

Abstract
It has been almost 25 years since the initial discovery that tau was the primary component of the neurofibrillary tangles (NFTs) in Alzheimer disease (AD) brain. Although AD is defined by both β-amyloid (Aβ) pathology (Aβ plaques) and tau pathology (NFTs), whether or not tau played a critical role in disease pathogenesis was a subject of discussion for many years. However, given the increasing evidence that pathological forms of tau can compromise neuronal function and that tau is likely an important mediator of Aβ toxicity, there is a growing awareness that tau is a central player in AD pathogenesis. In this review we begin with a brief history of tau, then provide an overview of pathological forms of tau, followed by a discussion of the differential degradation of tau by either the proteasome or autophagy and possible mechanisms by which pathological forms of tau may exert their toxicity. We conclude by discussing possible avenues for therapeutic intervention based on these emerging themes of tau’s role in AD.
Keywords: tau, Alzheimer disease, phosphorylation, truncation, oligomers, tau-based therapies, autophagy
Introduction
It has been more than 35 years since tau was first discovered as a protein that co-purified with microtubules through cycles of assembly and disassembly and was named for its ability to induce ‘tubule’ formation [1]. Two years later it was noted that tau was a phosphoprotein [2], and in 1984 it was demonstrated that tau in a more dephosphorylated state promotes microtubule assembly with greater efficiency, thus assigning a regulatory function to its phosphorylation [3]. Although these were intriguing findings at the time, interest in tau remained only moderate until it was demonstrated that tau made up the paired helical filaments which form the neurofibrillary tangles (NFTs) found in Alzheimer disease (AD) brain, and the tau that formed these filaments was abnormally phosphorylated [4–8]. These findings catapulted tau into the limelight. In 1988, the first sequence of tau (from mouse) was published [9], and in the following year the sequences of both bovine and human tau were elucidated [10, 11]. These findings set the stage for an explosion of studies on tau, particularly on tau phosphorylation and function [12]. However, tau studies began to wane when it became clear that the causes of the familial form of AD were autosomal dominant mutations in genes that affect the production of β-amyloid (Aβ). Aβ is a peptide generated from amyloid precursor protein (APP) that forms the core of the Aβ plaques, which are the other predominant pathological lesion in AD brain [13–16]. These and other findings demonstrating the toxicity and pathogenic properties of Aβ led to the notion that tau pathology was not central to the disease [17].
In 1998, it was first discovered that mutations in the tau gene caused rare autosomal dominant neurodegenerative diseases (collectively known as frontotemporal dementia with parkinsonism linked to chromosome 17 [FTDP-17]) in which there was pronounced tau pathology but no Aβ pathology [18, 19]. Following these findings, a growing list of sporadic neurodegenerative tauopathies marked by abnormal tau-containing lesions in the brain and without significant Aβ pathology, were characterized [20]. These findings strongly suggested that alterations in tau could result in neurodegeneration and hinted at the importance of tau in the pathogenic processes in AD. This idea was further supported by the finding that neurons in which tau had been knocked out were relatively resistant to Aβ-induced cell death compared to neurons from wild-type mice expressing normal tau levels [21].
It is now very clear that tau plays a critical role in the pathogenesis of AD. However, it is also becoming evident that NFTs are not the ‘toxic’ tau species but rather non-aggregated forms are likely the culprits. For example, in an inducible tauopathy model, attenuating expression of FTDP-17 mutant tau ameliorated behavioural deficits and neuronal loss although NFT numbers either remained unchanged or increased [22]. More recent studies have shown that knocking out tau significantly attenuates Aβ-induced behavioural deficits but does not affect Aβ levels in an APP mouse model of AD [23], and that Aβ-induced deficits in axonal transport do not occur in neurons from tau–/– mice [24]. These and other findings have contributed to a re-evaluation of the role of tau in neurodegeneration, including the consideration of a dual or synergistic relationship between tau and Aβ in AD pathogenesis [25, 26].
In this review, we will first present a brief overview of pathological modifications of tau and what may be the toxic species, followed by an overview of how tau is cleared from the cell and deficits that may occur in AD brain. We will then discuss possible targets of pathological tau that may contribute to disease progression and finally end with a presentation of potential ‘tau-centric’ therapeutic strategies for the treatment of AD.
Pathological modifications of tau
A defining feature of AD brain is tau pathology. Many different post-translational modifications of tau in AD brain have been described including phosphorylation, nitration, glycosylation, glycation, ubiquitylation and truncation [27, 28]. Given that abnormal phosphorylation is the most prominent modification of tau in AD brain, and truncation of tau also seems likely to play a key role in the evolution of tau inclusions, we will discuss these two modifications a bit further prior to exploring the role of tau in AD pathogenesis.
Tau phosphorylation
Tau is a phosphoprotein and phosphorylation plays a prominent role in regulating its physiological function. It is also clear that aberrant tau phosphorylation occurs in AD brain and is associated with pathogenesis. Previous reviews have provided full discussions of the kinases and phosphatases that regulate tau phosphorylation, as well as possible involvement of tau hyperphosphorylation in the pathogenesis of AD [29–33]. Therefore, in this review we will highlight several recent studies on the phosphorylation of specific sites on tau, and how they affect tau function and their involvement in AD pathogenesis. It also should be noted that tau phosphorylation was detected in brain biopsies from non-AD patients at many sites that are not normally phosphorylated in autopsied non-AD material, although the extent of phosphorylation was less than observed in autopsied AD brain samples. In this study it was suggested that the hypophosphorylation of autopsy-derived adult human tau was due to rapid dephosphorylation post-mortem. Overall these data indicate that most tau phospho-epitopes detected in AD brain are not unique to the disease state [34], although the extent to which they are phosphorylated is greater than what occurs in the normal state.
In AD, phosphorylation of tau at specific epitopes appears to be a progressive event with phosphorylation at Threonine 231 (T231) occurring early, at ‘pretangle’ stages [35, 36]. Phosphorylation at this epitope appears to contribute to conformational changes in tau that are detected by the antibody TG-3 and subsequently by Alz-50 [36, 37]. It has also been shown that phosphorylation of T231 significantly reduces the ability of tau to bind microtubules [38]. Glycogen synthase kinase 3β (GSK3β) is often considered to be a ‘tau kinase’ as it efficiently phosphorylates T231 and other AD relevant tau epitopes in vitro and in cells and is likely deregulated in AD brain [38, 39]. Therefore, GSK3β could contribute to the observed increase in pT231 in the disease state. Further, the peptidyl prolyl isomerase, Pin1, specifically binds to pT231 which results in a conformational change that can restore tau’s ability to bind to microtubules or facilitate dephosphorylation of this site by protein phosphatase 2A, which also increases tau-microtubule interactions [40]. Aged Pin1–/– mice exhibit increased phosphorylation of tau at T231 concomitant with increased tau aggregation and filament formation along with neurodegeneration [41], while soluble levels of Pin1 have been noted to decrease in AD brain [40]. Although Pin1 may act on other substrates in AD brain [40], these data suggest that increased phosphorylation of tau at T231 may play a role in the pathogenesis of AD.
Another phosphorylation site on tau that plays a pivotal role in regulating tau function is serine 262 (S262). S262 is located within the KXGS motif of the first microtubule binding repeat and is phosphorylated predominantly by the microtubule-associated protein microtubule affinity regulating kinase [42]. Phosphorylation of this site significantly decreases tau binding to microtubules [43], an effect that can be mimicked by pseudophosphorylation (mutation of serine or threonine to glutamate or aspartate to mimic phosphorylation) [44]. Increased phosphorylation at S262 was noted in pretangle neurons in AD brain suggesting that it was an early event in the pathogenic process. In flies, expression of tau with alanine mutations at both S262 and S356 (which is also part of a KXGS motif in the fourth microtubule binding domain) resulted in significantly less toxicity than expression of wild-type tau [45]. In another fly study co-expression of Aβ and tau resulted in neurodegeneration, while expression of Aβ with tau containing a serine to alanine mutation at S262 (S262A) did not, strongly suggesting that phosphorylation of S262 is required for Aβ-induced, tau-dependent toxicity [46]. Intriguingly, S262A tau was phosphorylated at S202 to a lesser extent than wild-type tau in this model [46]. Likewise S262A/S356A tau was also phosphorylated to a lesser extent than wild-type tau at S202 as well as the PHF-1 epitope (S396/S404) in the fly. However, when GSK3β was co-expressed with tau, both wild-type and S262A/S356A tau were efficiently phosphorylated at these same epitopes, although a significant reduction in the rough eye phenotype (indicative of neurodegeneration) was observed in flies expressing S262A/S356A tau compared to flies expressing wild-type tau [45]. These data suggest that the increased tau phosphorylation that results from increased GSK3β activity is not the mechanism involved in the neurodegenerative process, at least in this model system [45].
When considering the role of tau phosphorylation in the pathogenesis of AD it is becoming apparent that a specific complement of phosphorylated residues enhance neurotoxicity and that phosphorylation of any one single site is likely not sufficient to convert tau to a toxic species. In addition, as alluded to above, the phosphorylation of one epitope on tau can influence the phosphorylation of other epitopes [45–48]. Expressing tau pseudophosphorylated at S262 and T231 resulted in increased toxicity in PC12 cells compared to wild-type tau or tau that was pseudophosphorylated at just S262 [49]. In flies, pseudophosphorylation of 14 key Ser/Thr-Pro sites in tau significantly enhanced toxicity [50], while mutating these same sites to alanine significantly blocked tau-induced toxicity [51]. Strikingly, this study revealed that no particular individual phosphorylation site was responsible for enhancing tau toxicity, and that recovery was only obtained when all 14 sites were mutated; when individual Ser/Thr-Pro sites were mutated to alanine, there was no recovery from tau-induced toxicity. It remains to be established whether phosphorylation of specific sites impact neurons in a sublethal, pathological manner, or alternatively compromise certain cellular functions that may contribute to decreased cell survival over time, rather than causing acute neuronal death.
