

Abstract
The transcription factor p73, a member of the p53 family of proteins, is involved in the regulation of cell cycle progression and apoptosis. Due to alternative promoters and carboxy-terminal splicing, the P73 gene gives rise to a range of different isoforms. Interestingly, a particular increase in expression of the TAp73α isoform has been reported in various tumours. In addition, TAp73α has been shown to inhibit Bax activation and mitochondrial dysfunctions and thereby to confer small cell lung carcinoma (SCLC) cells resistance to drug-induced apoptosis. However, the precise mechanism by which TAp73α exerts its pro-survival effect is yet unclear. Here we report that TAp73α, but not TAp73β, regulates the expression of inducible Hsp72/HSPA1A. Hsp72 proved to be required for the survival effects of TAp73α as antisense knockdown of Hsp72 resulted in an abolishment of the anti-apoptotic effect of TAp73α in SCLC cells upon Etoposide treatment. Importantly, depletion of Hsp72 allowed activation of Bax, loss of mitochondrial membrane potential and lysosomal membrane permeabilization in SCLC cells even in the presence of TAp73α. Finally, we revealed that TAp73β counteracts the anti-apoptotic effect of TAp73α by preventing Hsp72 induction. Our results thus provide additional evidence for the potential oncogenic role of TAp73α, and extend the understanding of the mechanism for its anti-apoptotic effect.
Keywords: p73α, hsp72, HSPA1A, apoptosis, mitochondria, Bax
Introduction
The transcription factor p73 is a multifunctional protein, able to induce cell cycle arrest, apoptosis and differentiation [1–3]. The P73 gene gives rise to a vast number of isoforms due to alternative splicing and use of alternative promoters. Isoforms generated by the two promoters are called TA (for transactivation) and ΔN, the latter lacking the amino-terminal TA domain [4]. In addition, alternative splicing generates at least seven transcripts with different carboxy-terminals (α-η). However, TAp73α and TAp73β are the two main p73 isoforms expressed in human cells. TAp73α possess the longest carboxy-terminal extension and is the only TAp73 isoform to contain a sterile α motif (SAM) domain. TAp73β is shorter and lacks the extreme carboxy-terminal and SAM domain found in TAp73α[1, 5]. The ΔNp73 isoforms are regarded as potential oncogenes, capable of counteracting the effects of TAp73 isoforms and p53 [6]. In contrast to the P53 gene, frequently mutated or deleted in cancer, the P73 gene is very rarely found mutated in tumours. Instead, expression of p73 is greater in a range of tumours compared with normal tissues [7, 8]. Particularly an increased expression of the TAp73α isoform has been reported in medulloblastoma [9], B-cell chronic lymphocytic leukaemia [10], ovarian carcinomas [11], gastric adenocarcinoma [12], bladder cancer [13] and thyroid cancer [14]. Moreover, TAp73α was found to promote tumour resistance in response to treatment with DNA-damaging agents in an ovarian cancer cell line [15]. In line with these data, we previously reported that TAp73α inhibits drug-induced apoptosis in small cell lung carcinoma (SCLC) cells. The anti-apoptotic effect is exerted upstream of the mitochondria, at the level of Bax activation [16]. Interestingly, a protein known to inhibit Bax activation is the inducible heat shock protein HSPA1A/Hsp72/Hsp70–1 (referred to later as Hsp72) [17]. Hsp72 has also been shown to stabilize lysosomal membranes [18] and prevent other apoptotic events such as loss of mitochondrial membrane potential (ΔΨm) [19], and subsequent release of pro-apoptotic mitochondrial proteins [19], formation of the apoptosome complex [19–21], and caspase activation [19, 22]. ΔNp73α can via the activation of heat shock factor-1 (HSF-1) cause up-regulation of HSF-responsive genes, one major target gene being HSPA1A/HSP72 (referred to later as HSP72) [23]. This inspired us to investigate whether Hsp72 could account for the anti-apoptotic effects of TAp73α.
In the present study we reveal that TAp73α, but not TAp73β, is able to regulate the expression of Hsp72. Moreover, the anti-apoptotic effect of TAp73α in SCLC cells treated with Etoposide (VP16) was lost upon depletion of Hsp72 using antisense construct. Upon VP16 treatment, Hsp72 depletion allowed for Bax activation, loss of ΔΨm and lysosomal membrane permeabilization (LMP) to occur even in the presence of TAp73α. Meanwhile, the pro-apoptotic effects of TAp73β can be prevented by simultaneous co-expression with Hsp72. Finally, in line with other studies showing that the p53 family members interact and regulate the activity of each other, we found that the Hsp72-mediated anti-apoptotic effect of TAp73α can be counteracted by TAp73β. Thus, in addition to reported protective effects and possible tumorigenic functions of the TAp73α isoform, our findings uncover a potential mechanism that mediates its anti-apoptotic function.
Materials and methods
Cell culture, transfection and treatments
Human SCLC National Cancer Institute (NCI)-H82 [American Type Culture Collection (ATCC HTB-175)] and human embryonic kidney HEK-293 (ATCC CRL-1573) cell lines were used in this study. Cells were cultured at 37°C, 5% CO2 in Roswell Park Memorial Institute 1640 medium (NCI-H82), or Dulbecco’s modified Eagle’s medium (HEK-293), supplemented with 10% heat-inactivated foetal calf serum, 2 mM L-glutamine, penicillin (100 units/ml) and streptomycin (100 mg/ml). Twenty-four hours after setting in culture dishes with fresh medium, cells were transfected using Lipofectamine 2000 (Invitrogen Corporation, Carlsbad, CA, USA) according to the manufacturer’s protocol. The cell density was kept at levels allowing exponential growth.
