMed] [Google Scholar]
- 36.Fisher CD, Lickteig AJ, Augustine LM, et al. Hepatic cytochrome P450 enzyme alterations in humans with progressive stages of nonalcoholic fatty liver disease. Drug Metab Dispos. 2009;37:2087–94. doi: 10.1124/dmd.109.027466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berthiaume F, Barbe L, Mokuno Y, et al. Steatosis reversibly increases hepatocyte sensitivity to hypoxia-reoxygenation injury. J Surg Res. 2009;152:54–60. doi: 10.1016/j.jss.2007.12.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hasegawa T, Ito Y, Wijeweera J, et al. Reduced inflammatory response and increased microcirculatory disturbances during hepatic ischemia-reperfusion injury in steatotic livers of ob/ob mice. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1385–95. doi: 10.1152/ajpgi.00246.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Piguet AC, Stroka D, Zimmermann A, et al. Hypoxia aggravates non-alcoholic steatohepatitis in mice lacking hepatocellular PTEN. Clin Sci. 2010;118:401–10. doi: 10.1042/CS20090313. [DOI] [PubMed] [Google Scholar]
- 40.Savransky V, Bevans S, Nanayakkara A, et al. Chronic intermittent hypoxia causes hepatitis in a mouse model of diet-induced fatty liver. Am J Physiol Gastrointest Liver Physiol. 2007;293:G871–7. doi: 10.1152/ajpgi.00145.2007. [DOI] [PubMed] [Google Scholar]