Tau truncation
During the evolution of tau pathology in AD brain, tau appears to undergo sequential cleavage events [52]. Caspases, which are apparently elevated in AD brain [53–55], are likely involved in the proteolytic processing of tau. Several years ago it was shown that tau is cleaved by caspases at aspartic acid 421 (D421) in AD brain and appeared to be generated early in the pathogenic process [56, 57]. Tau truncated at D421 is more fibrillogenic than full length tau [56, 57] and early studies suggested that tau truncated at D421 significantly increased cell death [58, 59]. Recently it was demonstrated, using multiphoton imaging, that there are transient increases in caspase activity in neurons in a mouse tauopathy model, and this is followed by the formation of NFTs without acute cell death. Additionally, expression of tau truncated at D421 in wild-type mouse brain also resulted in the formation of similar tau aggregates [60]. These data clearly indicate that caspase activation and tau cleavage precede the formation of tau fibrils and that caspase activation does not necessarily lead to acute cell death. Indeed, it is well documented that caspases play a number of roles in physiological processes [61–64]. Nonetheless, in AD inappropriate, albeit transient, increases in caspase activity and tau cleavage may compromise neuronal function. For example, inducible expression of tau truncated at D421 in a cortical neuronal cell line results in significant effects on mitochondrial function without profound cell death [65]. However, when the cells are subjected to stressors, the presence of tau truncated at D421 results in a significant decrease in cell survival compared to cells expressing wild-type tau [66]. Flies expressing tau truncated at D421 also displayed greater neuronal toxicity compared to what was observed in flies expressing full-length wild-type tau [67].
In addition to being cleaved by caspases at D421, tau may also be cleaved at D13 and D402 in AD brain by caspase 6 [68–70]. In one study the level of tau cleaved at D402 in NFTs and neuropil threads was associated with lower global cognitive scores in cases with no cognitive impairment, indicating that it may be an early event in the development of AD [68]. Finally, at later stages in the evolution of tau pathology tau undergoes further processing including truncation at glutamic acid (E) 391 which results in the formation of the MN423 epitope, along with further processing of the amino terminal [71]. These latter cleavage events appear after tau has formed the NFTs and seem to be part of the maturation process of the tangles [52, 71].
NFTs are not the primary toxic species
A final issue that needs to be addressed is: What is the toxic form of tau in AD? Although initially it was thought that the NFTs were the toxic species, it is becoming apparent that the mature fibrillar tangles are unlikely to be the primary mediators of toxicity, although at later stages of the disease they compromise cell viability. For example, cytotoxicity of an aggregation prone mutant tau occurred prior to the appearance of thioflavin S+ tau aggregates in an inducible cell model [72] and expression of tau in flies results in progressive neurodegeneration without NFT formation [73]. It has also been shown that in a repressible tau transgenic mouse model, turning off tau expression resulted in the attenuation of tau-induced memory impairment and neuronal loss, but NFTs continued to accumulate [22]. Furthermore, inhibition of tau hyperphosphorylation in the JNPL3 mouse model which expresses tau with an FTDP-17 mutation attenuated the severe motor deficits and reduced the amount of soluble tau aggregates without affecting NFT counts [74]. Another indication that NFTs are not the mediators of toxicity is that depletion of endogenous wild-type tau prevented behavioural abnormalities in an APP transgenic mouse model that does not develop NFTs [23]. Therefore, NFTs are likely not the major cause of neuronal toxicity and cognitive deficits, and in fact may play a protective role in the initial stages of the disease. Indeed, it has been suggested that the formation of disease relevant protein aggregates may in general be protective by sequestering dispersed small aggregates, oligomers or misfolded proteins to minimize their toxicity and eventually facilitate their clearance by proteasomal activity or autophagy [75, 76]. Although this concept is supported by data from other neurodegenerative disease cell models [77, 78] it remains to be validated with respect to tau and AD.
Tau turnover and possible deficits in Alzheimer disease
AD results in deficits in numerous physiological processes, including degradative mechanisms. Proteasome activity in AD brain is lower than that in control brain, and this lowered activity is independent of the expression of proteasome subunits [79]. In addition, autophagic processes in AD brain are suggested to be compromised either as a result of decreased levels of macroautophagy regulatory proteins such as Beclin-1 [80], or impaired clearance of autophagic vacuoles by lysosomes [81]. Evidence of general degradative dysfunction in AD, along with co-labelling of tau with degradative markers (i.e. ubiquitin) [82], has led to a more detailed study of tau turnover in general and how it may be compromised in AD brain [83–85]. Tau, like other aggregative proteins involved in neurodegenerative diseases, has been described as a substrate for both proteasomal [86, 87] and autophagic [83] degradation and therefore its turnover may be impaired in the disease state. It should be noted that the majority of studies examining tau turnover have been done with exogenous, rather than endogenously expressed tau, a caveat that needs to be considered when evaluating the data.
Tau’s susceptibility to proteasomal degradation has been reported to be influenced by phosphorylation, specifically at S199/S202/S205 with phosphorylation being required for tau’s recognition by the E3 ligase C-terminus of Hsp70 interacting protein (CHIP) and subsequent proteasomal degradation [86]. However, another report [87] asserted the opposite: recognition of tau by CHIP occurred independent of its phosphorylation. Yet another report [88] showed that the ability of tau to be ubiquitylated by CHIP was dependent on the presence of exon 10 in the microtubule binding domain. Despite these differences, all three studies provided evidence that tau was ubiquitylated by CHIP. However, questions arise upon close examination of the findings that were presented. In separate studies, CHIP was found to catalyse the formation of forked ubiquitin chains in vitro on multiple substrates that consist of a mixture of lysine (K)11, K48 and K63 ubiquitin linkages and are insensitive to proteasome degradation [89]. This suggests that in the absence of other unidentified cofactors, CHIP behaves indiscriminately in in vitro reaction conditions. Second, degradation of the putative CHIP substrate nNOS is unaffected in cells derived from a CHIP-null mouse [90], which suggests that CHIP is dispensable in the degradation of its substrates, and/or plays a role related to, but distinct from, acting directly as an E3 ligase towards its substrates. In fact, a recent study provided evidence that tau and tau truncated at D421 were not ubiquitylated by CHIP in situ, despite evidence of CHIP-mediated ubiquitylation in an in vitro reaction [91]. Overall, it is apparent that further studies are required to demonstrate definitively that tau is a bona fide substrate of CHIP in situ.
Although the proteasome primarily degrades ubiquitylated substrates, there is a growing awareness that it also can degrade proteins in an ubiquitin-independent manner and there is good evidence that tau’s degradation by the proteasome can occur in the absence of direct ubiquitylation. Using SH-SY5Y neuroblastoma cells and in vitro techniques, David and colleagues [92] reported that clearance of full-length tau (without amino-terminal inserts) was sensitive to the proteasome inhibitor lactacystin, and that tau removal occurred independent of ubiquitylation. Similar results were reported with full-length tau in a cortical cell model [91]. A more recent study [93] also determined that phosphorylated tau is degraded by the proteasome independent of ubiquitylation through coexpression of dominant negative ubiquitin mutants with tau. The presence of these dominant-negative ubiquitin mutants was found not to affect the degradation of tau. Interestingly, the authors also found that phosphorylated tau co-immunoprecipitated with the chaperone Hsp70, but not with CHIP [93].
In addition to degradation by the proteasome, there is evidence that tau is processed by the lysosome, and more specifically, by autophagy. Treatment of a stable cell line expressing human tau with either the lysomotrophic drug chloroquine, the macroautophagy inhibitor 3-methyladenine (3-MA), or cathepsin inhibitors resulted in a delay to the degradation of tau and promoted formation of high molecular weight species [83]. In a separate study, D421-cleaved tau was produced in SK-N-SH neuroblastoma cells by treatment with prostaglandin J2, and the D421-tau cleavage product was degraded in a cathepsin-dependent manner [94].
Using a highly fibrillar internal fragment of tau, another study elucidated a pathway by which either inhibition of macroautophagy using 3-MA, or neutralization of the lysosome using NH4Cl, slows the clearance of most forms of soluble tau, particularly pS262 tau [95]. However, this treatment resulted in the formation of a smaller cleavage product that engaged the machinery for chaperone-mediated autophagy, yet was not translocated into the lysosome. Despite this finding of involvement of macroautophagy and chaperone-mediated autophagy in the clearance of a small cleavage product of tau [95], further work is needed to determine the relative involvement of the autophagy pathway in the turnover of tau.
How to reconcile the equally compelling work describing both proteasomal and autophagic degradation of tau? The existence of an aggregative protein involved in neurodegeneration that is susceptible to either proteasomal or autophagic degradation depending on post-translational or aggregative state, is not unique. Huntingtin [96, 97], α-synuclein [98] and ataxin-3 [99, 100] have all been described as substrates of the proteasome as well as autophagy. Also, recent work examining mechanisms behind the selective autophagic clearance of aggregation-prone proteins involved in neurodegeneration describes K63-linked ubiquitylation (rather than the more prevalent K48-linked ubiquitylation) of both tau and superoxide dismutase 1 (SOD1) leading to their autophagic clearance [101]. Interestingly, a mechanism for K63-linked ubiquitylation has been proposed for tau involving the E3 ligase TRAF6 [102]; the authors describe binding of K63-ubiquitylated tau to the shuttling factor p62, which has been linked to both the proteasome and autophagy [103]. Therefore it remains likely that if this mechanism for selective autophagy also occurs in AD brain, tau may be degraded by either the proteasome or autophagy, perhaps depending on conformational and aggregative state. In fact, work from our own lab has demonstrated that tau cleaved at D421 is more susceptible to autophagy-mediated clearance than full-length tau, which is prone to non-ubiquitin dependent degradation by the proteasome [91]. In agreement with our findings it was recently demonstrated that deletion of cathepsin D (a lysosomal protease and thus involved in autophagy) in flies resulted in the specific accumulation of tau truncated at D421 [67]. D421-tau has been reported to be more highly fibrillogenic than full-length tau [104], which may be a determining factor in the predominantly autophagic degradation of this form of tau, as aggregate-prone proteins are highly susceptible to autophagy [105]. It is also interesting to note that tau phosphorylated or pseudophosphorylated at S262 (which significantly changes the conformation of the protein [44]) appears to be degraded through the autophagy pathway and not the proteasomal pathway [95].
Tau toxicity in Alzheimer disease
Although for many years ‘Aβ-centric’ hypotheses dominated AD research [17], the importance of tau in the pathogenesis of AD is now much more appreciated [26], and seems to increase as new findings emerge. An early strong indicator of the importance of tau in Aβ-induced neurotoxicity came from a study in which neurons from tau–/– mice were found to be very resistant to Aβ-induced neurite degeneration compared to neurons from wild-type mice or mice expressing human tau [21]. Complementing this study, reduction of tau levels in a human mutant APP mouse model by crossing them with tau–/– mice resulted in marked improvements in performance in spatial learning, and reduced premature mortality, hyperactivity and excitotoxicity [23]. It was also recently demonstrated that reduction or elimination of tau was sufficient to prevent exogenous Aβ-induced axonal transport deficits in mouse primary neurons [24]. Finally, expression of a dominant negative form of tau in mice expressing human mutant APP resulted in the sequestration of Fyn kinase away from synapses, which prevented phosphorylation of the N-methyl D aspartate (NMDA) receptor and coincided with an attenuation of memory deficits. Similar results were obtained when tau was knocked out in these mice [106]. Overall these data strongly indicate that tau is an important mediator of Aβ-induced excitoxicity, although its exact mechanism of action is unclear.
Tau was originally discovered as a microtubule-associated protein and the majority of studies involving tau have focused on its ability to modulate microtubules; a function that is regulated by its phosphorylation state. Abnormal phosphorylation of tau, a prominent feature of AD brain, decreases its microtubule binding ability, which may destabilize microtubules and result in cellular damage [8, 107]. This ‘loss of function’ model could explain several aspects of tau toxicity; however, recent research points to possible ‘gain of function’ mechanisms of tau toxicity that may not be directly dependent on its microtubule binding ability [32]. For example, tau at approximately physiological concentrations did not inhibit axonal transport; however, tau filaments at the same concentration were found to be inhibitory and this inhibition was not due to direct microtubule binding [108]. Inhibition of anterograde axonal transport is now thought to be associated with the ability of tau to bind kinesin [109, 110]. Whether tau inhibits the interaction of cargoes with kinesin [109] or disrupts the attachment of kinesin to microtubules [111, 112] is still under investigation.
Tau-induced impairment of axonal transport has been implicated in the mislocalization of proteins and organelles to the soma. Pathological forms of tau have repeatedly been shown to cause abnormal somatic localization of mitochondria [112–115], which are the main source of energy production and calcium buffering in cells. Thus, sequestration of mitochondria away from axons, which contain areas of high energy demand and calcium influx, such as nodes of Ranvier and synapses, has a profound impact on cellular homeostasis [116]. For this reason, tau-induced mitochondrial mislocalization may be a key component in neurodegenerative processes [108, 112]. A recent paper provided some insight into a possible ‘gain of function’ mechanism by which pathologically modified tau may disrupt axonal transport of mitochondria and other kinesin cargos. These studies were carried out using an FTDP-17 tau mouse model, and the hyperphosphorylated tau was found to selectively interact with c-Jun N-terminal kinase interacting protein 1 (JIP1), which regulates the association of cargo with the kinesin motor complex. This binding of JIP1 by hyperphosphorylated FTDP-17 tau caused JIP1 to mislocalize to neuronal cell bodies which was likely a contributing factor to the axonal transport deficits observed in these mice [116, 117].
Beyond impairment of mitochondrial transport, tau may disrupt several other aspects of mitochondria under pathological conditions. There is significant evidence indicating that mitochondria are compromised early in AD brain tissue [118–120], which has been replicated in experimental models exhibiting Aβ and tau pathologies [25, 121–126]. Abnormalities exist in mitochondrial fission/fusion and respiration protein levels in AD brain [120, 127], which may explain mitochondrial morphological alterations observed in several areas of AD patient brains [118] and apparent deficits in energy metabolism observed via PET scan [128]. Interestingly, stable expression of D421 truncated tau in cortical cell lines induced fragmentation of mitochondria [65], possibly replicating the imbalance of mitochondrial fission and fusion in AD brain. Reductions in several components of the tricarboxylic acid (TCA) cycle and the oxidative phosphorylation (OXPHOS) system have also been reported in AD transgenic mouse models [124, 125]. In fact, a recent study provided evidence that tau can synergize with Aβ at the level of mitochondria resulting in enhanced mitochondrial damage. This study compared mitochondrial measures between tau aggregate forming P301Ltau mice, Aβ plaque forming APPSWEPS2N141I mice, and triple transgenic (3xTg) mice which combine these mutations and pathologies. Proteomic analysis of 3xTg mice showed vast deregulation of several OXPHOS proteins related to complex I and IV, whereas tau transgenic mice only showed deregulation of complex I and Aβ plaque forming mice only showed deregulation of complex IV. Compared to either of the single pathology mice, 3xTg mice showed more severe or earlier onset of mitochondrial deficits, loss of mitochondrial membrane potential, and increased reactive oxygen species (ROS) production [25, 126]. This suggests that tau may exert its toxicity through mitochondria, and may permit or facilitate Aβ-induced mitochondrial damage. Figure 1 summarizes some of the possible targets of pathological forms of tau.
Fig 1.
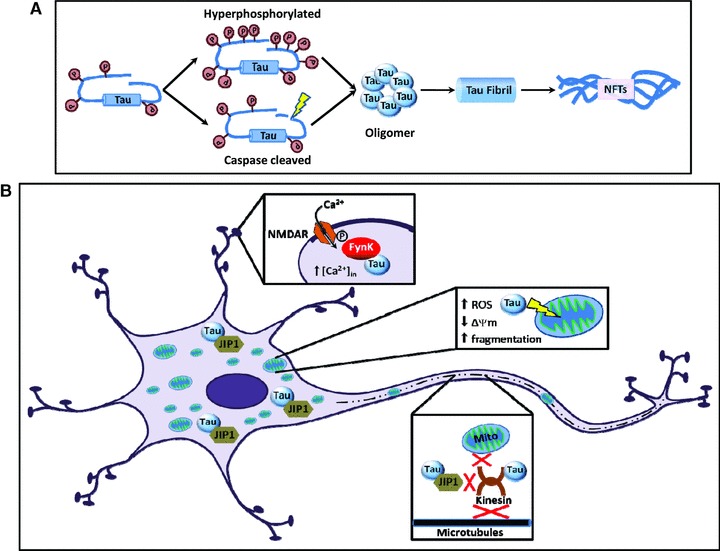
Cellular targets of toxic tau. (A) Tau undergoes phosphorylation in physiological conditions. However, in pathological conditions, tau becomes hyperphosphorylated and/or cleaved, which facilitates aggregation and increases the toxicity of tau. In the continuum of the aggregation process, pathologically modified tau monomers first form oligomers, which further aggregate into fibrils and finally into NFTs. Recent studies suggest that monomeric or oligomeric species of tau are the more toxic than aggregated forms [22]. (B) Tau may manifest its toxicity by enabling or facilitating Aβ-induced excitotoxicity, mitochondrial damage and/or by disrupting axonal transport. Pathological tau may participate in the localization of Fyn kinase to the post-synaptic compartment, where it phosphorylates NMDAR subunits, causing increased inward Ca2+ conductance and leading to excitotoxicity [106]. Tau also contributes to mitochondrial dysfunction (i.e. decreased Δψm, increased ROS, fragmentation) [65] and disrupts anterograde axonal transport of mitochondria and possibly other synaptic elements by binding to JIP1, which regulates the binding of cargos to the kinesin motor complex [116]. Transport deficits may cause energy depletion at synapses, thereby impairing synaptic transmission.
Potential ‘tau-centric’ therapeutic strategies
There is a growing interest in developing pharmacological agents directed at pathological forms of tau. These treatment strategies are largely in the early stages of development; nonetheless several studies have already yielded exciting and encouraging results. In this section, we highlight several of the emerging treatment strategies that are being explored. However, it should be noted that with many of the different approaches discussed herein the pharmacokinetic properties, bioavailability and possible side effects of these ‘tau-directed’ therapies are extremely important issues that remain to be fully addressed.
Tau aggregation inhibitors
It is well established that the NFT load can be used as a marker to clinically classify the stages of disease progression [129]. Also, the amount and progression of NFTs correlates positively with the extent of cognitive impairment [130]. Based on these and other findings it was originally suggested that NFTs were significant contributors to the pathogenic process in AD; however, more recently this has been called into question [22, 131]. Multiple potentially pathological forms of tau exist in the brains of AD patients, and there is significant debate over which forms are the toxic species. Nonetheless, there has been an effort to develop tau aggregate inhibitors as a therapeutic strategy for the treatment of AD with the presupposition that aggregated tau in some form (oligomeric, soluble or insoluble) is toxic. As mentioned previously, there are data to support the concept that soluble, aggregated tau species are toxic. For example, in a squid axoplasm model, monomeric tau at physiological concentrations had no effect on axonal transport, while tau filaments at the same concentrations selectively inhibited anterograde transport processes suggesting that pathological tau aggregation may be a contributing factor to neurodegeneration [108].
A number of labs have been investigating potential tau aggregation inhibitors, and in a recent review small-molecule inhibitors of tau aggregation were thoroughly described [132]. Therefore in this review we will just highlight a few key studies. Methylene blue is a phenothiazine that, along with other phenothiazine derivatives, has been used to treat a number of different medical conditions [133]. One important effect of methylene blue is the inhibition of aggregation of proteins that take on β-sheet conformation in vitro, including tau [134, 135]. Use of methylene blue in vivo has yielded mixed results. In a mouse tauopathy model, 1 mM methylene blue was administered to the right hippocampus using a mini-osmotic pump. This resulted in a reduction in total tau levels that were paralleled by a decrease in phosphorylated (S202/T205) tau along with improved behaviour but without any effect on pathology [136]. When the mice were administered a therapeutically relevant dose of 10 mg/kg of methylene blue in the drinking water for 12 weeks there was variable improvement in behaviour and decreases in soluble tau levels, with no change in tau tangle pathology. Interestingly, when the levels of methylene blue in the cerebellum were measured, they were found to positively correlate with Morris water maze performance and inversely correlate with soluble tau levels. These results are in agreement with previous findings showing that NFTs are likely not associated with functional deficits but that reducing soluble tau levels is beneficial [136]. These results also suggest that the putative therapeutic effect of methylene blue may not be associated with its ability to inhibit aggregate formation. In Zebrafish, methylene blue inhibited the aggregation of a mutant form of huntingtin, but had no effect on tau pathology when an FTDP-17 mutant tau was expressed [137], which is similar to what was observed in the mouse model. However, in contrast to the mouse model, methylene blue treatment of the Zebrafish did not prevent or reverse the functional deficits that resulted from the expression of the FTDP-17 mutant tau or the mutant huntingtin [137].
N3 is a benzothiazole derivative that has been shown to protect against tau aggregation and tau-mediated neurodegeneration concurrent with neuroprotection in an in vivo model [138]. Based on in vitro assays, it was proposed that N3 was a direct tau fibrillization inhibitor and that this mechanism contributed to its neuroprotective effects. However, further studies demonstrated that N3 disrupts the binding of tau filaments to Thioflavin S, but does not directly antagonize the formation of tau fibrils. These studies again strongly suggest that the neuroprotective effects of the compound are not due to a reduction in tau aggregation [139]. Taken together, the results of these and other studies suggest that the reduction of tau fibril formation in vitro by certain small molecules may not be related to their therapeutic effects in vivo.
Microtubule stabilizing agents
Given that, in AD brain, tau is hyperphosphorylated and less functional as a microtubule stabilizer, therapies that compensate for this loss of tau function and stabilize microtubules are being considered. A reduction in microtubule integrity has been proposed to be an important factor in synaptic dysfunction [140]. In brain slice preparations a derivative of the microtubule stabilizing agent taxol protected against synaptic loss in response to lysosomal stress [140]. These findings are of interest, but unfortunately these taxol-based derivatives often show low blood–brain barrier permeability [141]. However, intranasal administration of an octapeptide (NAP), which stabilizes microtubules, in an AD mouse model reduced both tau and Aβ pathology indicating that it has therapeutic potential [142]. In addition, brain penetrant microtubule-stabilizing compounds have been identified which may allow for an in vivo analysis of the efficacy of this type of approach in the treatment of AD in appropriate animal models [141].
Tau immunotherapy
Although anti-Aβ immunotherapies yielded very promising results in AD animal models, results from clinical trials have been mixed. For example, Aβ levels and plaque loads can be reduced by Aβ immunotherapies, but improvements in cognition and disease progression have not been substantial [143]. Clinical trials using Aβ vaccines (such as Bapineuzumab; a humanized monoclonal antibody to Aβ) are still ongoing at this time; however, the results thus far suggest that other targets may need to be considered.
Over the past several years, there has been a growing interest in immunotherapies targeted at reducing tau levels as a strategy for treating AD [144]. One of the possible immunotherapeutic approaches being considered is the selective reduction of pathological forms of tau. Asuni and colleagues demonstrated that immunization of mice expressing P301L-tau (JNPL3 mice) with a small phospho-tau peptide (amino acids 379–408 with S396/S404 phosphorylated) resulted in the production of antibodies that entered the brain, which reduced the extent and slowed the progression of the behavioural phenotype [145]. This group recently extended these studies by creating a new mouse model with accelerated development of tau pathology and immunizing with the same phospho-tau peptide. Using this model they demonstrated that immunization with this tau peptide resulted in a reduction in soluble and insoluble tau phosphorylated at S396/S404 and a significant attenuation of cognitive impairment [146]. A study from another group also demonstrated that immunization of other tauopathy mouse models with phospho-tau peptides also reduced tau pathology without significant side effects [147]. However, an earlier study found that immunization of mice with full length recombinant tau resulted in neurological deficits with neuropathology [148]. Given this finding and the fact that an active immunization protocol with Aβ resulted in an adverse response in a number of the human cases [149], it would seem that a passive immunization protocol may be a better therapeutic approach. In addition, the optimal AD relevant form of tau to be targeted for immunotherapy needs to be considered. Although the studies cited above focused on phospho-tau, there is a growing awareness that oligomeric tau forms may be the more toxic species, and therefore would make an appropriate target for tau immunotherapy [144]. In a recent study, relatively pure, homogenous populations of neurotoxic tau oligomers were prepared [150], thus providing a potential immunogen for the generation of antibodies to prefilament forms of tau.
Finally, there is a growing awareness that pathological forms of tau, as well as other disease relevant proteins such as α-synuclein (Parkinson’s disease) and mutant huntingtin (Huntington’s disease) may be capable of being spread from cell to cell and thus facilitate disease progression [151–154]. In addition, extracellular tau may also induce toxicity. Therefore, it has been proposed that extracellular tau may be a potential target for immunotherapy in the future [155].
Autophagy activators
Reducing the burden of aggregated proteins through up-regulation of autophagy has been proposed as a potential therapeutic approach for various neurodegenerative diseases [156]. Numerous studies have focused on the use of rapamycin, a well characterized clinically approved drug that up-regulates autophagy through inhibition of the central regulatory protein mammalian target of rapamycin (mTOR). In cell models, rapamycin mediates the reduction of aggregates composed of both polyglutamine and polyalanine expansions [157, 158]. In Drosophila models expressing wild-type or mutant forms of tau, rapamycin reduced toxicity and it was suggested that this was due to a reduction in insoluble tau [158]. Rapamycin also reduced both Aβ and tau pathology in the 3xTg AD mouse model, though it is unclear in this study if reductions of tau pathology occur as a direct result of rapamycin treatment, or as a downstream effect of Aβ reduction [159].
Though the use of rapamycin as an autophagic-inducing agent has shown promise in effecting the clearance of insoluble and pathologically modified tau in cell and animal models, questions about potential pharmacological side effects remain. mTOR is a central signalling molecule that regulates the cell’s responses to nutritional and environmental stress. Many cellular processes besides autophagy are regulated by mTOR, including protein transcription and translation. The activity of mTOR is modulated by a variety of inputs such as varied nutrient conditions in the cell’s microenvironment, cell stress and presence or absence of growth factors [160]. As such, in addition to up-regulating autophagy, inhibition of mTOR by rapamycin can also inhibit protein synthesis. It has been suggested that inhibition of protein translation may in fact be a reason for rapamycin’s observed clearance of mutant huntingtin aggregates; in this study, the authors note Atg5-independent clearance of mutant huntingtin alongside global suppression of protein translation [161]. In addition to this finding, it has also been suggested that in isolated neurons, rapamycin is ineffective in promoting the conversion of LC3-I to LC3-II, a key indicator of autophagy up-regulation [162].
The myriad signalling responsibilities of mTOR, and the above-mentioned concerns over rapamycin’s efficacy towards autophagy up-regulation in neurons have sparked searches for new specific autophagy inducing factors. A small molecule screen carried out to determine new effectors of autophagy identified multiple mTOR-independent candidates, some of which were effective in promoting both the clearance of huntingtin aggregates as well as decreasing levels of mutant α-synuclein in mammalian cell and Drosophila models of protein aggregation [163]. In addition, the disaccharide trehalose (found in multiple non-mammalian organisms) has been shown to reduce both mutant huntingtin aggregate burden and accumulation of mutant α-synuclein in an mTOR-independent manner [164]. Trehalose also promoted the clearance of phosphorylated and aggregated tau in a mouse model that was both parkin-null and expressed tau with multiple FTDP-17 mutations [165]. Concurrent with decreases in tau pathology, the study described improvements in behavioural deficits, as well as decreases in both astrogliosis and dopaminergic neuron death [165]. The findings of the above studies are quite promising, though further study of additional pharmacological effects of mTOR-independent autophagy is needed.
Mitochondria targeted therapies
Given that one of the primary targets of pathological forms of tau may be the mitochondria, therapies that increase mitochondrial function in concert with therapies that target tau may be a reasonable strategy. Indeed, mitochondria have been increasingly recognized as a beneficial therapeutic target in neurodegenerative diseases, including AD [166]. Many proposed therapies target mitochondrial ROS production and cellular oxidative stress which result from malfunctioning mitochondria and compromise cell viability.
Due to the well-documented oxidative damage associated with neurodegenerative diseases, antioxidant molecules, such as vitamins C and E, glutathione, Coenzyme Q10 (ubiquinone), and a mitochondrial targeted derivative of ubiquinone, MitoQ, have been popular therapeutic candidates. Vitamins C and E show neuroprotective effects in AD mouse models including decreased Aβ plaque load and enhanced cognitive function [167, 168]. However, conflicting results exist as to the efficacy of these antioxidants in AD patients, which in part may be due to differences in study design and the characteristics of the subjects [169]. Ubiquinone is an important component of the electron transport chain that can act as a potent lipid oxidant scavenger. Therapeutic ubiquinone has shown positive results in animal models of AD and Parkinson’s disease [170, 171] and appears to show some efficacy in clinical trials with Parkinson’s disease [172, 173] and Huntington’s disease [174, 175] patients. MitoQ is a lipophilic cation analogue of ubiquinone, which accumulates specifically in mitochondria in a mitochondrial membrane potential dependent manner and can be recycled by the electron transport chain [176]. Currently, MitoQ clinical trials for Parkinson’s disease have yielded encouraging results suggesting that MitoQ may prove to be a useful therapeutic molecule [177]. However, using these molecules as therapies has several drawbacks such as poor translocation across the blood–brain barrier and the non-universal contribution of these oxidants to cellular oxidative damage. Therefore, treating with a single antioxidant may be beneficial but will not abate all oxidative stress [166].
Summary and conclusions
Studies over the past decade have brought about a maturation of our understanding of the role of tau in AD pathogenesis. These discoveries have breathed new life into a number of encouraging avenues for future tau research including: (1) investigation of soluble, non-aggregated forms of tau as a primary disease agent, (2) exploring the role of tau as an enhancer, or even gatekeeper, of Aβ-induced degeneration, (3) elucidating the mechanisms/pathways regulating the degradation of tau as determined by its post-translational state and (4) examining the mechanisms by which pathological forms of tau may negatively impact mitochondrial biology. Going forward, studies focused in these areas as well as further mechanistic dissection of the contribution of different tau isoforms in the disease process, will provide us with foundational information for the development of therapies for the treatment of AD. Figure 2 provides a summary of possible tau-directed therapeutic strategies.
Fig 2.
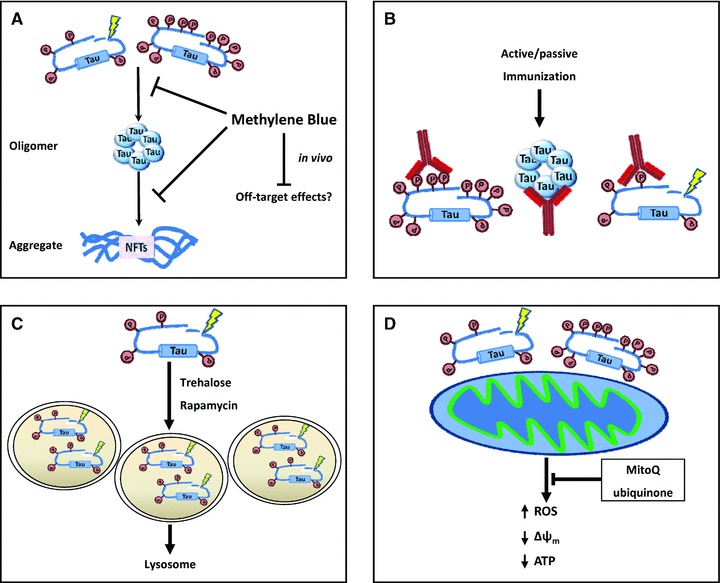
Treatment strategies to reduce the impact of tau pathology. (A) In vitro, the formation of tau oligomers and aggregates is disrupted by the use of phenothiazine derivatives such as methylene blue. This results in a reduction of phosphorylated and total tau levels, though in vivo the effect of methylene blue treatment on the formation of aggregates is debated (see text). (B) Active immunization of tau in in vivo models has been shown to improve behavioural measures and reduce levels of phosphorylated and total tau. (C) Activators of autophagy have been shown to increase the degradation of pathological forms of tau, and improve pathological measures in mouse models of AD. (D) The detrimental effects of expressing pathological forms of tau on mitochondrial function may be slowed or reversed with antioxidants such as mitoQ, ubiquinone and other antioxidants.
Although alterations in tau are likely a significant component of AD pathogenesis, it is unlikely that a monotherapeutic approach will be highly efficacious in slowing or halting disease progression given the complex nature of the disease. Therefore, combining therapies is likely a strategy that needs to be pursued. Conceptually, combinatorial approaches for treatment of AD are already being used (e.g. treating with both cholinesterase inhibitors and NMDA antagonists) and more are being considered [178–180]. Overall, it is clear that understanding the role of tau in AD pathogenesis is fundamental to the development of successful therapeutic strategies for the treatment of AD.
Acknowledgments
The work cited from the authors’ lab was supported by NIH grants NS051279 and NS041744 and a grant from the Alzheimer’s Association.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Weingarten MD, Lockwood AH, Hwo SY, et al. A protein factor essential for microtubule assembly. Proc Natl Acad Sci USA. 1975;72:1858–62. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cleveland DW, Hwo SY, Kirschner MW. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J Mol Biol. 1977;116:227–47. doi: 10.1016/0022-2836(77)90214-5. [DOI] [PubMed] [Google Scholar]
- 3.Lindwall G, Cole RD. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem. 1984;259:5301–5. [PubMed] [Google Scholar]
- 4.Grundke-Iqbal I, Iqbal K, Quinlan M, et al. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem. 1986;261:6084–9. [PubMed] [Google Scholar]
- 5.Grundke-Iqbal I, Iqbal K, Tung YC, et al. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. 1986;83:4913–7. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kosik KS, Joachim CL, Selkoe DJ. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci USA. 1986;83:4044–8. doi: 10.1073/pnas.83.11.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee VM, Balin BJ, Otvos L, Jr, et al. A68: a major subunit of paired helical filaments and derivatized forms of normal tau. Science. 1991;251:675–8. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- 8.Ihara Y, Nukina N, Miura R, et al. Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer’s disease. J Biochem. 1986;99:1807–10. doi: 10.1093/oxfordjournals.jbchem.a135662. [DOI] [PubMed] [Google Scholar]
- 9.Lee G, Cowan N, Kirschner M. The primary structure and heterogeneity of tau protein from mouse brain. Science. 1988;239:285–8. doi: 10.1126/science.3122323. [DOI] [PubMed] [Google Scholar]
- 10.Goedert M, Spillantini MG, Potier MC, et al. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 1989;8:393–9. doi: 10.1002/j.1460-2075.1989.tb03390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Himmler A. Structure of the bovine tau gene: alternatively spliced transcripts generate a protein family. Mol Cell Biol. 1989;9:1389–96. doi: 10.1128/mcb.9.4.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stoothoff WH, Johnson GV. Tau phosphorylation: physiological and pathological consequences. Biochim Biophys Acta. 2005;1739:280–97. doi: 10.1016/j.bbadis.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 13.Goate A, Chartier-Harlin MC, Mullan M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349:704–6. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 14.Levy-Lahad E, Wasco W, Poorkaj P, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269:973–7. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 15.Rogaev EI, Sherrington R, Rogaeva EA, et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature. 1995;376:775–8. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 16.Sherrington R, Rogaev EI, Liang Y, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375:754–60. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 17.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 18.Clark LN, Poorkaj P, Wszolek Z, et al. Pathogenic implications of mutations in the tau gene in pallido-ponto-nigral degeneration and related neurodegenerative disorders linked to chromosome 17. Proc Natl Acad Sci USA. 1998;95:13103–7. doi: 10.1073/pnas.95.22.13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–5. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 20.Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–59. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 21.Rapoport M, Dawson HN, Binder LI, et al. Tau is essential to beta -amyloid-induced neurotoxicity. Proc Natl Acad Sci USA. 2002;99:6364–9. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Santacruz K, Lewis J, Spires T, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–81. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roberson ED, Scearce-Levie K, Palop JJ, et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316:750–4. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 24.Vossel KA, Zhang K, Brodbeck J, et al. Tau reduction prevents A{beta}-induced defects in axonal transport. Science. 2010 doi: 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rhein V, Song X, Wiesner A, et al. Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc Natl Acad Sci USA. 2009;106:20057–62. doi: 10.1073/pnas.0905529106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Small SA, Duff K. Linking Abeta and tau in late-onset Alzheimer’s disease: a dual pathway hypothesis. Neuron. 2008;60:534–42. doi: 10.1016/j.neuron.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alonso AC, Li B, Grundke-Iqbal I, et al. Mechanism of tau-induced neurodegeneration in Alzheimer disease and related tauopathies. Curr Alzheimer Res. 2008;5:375–84. doi: 10.2174/156720508785132307. [DOI] [PubMed] [Google Scholar]
- 28.Reynolds MR, Berry RW, Binder LI. Nitration in neurodegeneration: deciphering the “Hows”“nYs”. Biochemistry. 2007;46:7325–36. doi: 10.1021/bi700430y. [DOI] [PubMed] [Google Scholar]
- 29.Chun W, Johnson GV. The role of tau phosphorylation and cleavage in neuronal cell death. Front Biosci. 2007;12:733–56. doi: 10.2741/2097. [DOI] [PubMed] [Google Scholar]
- 30.Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. 2009;15:112–9. doi: 10.1016/j.molmed.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 31.Iqbal K, Grundke-Iqbal I. Alzheimer neurofibrillary degeneration: significance, etiopathogenesis, therapeutics and prevention. J Cell Mol Med. 2008;12:38–55. doi: 10.1111/j.1582-4934.2008.00225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mi K, Johnson GV. The role of tau phosphorylation in the pathogenesis of Alzheimer’s disease. Curr Alzheimer Res. 2006;3:449–63. doi: 10.2174/156720506779025279. [DOI] [PubMed] [Google Scholar]
- 33.Dolan PJ, Johnson GV. The role of tau kinases in Alzheimer’s disease. Curr Opin Drug Discov Devel. 13:595–603. [PMC free article] [PubMed] [Google Scholar]
- 34.Matsuo ES, Shin RW, Billingsley ML, et al. Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer’s disease paired helical filament tau. Neuron. 1994;13:989–1002. doi: 10.1016/0896-6273(94)90264-x. [DOI] [PubMed] [Google Scholar]
- 35.Augustinack JC, Schneider A, Mandelkow EM, et al. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol. 2002;103:26–35. doi: 10.1007/s004010100423. [DOI] [PubMed] [Google Scholar]
- 36.Luna-Munoz J, Chavez-Macias L, Garcia-Sierra F, et al. Earliest stages of tau conformational changes are related to the appearance of a sequence of specific phospho-dependent tau epitopes in Alzheimer’s disease. J Alzheimers Dis. 2007;12:365–75. doi: 10.3233/jad-2007-12410. [DOI] [PubMed] [Google Scholar]
- 37.Luna-Munoz J, Garcia-Sierra F, Falcon V, et al. Regional conformational change involving phosphorylation of tau protein at the Thr231, precedes the structural change detected by Alz-50 antibody in Alzheimer’s disease. J Alzheimers Dis. 2005;8:29–41. doi: 10.3233/jad-2005-8104. [DOI] [PubMed] [Google Scholar]
- 38.Cho JH, Johnson GV. Primed phosphorylation of tau at Thr231 by glycogen synthase kinase 3beta (GSK3beta) plays a critical role in regulating tau’s ability to bind and stabilize microtubules. J Neurochem. 2004;88:349–58. doi: 10.1111/j.1471-4159.2004.02155.x. [DOI] [PubMed] [Google Scholar]
- 39.Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase 3 (GSK3) Trends Biol Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 40.Balastik M, Lim J, Pastorino L, et al. Pin1 in Alzheimer’s disease: multiple substrates, one regulatory mechanism. Biochim Biophys Acta. 2007;1772:422–9. doi: 10.1016/j.bbadis.2007.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liou YC, Sun A, Ryo A, et al. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature. 2003;424:556–61. doi: 10.1038/nature01832. [DOI] [PubMed] [Google Scholar]
- 42.Drewes G, Trinczek B, Illenberger S, et al. Microtubule-associated protein/ microtubule affinity-regulating kinase (p110mark). A novel protein kinase that regulates tau-microtubule interactions and dynamic instability by phosphorylation at the Alzheimer-specific site serine 262. J Biol Chem. 1995;270:7679–88. doi: 10.1074/jbc.270.13.7679. [DOI] [PubMed] [Google Scholar]
- 43.Biernat J, Gustke N, Drewes G, et al. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron. 1993;11:153–63. doi: 10.1016/0896-6273(93)90279-z. [DOI] [PubMed] [Google Scholar]
- 44.Fischer D, Mukrasch MD, Biernat J, et al. Conformational changes specific for pseudophosphorylation at serine 262 selectively impair binding of tau to microtubules. Biochemistry. 2009;48:10047–55. doi: 10.1021/bi901090m. [DOI] [PubMed] [Google Scholar]
- 45.Chatterjee S, Sang TK, Lawless GM, et al. Dissociation of tau toxicity and phosphorylation: role of GSK-3beta, MARK and Cdk5 in a Drosophila model. Hum Mol Genet. 2009;18:164–77. doi: 10.1093/hmg/ddn326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iijima K, Gatt A, Iijima-Ando K. Tau Ser262 phosphorylation is critical for Abeta42-induced tau toxicity in a transgenic Drosophila model of Alzheimer’s disease. Hum Mol Genet. 2010;19:2947–57. doi: 10.1093/hmg/ddq200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ding H, Matthews TA, Johnson GV. Site-specific phosphorylation and caspase cleavage differentially impact tau-microtubule interactions and tau aggregation. J Biol Chem. 2006;281:19107–14. doi: 10.1074/jbc.M511697200. [DOI] [PubMed] [Google Scholar]
- 48.Steinhilb ML, Dias-Santagata D, Fulga TA, et al. Tau phosphorylation sites work in concert to promote neurotoxicity in vivo. Mol Biol Cell. 2007;18:5060–8. doi: 10.1091/mbc.E07-04-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alonso AD, Di Clerico J, Li B, et al. Phosphorylation of tau at Thr212, Thr231, and Ser262 combined causes neurodegeneration. J Biol Chem. 2010;285:30851–60. doi: 10.1074/jbc.M110.110957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dias-Santagata D, Fulga TA, Duttaroy A, et al. Oxidative stress mediates tau-induced neurodegeneration in Drosophila. J Clin Invest. 2007;117:236–45. doi: 10.1172/JCI28769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Steinhilb ML, Dias-Santagata D, Mulkearns EE, et al. S/P and T/P phosphorylation is critical for tau neurotoxicity in Drosophila. J Neurosci Res. 2007;85:1271–8. doi: 10.1002/jnr.21232. [DOI] [PubMed] [Google Scholar]
- 52.Basurto-Islas G, Luna-Munoz J, Guillozet-Bongaarts AL, et al. Accumulation of aspartic acid421- and glutamic acid391-cleaved tau in neurofibrillary tangles correlates with progression in Alzheimer disease. J Neuropathol Exp Neurol. 2008;67:470–83. doi: 10.1097/NEN.0b013e31817275c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rohn TT, Head E, Nesse WH, et al. Activation of caspase-8 in the Alzheimer’s disease brain. Neurobiol Dis. 2001;8:1006–16. doi: 10.1006/nbdi.2001.0449. [DOI] [PubMed] [Google Scholar]
- 54.Rohn TT, Rissman RA, Davis MC, et al. Caspase-9 activation and caspase cleavage of tau in the Alzheimer’s disease brain. Neurobiol Dis. 2002;11:341–54. doi: 10.1006/nbdi.2002.0549. [DOI] [PubMed] [Google Scholar]
- 55.Stadelmann C, Deckwerth TL, Srinivasan A, et al. Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer’s disease. Evidence for apoptotic cell death. Am J Pathol. 1999;155:1459–66. doi: 10.1016/S0002-9440(10)65460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gamblin TC, Chen F, Zambrano A, et al. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc Natl Acad Sci USA. 2003;100:10032–7. doi: 10.1073/pnas.1630428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rissman RA, Poon WW, Blurton-Jones M, et al. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest. 2004;114:121–30. doi: 10.1172/JCI20640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chung CW, Song YH, Kim IK, et al. Proapoptotic effects of tau cleavage product generated by caspase-3. Neurobiol Dis. 2001;8:162–72. doi: 10.1006/nbdi.2000.0335. [DOI] [PubMed] [Google Scholar]
- 59.Fasulo L, Ugolini G, Cattaneo A. Apoptotic effect of caspase-3 cleaved tau in hippocampal neurons and its potentiation by tau FTDP-mutation N279K. J Alzheimers Dis. 2005;7:3–13. doi: 10.3233/jad-2005-7102. [DOI] [PubMed] [Google Scholar]
- 60.de Calignon A, Fox LM, Pitstick R, et al. Caspase activation precedes and leads to tangles. Nature. 2010;464:1201–4. doi: 10.1038/nature08890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li Z, Jo J, Jia JM, et al. Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell. 2010;141:859–71. doi: 10.1016/j.cell.2010.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McLaughlin B, Hartnett KA, Erhardt JA, et al. Caspase 3 activation is essential for neuroprotection in preconditioning. Proc Natl Acad Sci USA. 2003;100:715–20. doi: 10.1073/pnas.0232966100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schwerk C, Schulze-Osthoff K. Non-apoptotic functions of caspases in cellular proliferation and differentiation. Biochem Pharmacol. 2003;66:1453–8. doi: 10.1016/s0006-2952(03)00497-0. [DOI] [PubMed] [Google Scholar]
- 64.Zeuner A, Eramo A, Peschle C, et al. Caspase activation without death. Cell Death Differ. 1999;6:1075–80. doi: 10.1038/sj.cdd.4400596. [DOI] [PubMed] [Google Scholar]
- 65.Quintanilla RA, Matthews-Roberson TA, Dolan PJ, et al. Caspase-cleaved tau expression induces mitochondrial dysfunction in immortalized cortical neurons: implications for the pathogenesis of Alzheimer disease. J Biol Chem. 2009;284:18754–66. doi: 10.1074/jbc.M808908200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Matthews-Roberson TA, Quintanilla RA, Ding H, et al. Immortalized cortical neurons expressing caspase-cleaved tau are sensitized to endoplasmic reticulum stress induced cell death. Brain Res. 2008;1234:206–12. doi: 10.1016/j.brainres.2008.07.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Khurana V, Elson-Schwab I, Fulga TA, et al. Lysosomal dysfunction promotes cleavage and neurotoxicity of tau in vivo. PLoS Genet. 2010;6:e1001026. doi: 10.1371/journal.pgen.1001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Albrecht S, Bourdeau M, Bennett D, et al. Activation of caspase-6 in aging and mild cognitive impairment. Am J Pathol. 2007;170:1200–9. doi: 10.2353/ajpath.2007.060974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guo H, Albrecht S, Bourdeau M, et al. Active caspase-6 and caspase-6-cleaved tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer’s disease. Am J Pathol. 2004;165:523–31. doi: 10.1016/S0002-9440(10)63317-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Horowitz PM, Patterson KR, Guillozet-Bongaarts AL, et al. Early N-terminal changes and caspase-6 cleavage of tau in Alzheimer’s disease. J Neurosci. 2004;24:7895–902. doi: 10.1523/JNEUROSCI.1988-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Binder LI, Guillozet-Bongaarts AL, Garcia-Sierra F, et al. Tau, tangles, and Alzheimer’s disease. Biochim Biophys Acta. 2005;1739:216–23. doi: 10.1016/j.bbadis.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 72.Khlistunova I, Biernat J, Wang Y, et al. Inducible expression of tau repeat domain in cell models of tauopathy: aggregation is toxic to cells but can be reversed by inhibitor drugs. J Biol Chem. 2006;281:1205–14. doi: 10.1074/jbc.M507753200. [DOI] [PubMed] [Google Scholar]
- 73.Wittmann CW, Wszolek MF, Shulman JM, et al. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001;293:711–4. doi: 10.1126/science.1062382. [DOI] [PubMed] [Google Scholar]
- 74.Le Corre S, Klafki HW, Plesnila N, et al. An inhibitor of tau hyperphosphorylation prevents severe motor impairments in tau transgenic mice. Proc Natl Acad Sci USA. 2006;103:9673–8. doi: 10.1073/pnas.0602913103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Arrasate M, Mitra S, Schweitzer ES, et al. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–10. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 76.Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–6. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- 77.Tanaka M, Kim YM, Lee G, et al. Aggresomes formed by alpha-synuclein and synphilin-1 are cytoprotective. J Biol Chem. 2004;279:4625–31. doi: 10.1074/jbc.M310994200. [DOI] [PubMed] [Google Scholar]
- 78.Taylor JP, Tanaka F, Robitschek J, et al. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum Mol Genet. 2003;12:749–57. doi: 10.1093/hmg/ddg074. [DOI] [PubMed] [Google Scholar]
- 79.Keller JN, Hanni KB, Markesbery WR. Impaired proteasome function in Alzheimer’s disease. J Neurochem. 2000;75:436–9. doi: 10.1046/j.1471-4159.2000.0750436.x. [DOI] [PubMed] [Google Scholar]
- 80.Pickford F, Masliah E, Britschgi M, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118:2190–9. doi: 10.1172/JCI33585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Boland B, Kumar A, Lee S, et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci. 2008;28:6926–37. doi: 10.1523/JNEUROSCI.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bancher C, Brunner C, Lassmann H, et al. Tau and ubiquitin immunoreactivity at different stages of formation of Alzheimer neurofibrillary tangles. Prog Clin Biol Res. 1989;317:837–48. [PubMed] [Google Scholar]
- 83.Hamano T, Gendron TF, Causevic E, et al. Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur J Neurosci. 2008;27:1119–30. doi: 10.1111/j.1460-9568.2008.06084.x. [DOI] [PubMed] [Google Scholar]
- 84.Keck S, Nitsch R, Grune T, et al. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J Neurochem. 2003;85:115–22. doi: 10.1046/j.1471-4159.2003.01642.x. [DOI] [PubMed] [Google Scholar]
- 85.Tseng BP, Green KN, Chan JL, et al. Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol Aging. 2008;29:1607–18. doi: 10.1016/j.neurobiolaging.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shimura H, Miura-Shimura Y, Kosik KS. Binding of tau to heat shock protein 27 leads to decreased concentration of hyperphosphorylated tau and enhanced cell survival. J Biol Chem. 2004;279:17957–62. doi: 10.1074/jbc.M400351200. [DOI] [PubMed] [Google Scholar]
- 87.Petrucelli L, Dickson D, Kehoe K, et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet. 2004;13:703–14. doi: 10.1093/hmg/ddh083. [DOI] [PubMed] [Google Scholar]
- 88.Hatakeyama S, Matsumoto M, Kamura T, et al. U-box protein carboxyl terminus of Hsc70-interacting protein (CHIP) mediates poly-ubiquitylation preferentially on four-repeat tau and is involved in neurodegeneration of tauopathy. J Neurochem. 2004;91:299–307. doi: 10.1111/j.1471-4159.2004.02713.x. [DOI] [PubMed] [Google Scholar]
- 89.Kim HT, Kim KP, Lledias F, et al. Certain pairs of ubiquitin-conjugating enzymes (E2s) and ubiquitin-protein ligases (E3s) synthesize nondegradable forked ubiquitin chains containing all possible isopeptide linkages. J Biol Chem. 2007;282:17375–86. doi: 10.1074/jbc.M609659200. [DOI] [PubMed] [Google Scholar]
- 90.Morishima Y, Wang AM, Yu Z, et al. CHIP deletion reveals functional redundancy of E3 ligases in promoting degradation of both signaling proteins and expanded glutamine proteins. Hum Mol Genet. 2008;17:3942–52. doi: 10.1093/hmg/ddn296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dolan PJ, Johnson GV. A caspase cleaved form of tau is preferentially degraded through the autophagy pathway. J Biol Chem. 2010;285:21978–87. doi: 10.1074/jbc.M110.110940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.David DC, Layfield R, Serpell L, et al. Proteasomal degradation of tau protein. J Neurochem. 2002;83:176–85. doi: 10.1046/j.1471-4159.2002.01137.x. [DOI] [PubMed] [Google Scholar]
- 93.Carrettiero DC, Hernandez I, Neveu P, et al. The cochaperone BAG2 sweeps paired helical filament- insoluble tau from the microtubule. J Neurosci. 2009;29:2151–61. doi: 10.1523/JNEUROSCI.4660-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Arnaud LT, Myeku N, Figueiredo-Pereira ME. Proteasome-caspase-cathepsin sequence leading to tau pathology induced by prostaglandin J2 in neuronal cells. J Neurochem. 2009;110:328–42. doi: 10.1111/j.1471-4159.2009.06142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang Y, Martinez-Vicente M, Kruger U, et al. Synergy and antagonism of macroautophagy and chaperone-mediated autophagy in a cell model of pathological tau aggregation. Autophagy. 2009;6:182–3. doi: 10.4161/auto.6.1.10815. [DOI] [PubMed] [Google Scholar]
- 96.Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11:1107–17. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- 97.Bauer PO, Nukina N. Enhanced degradation of mutant huntingtin by rho kinase inhibition is mediated through activation of proteasome and macroautophagy. Autophagy. 2009;5:747–8. doi: 10.4161/auto.5.5.8704. [DOI] [PubMed] [Google Scholar]
- 98.Webb JL, Ravikumar B, Atkins J, et al. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–13. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 99.Doss-Pepe EW, Stenroos ES, Johnson WG, et al. Ataxin-3 interactions with rad23 and valosin-containing protein and its associations with ubiquitin chains and the proteasome are consistent with a role in ubiquitin-mediated proteolysis. Mol Cell Biol. 2003;23:6469–83. doi: 10.1128/MCB.23.18.6469-6483.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Menzies FM, Huebener J, Renna M, et al. Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain. 2010;133:93–104. doi: 10.1093/brain/awp292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tan JM, Wong ES, Kirkpatrick DS, et al. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet. 2008;17:431–9. doi: 10.1093/hmg/ddm320. [DOI] [PubMed] [Google Scholar]
- 102.Babu JR, Geetha T, Wooten MW. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J Neurochem. 2005;94:192–203. doi: 10.1111/j.1471-4159.2005.03181.x. [DOI] [PubMed] [Google Scholar]
- 103.Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–45. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 104.Berry RW, Abraha A, Lagalwar S, et al. Inhibition of tau polymerization by its carboxy-terminal caspase cleavage fragment. Biochemistry. 2003;42:8325–31. doi: 10.1021/bi027348m. [DOI] [PubMed] [Google Scholar]
- 105.Williams A, Jahreiss L, Sarkar S, et al. Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr Top Dev Biol. 2006;76:89–101. doi: 10.1016/S0070-2153(06)76003-3. [DOI] [PubMed] [Google Scholar]
- 106.Ittner LM, Ke YD, Delerue F, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell. 2010;142:387–97. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 107.Gustke N, Steiner B, Mandelkow EM, et al. The Alzheimer-like phosphorylation of tau protein reduces microtubule binding and involves Ser-Pro and Thr-Pro motifs. FEBS Lett. 1992;307:199–205. doi: 10.1016/0014-5793(92)80767-b. [DOI] [PubMed] [Google Scholar]
- 108.LaPointe NE, Morfini G, Pigino G, et al. The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. J Neurosci Res. 2009;87:440–51. doi: 10.1002/jnr.21850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dubey M, Chaudhury P, Kabiru H, et al. Tau inhibits anterograde axonal transport and perturbs stability in growing axonal neurites in part by displacing kinesin cargo: neurofilaments attenuate tau-mediated neurite instability. Cell Motil Cytoskeleton. 2008;65:89–99. doi: 10.1002/cm.20243. [DOI] [PubMed] [Google Scholar]
- 110.Cuchillo-Ibanez I, Seereeram A, Byers HL, et al. Phosphorylation of tau regulates its axonal transport by controlling its binding to kinesin. FASEB J. 2008;22:3186–95. doi: 10.1096/fj.08-109181. [DOI] [PubMed] [Google Scholar]
- 111.Trinczek B, Ebneth A, Mandelkow EM, et al. Tau regulates the attachment/detachment but not the speed of motors in microtubule-dependent transport of single vesicles and organelles. J Cell Sci. 1999;112(Pt 14):2355–67. doi: 10.1242/jcs.112.14.2355. [DOI] [PubMed] [Google Scholar]
- 112.Vershinin M, Carter BC, Razafsky DS, et al. Multiple-motor based transport and its regulation by tau. Proc Natl Acad Sci USA. 2007;104:87–92. doi: 10.1073/pnas.0607919104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tatebayashi Y, Haque N, Tung YC, et al. Role of tau phosphorylation by glycogen synthase kinase-3beta in the regulation of organelle transport. J Cell Sci. 2004;117:1653–63. doi: 10.1242/jcs.01018. [DOI] [PubMed] [Google Scholar]
- 114.Ebneth A, Godemann R, Stamer K, et al. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer’s disease. J Cell Biol. 1998;143:777–94. doi: 10.1083/jcb.143.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Morel M, Authelet M, Dedecker R, et al. Glycogen synthase kinase-3beta and the p25 activator of cyclin dependent kinase 5 increase pausing of mitochondria in neurons. Neuroscience. 2010;167:1044–56. doi: 10.1016/j.neuroscience.2010.02.077. [DOI] [PubMed] [Google Scholar]
- 116.Ittner LM, Ke YD, Gotz J. Phosphorylated tau interacts with c-Jun N-terminal kinase-interacting protein 1 (JIP1) in Alzheimer disease. J Biol Chem. 2009;284:20909–16. doi: 10.1074/jbc.M109.014472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ittner LM, Fath T, Ke YD, et al. Parkinsonism and impaired axonal transport in a mouse model of frontotemporal dementia. Proc Natl Acad Sci USA. 2008;105:15997–6002. doi: 10.1073/pnas.0808084105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Baloyannis SJ. Mitochondrial alterations in Alzheimer’s disease. J Alzheimers Dis. 2006;9:119–26. doi: 10.3233/jad-2006-9204. [DOI] [PubMed] [Google Scholar]
- 119.Bubber P, Haroutunian V, Fisch G, et al. Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann Neurol. 2005;57:695–703. doi: 10.1002/ana.20474. [DOI] [PubMed] [Google Scholar]
- 120.Wang X, Su B, Lee HG, et al. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci. 2009;29:9090–103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Manczak M, Anekonda TS, Henson E, et al. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–49. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 122.Wang X, Su B, Siedlak SL, et al. Amyloid-{beta} overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci USA. 2008;105:19318–23. doi: 10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Singh P, Suman S, Chandna S, et al. Possible role of amyloid-beta, adenine nucleotide translocase and cyclophilin-D interaction in mitochondrial dysfunction of Alzheimer’s disease. Bioinformation. 2009;3:440–5. doi: 10.6026/97320630003440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yao J, Irwin RW, Zhao L, et al. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2009;106:14670–5. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hauptmann S, Scherping I, Drose S, et al. Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol Aging. 2009;30:1574–86. doi: 10.1016/j.neurobiolaging.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 126.Eckert A, Schulz KL, Rhein V, et al. Convergence of Amyloid-beta and tau pathologies on mitochondria in vivo. Mol Neurobiol. 2010;41:107–14. doi: 10.1007/s12035-010-8109-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chandrasekaran K, Hatanpaa K, Rapoport SI, et al. Decreased expression of nuclear and mitochondrial DNA-encoded genes of oxidative phosphorylation in association neocortex in Alzheimer disease. Brain Res Mol Brain Res. 1997;44:99–104. doi: 10.1016/s0169-328x(96)00191-x. [DOI] [PubMed] [Google Scholar]
- 128.Grady CL, Haxby JV, Horwitz B, et al. Longitudinal study of the early neuropsychological and cerebral metabolic changes in dementia of the Alzheimer type. J Clin Exp Neuropsychol. 1988;10:576–96. doi: 10.1080/01688638808402796. [DOI] [PubMed] [Google Scholar]
- 129.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 130.Grober E, Dickson D, Sliwinski MJ, et al. Memory and mental status correlates of modified Braak staging. Neurobiol Aging. 1999;20:573–9. doi: 10.1016/s0197-4580(99)00063-9. [DOI] [PubMed] [Google Scholar]
- 131.Gotz J, Ittner LM, Fandrich M, et al. Is tau aggregation toxic or protective: a sensible question in the absence of sensitive methods. J Alzheimers Dis. 2008;14:423–9. doi: 10.3233/jad-2008-14410. [DOI] [PubMed] [Google Scholar]
- 132.Bulic B, Pickhardt M, Schmidt B, et al. Development of tau aggregation inhibitors for Alzheimer’s disease. Angew Chem Int Ed Engl. 2009;48:1740–52. doi: 10.1002/anie.200802621. [DOI] [PubMed] [Google Scholar]
- 133.Mosnaim AD, Ranade VV, Wolf ME, et al. Phenothiazine molecule provides the basic chemical structure for various classes of pharmacotherapeutic agents. Am J Ther. 2006;13:261–73. doi: 10.1097/01.mjt.0000212897.20458.63. [DOI] [PubMed] [Google Scholar]
- 134.Wischik CM, Edwards PC, Lai RY, et al. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci USA. 1996;93:11213–8. doi: 10.1073/pnas.93.20.11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Taniguchi S, Suzuki N, Masuda M, et al. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J Biol Chem. 2005;280:7614–23. doi: 10.1074/jbc.M408714200. [DOI] [PubMed] [Google Scholar]
- 136.O’Leary JC, 3rd, Li Q, Marinec P, et al. Phenothiazine-mediated rescue of cognition in tau transgenic mice requires neuroprotection and reduced soluble tau burden. Mol Neurodegener. 2010;5:45. doi: 10.1186/1750-1326-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.van Bebber F, Paquet D, Hruscha A, et al. Methylene blue fails to inhibit tau and polyglutamine protein dependent toxicity in zebrafish. Neurobiol Dis. 2010;39:265–71. doi: 10.1016/j.nbd.2010.03.023. [DOI] [PubMed] [Google Scholar]
- 138.Hall GF, Lee S, Yao J. Neurofibrillary degeneration can be arrested in an in vivo cellular model of human tauopathy by application of a compound which inhibits tau filament formation in vitro. J Mol Neurosci. 2002;19:253–60. doi: 10.1385/jmn:19:3:251. [DOI] [PubMed] [Google Scholar]
- 139.Honson NS, Jensen JR, Abraha A, et al. Small-molecule mediated neuroprotection in an in situ model of tauopathy. Neurotox Res. 2009;15:274–83. doi: 10.1007/s12640-009-9028-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Butler D, Bendiske J, Michaelis ML, et al. Microtubule-stabilizing agent prevents protein accumulation-induced loss of synaptic markers. Eur J Pharmacol. 2007;562:20–7. doi: 10.1016/j.ejphar.2007.01.053. [DOI] [PubMed] [Google Scholar]
- 141.Brunden KR, Yao Y, Potuzak JS, et al. The characterization of microtubule-stabilizing drugs as possible therapeutic agents for Alzheimer’s disease and related tauopathies. Pharmacol Res. 2010 doi: 10.1016/j.phrs.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Matsuoka Y, Gray AJ, Hirata-Fukae C, et al. Intranasal NAP administration reduces accumulation of amyloid peptide and tau hyperphosphorylation in a transgenic mouse model of Alzheimer’s disease at early pathological stage. J Mol Neurosci. 2007;31:165–70. doi: 10.1385/jmn/31:02:165. [DOI] [PubMed] [Google Scholar]
- 143.von Bernhardi R. Immunotherapy in Alzheimer’s disease: where do we stand? Where should we go. J Alzheimers Dis. 2010;19:405–21. doi: 10.3233/JAD-2010-1248. [DOI] [PubMed] [Google Scholar]
- 144.Kayed R, Jackson GR. Prefilament tau species as potential targets for immunotherapy for Alzheimer disease and related disorders. Curr Opin Immunol. 2009;21:359–63. doi: 10.1016/j.coi.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 145.Asuni AA, Boutajangout A, Quartermain D, et al. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci. 2007;27:9115–29. doi: 10.1523/JNEUROSCI.2361-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau prevents cognitive decline in a new tangle mouse model. J Neurosci. 2010;30:16559–66. doi: 10.1523/JNEUROSCI.4363-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Boimel M, Grigoriadis N, Lourbopoulos A, et al. Efficacy and safety of immunization with phosphorylated tau against neurofibrillary tangles in mice. Exp Neurol. 2010;224:472–85. doi: 10.1016/j.expneurol.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 148.Rosenmann H, Grigoriadis N, Karussis D, et al. Tauopathy-like abnormalities and neurologic deficits in mice immunized with neuronal tau protein. Arch Neurol. 2006;63:1459–67. doi: 10.1001/archneur.63.10.1459. [DOI] [PubMed] [Google Scholar]
- 149.Brody DL, Holtzman DM. Active and passive immunotherapy for neurodegenerative disorders. Annu Rev Neurosci. 2008;31:175–93. doi: 10.1146/annurev.neuro.31.060407.125529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lasagna-Reeves CA, Castillo-Carranza DL, Guerrero-Muoz MJ, et al. Preparation and characterization of neurotoxic tau oligomers. Biochemistry. 2010;49:10039–41. doi: 10.1021/bi1016233. [DOI] [PubMed] [Google Scholar]
- 151.Clavaguera F, Bolmont T, Crowther RA, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–13. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Desplats P, Lee HJ, Bae EJ, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci USA. 2009;106:13010–5. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284:12845–52. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ren PH, Lauckner JE, Kachirskaia I, et al. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol. 2009;11:219–25. doi: 10.1038/ncb1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Avila J. Intracellular and extracellular tau. Front Neurosci. 2010;4:49. doi: 10.3389/fnins.2010.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Renna M, Jimenez-Sanchez M, Sarkar S, et al. Chemical inducers of autophagy that enhance the clearance of mutant proteins in neurodegenerative diseases. J Biol Chem. 2010;285:11061–7. doi: 10.1074/jbc.R109.072181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–95. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 158.Berger Z, Ravikumar B, Menzies FM, et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet. 2006;15:433–42. doi: 10.1093/hmg/ddi458. [DOI] [PubMed] [Google Scholar]
- 159.Caccamo A, Majumder S, Richardson A, et al. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and tau: effects on cognitive impairments. J Biol Chem. 2010;285:13107–20. doi: 10.1074/jbc.M110.100420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–84. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 161.King MA, Hands S, Hafiz F, et al. Rapamycin inhibits polyglutamine aggregation independently of autophagy by reducing protein synthesis. Mol Pharmacol. 2008;73:1052–63. doi: 10.1124/mol.107.043398. [DOI] [PubMed] [Google Scholar]
- 162.Tsvetkov AS, Mitra S, Finkbeiner S. Protein turnover differences between neurons and other cells. Autophagy. 2009;5 doi: 10.4161/auto.5.7.9291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sarkar S, Perlstein EO, Imarisio S, et al. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat Chem Biol. 2007;3:331–8. doi: 10.1038/nchembio883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sarkar S, Davies JE, Huang Z, et al. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2007;282:5641–52. doi: 10.1074/jbc.M609532200. [DOI] [PubMed] [Google Scholar]
- 165.Rodriguez-Navarro JA, Rodriguez L, Casarejos MJ, et al. Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol Dis. 2010;39:423–38. doi: 10.1016/j.nbd.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 166.Du H, Yan SS. Mitochondrial medicine for neurodegenerative diseases. Int J Biochem Cell Biol. 2010;42:560–72. doi: 10.1016/j.biocel.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Conte V, Uryu K, Fujimoto S, et al. Vitamin E reduces amyloidosis and improves cognitive function in Tg2576 mice following repetitive concussive brain injury. J Neurochem. 2004;90:758–64. doi: 10.1111/j.1471-4159.2004.02560.x. [DOI] [PubMed] [Google Scholar]
- 168.Harrison FE, Hosseini AH, McDonald MP, et al. Vitamin C reduces spatial learning deficits in middle-aged and very old APP/PSEN1 transgenic and wild-type mice. Pharmacol Biochem Behav. 2009;93:443–50. doi: 10.1016/j.pbb.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Boothby LA, Doering PL. Vitamin C and vitamin E for Alzheimer’s disease. Ann Pharmacother. 2005;39:2073–80. doi: 10.1345/aph.1E495. [DOI] [PubMed] [Google Scholar]
- 170.Beal MF, Matthews RT, Tieleman A, et al. Coenzyme Q10 attenuates the 1-methyl-4-phenyl-1,2,3,tetrahydropyridine (MPTP) induced loss of striatal dopamine and dopaminergic axons in aged mice. Brain Res. 1998;783:109–14. doi: 10.1016/s0006-8993(97)01192-x. [DOI] [PubMed] [Google Scholar]
- 171.Yang X, Yang Y, Li G, et al. Coenzyme Q10 attenuates beta-amyloid pathology in the aged transgenic mice with Alzheimer presenilin 1 mutation. J Mol Neurosci. 2008;34:165–71. doi: 10.1007/s12031-007-9033-7. [DOI] [PubMed] [Google Scholar]
- 172.Shults CW, Oakes D, Kieburtz K, et al. Effects of coenzyme Q10 in early Parkinson disease: evidence of slowing of the functional decline. Arch Neurol. 2002;59:1541–50. doi: 10.1001/archneur.59.10.1541. [DOI] [PubMed] [Google Scholar]
- 173.Chen JJ. Parkinson’s disease: health-related quality of life, economic cost, and implications of early treatment. Am J Manag Care. 2010;16:S87–93. [PubMed] [Google Scholar]
- 174.Ferrante KL, Shefner J, Zhang H, et al. Tolerance of high-dose (3,000 mg/day) coenzyme Q10 in ALS. Neurology. 2005;65:1834–6. doi: 10.1212/01.wnl.0000187070.35365.d7. [DOI] [PubMed] [Google Scholar]
- 175.Hyson HC, Kieburtz K, Shoulson I, et al. Safety and tolerability of high-dosage coenzyme Q10 in Huntington’s disease and healthy subjects. Mov Disord. 2010;25:1924–8. doi: 10.1002/mds.22408. [DOI] [PubMed] [Google Scholar]
- 176.James AM, Sharpley MS, Manas AR, et al. Interaction of the mitochondria-targeted antioxidant MitoQ with phospholipid bilayers and ubiquinone oxidoreductases. J Biol Chem. 2007;282:14708–18. doi: 10.1074/jbc.M611463200. [DOI] [PubMed] [Google Scholar]
- 177.Snow BJ, Rolfe FL, Lockhart MM, et al. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov Disord. 2010;25:1670–4. doi: 10.1002/mds.23148. [DOI] [PubMed] [Google Scholar]
- 178.Pratico D, Sung S. Lipid peroxidation and oxidative imbalance: early functional events in Alzheimer’s disease. J Alzheimers Dis. 2004;6:171–5. doi: 10.3233/jad-2004-6209. [DOI] [PubMed] [Google Scholar]
- 179.Schmitt B, Bernhardt T, Moeller HJ, et al. Combination therapy in Alzheimer’s disease: a review of current evidence. CNS Drugs. 2004;18:827–44. doi: 10.2165/00023210-200418130-00001. [DOI] [PubMed] [Google Scholar]
- 180.Sobow T. Combination treatments in Alzheimer’s disease: risks and benefits. Expert Rev Neurother. 2010;10:693–702. doi: 10.1586/ern.10.43. [DOI] [PubMed] [Google Scholar]