Reagents and antibodies
Etoposide (VP16) was obtained from Bristol-Myers (New York, NY, USA), Hoechst 33342 and tetramethylrhodamine ethyl esters (TMRE) were purchased from Invitrogen Corporation (Carlsbad, CA, USA). Following primary antibodies were used in this study; rabbit polyclonal anti-Hsp72 antibody (BioSite Incorporated, San Diego, CA, USA), mouse monoclonal anti-active Bax 6A7 (BD Biosciences, San Jose, CA, USA), mouse monoclonal anti-p73α/β (Ab-4) antibody (Thermo Fisher Scientific, Runcorn, Cheshire, UK), rabbit polyclonal anti-glyceraldehyde-3-phosphate dehydrogenase (G3PDH) antibody (Trevigen, Gaithersburg, MD, USA), rabbit anti-HSF1 antibody, rabbit anti-Hsp90 antibody, mouse monoclonal anti-Hsp27 antibody and rabbit anti-Hsc70 antibody (all from Cell Signaling Technology, Inc., Danvers, MA, USA). For immunoblot experiments horseradish peroxidase-conjugated (HRP) goat anti-rabbit or goat antimouse IgG (Pierce Biotechnology, Rockford, IL, USA) were used. In immunofluorescence and fluorescence activated cell sorting (FACS) experiments secondary Alexa Fluor 594-conjugated goat anti-rabbit IgG and goat antimouse IgG (Invitrogen Corporation) were used.
Plasmids and small interfering RNAs
Expression vectors encoding human p73 isoforms (TAp73α, TAp73β, ΔNp73α) were gifts from Dr. G. Melino and have been described [24]. The expression level of each p73 variant used in this study was examined using immunoblotting. At protein level there was no significant difference in their expression. Dr. T. Perlmann kindly provided us with plasmid encoding β-galactosidase (pCMX-βgal). The promoter-luciferase construct HSP72-luc, containing 0.8 kb of the HSP70.1(HSPA1A) gene, was a gift from Dr. N. Mivechi and has been described [25]. Enhanced green fluorescent protein (EGFP) plasmid was from Clontech Laboratories, Inc. (Mountain View, CA, USA). Hsp72 expression plasmid and antisense Hsp72 vector (asHsp72), both cloned into pCDNA3 vectors, were gifts from Dr. M. Jäättelä and has been described [26]. Non-targeting control and HSP72/HSPA1A targeting small interfering RNAs (siRNAs) (ON-TARGETplus SMART pool L-005168, Human HSPA1A, nM-005345) were obtained from Fisher Scientific UK Ltd. (Loughborough, Leicestershire, UK).
Reporter gene assays
Transfections were performed in 24-well plates with Lipofectamine 2000, according to the manufacturer’s protocol. Each well was transfected with 100 ng of HSP72-luc reporter plasmid, 300 ng of empty (control) vector or p73 expression vector and 100 ng of pCMX-β-gal reference plasmid containing a bacterial β-galactosidase gene. Cells were harvested 24 hrs after transfection, lysed and extracts were assayed for luciferase and β-galactosidase activities in a microplate luminometer/photometer reader (Orion Microplate Luminometer; Berthold detection systems; Berthold Technologies, Oak Ridge, TN, USA).
RNA isolation and PCR analysis
Twenty-four hours after transfection with empty (control) vector or plasmids encoding p73 isoforms, cells were resuspended in RNA-Bee (BioSite Incorporated) and RNA isolated using chloroform extraction. For first-strand cDNA synthesis 1 μg of RNA was mixed with Oligo(dT) primers, dNTP mix, 5X first-strand buffer, Ditiotreitol, RNAseOUT and SuperScript II RT, all according to manufacturer’s protocol (Invitrogen Corporation). PCR reaction were performed on cDNA using the following primers for HSP72, forward; 5′-gtg cag tcg gac atg aag ca-3′ and reverse; 5′-cag gat gga cac gtc gaa gg-3′ (as described in [27]), P73, forward; 5′ -GTG GAA GGC AAT AAT CTC TCG CAG-3′ and reverse; 5′-G GTT GAC GGA GAG CAG CTT GTT CAT-3′, and G3PDH, forward; 5′-ATG GCC TTC CGT GTC CCC ACT G-3′ and reverse; 5′-TGA GTG TGG CAG GGA CTC CCC A-3′, at an annealing temperature of 55°C. PCR products were resolved on a 1% agarose gel and visualized using a UV transilluminator. Quantification of DNA band intensity was made using ImageJ software according to the supplier’s instructions.
Immunoblotting
Twenty-four hours after transfection, cells were lysed and resuspended in Laemmli’s loading buffer and boiled for 3 min. Samples were resolved on 10% SDS-PAGE and blotted onto nitrocellulose membranes (GE Healthcare, Uppsala, Sweden). Membranes were then probed with rabbit anti-Hsp72 antibody, rabbit anti-HSF1 antibody, rabbit anti-Hsp90 antibody, mouse anti-Hsp27 antibody, rabbit anti-Hsc70 antibody, mouse anti-p73 antibody and/or rabbit anti-G3PDH antibody. Primary antibody binding was detected using HRP-conjugated goat antimouse or goat anti-rabbit IgG (Pierce Biotechnology). After repeated washing in Tris-buffered saline, bands were visualized by enhanced chemiluminescence (ECL Plus) following the manufacturer’s instructions (GE Healthcare). Quantification of protein band intensity was made using ImageJ software.
Immunofluorescence and confocal imaging
One day after co-transfection of EGFP with either empty (control) vector or plasmids encoding p73 isoforms, Hsp72 and/or asHsp72, cells were harvested, fixed in 4% paraformaldehyde and blocked/permeabilized in phosphate-buffered solution (PBS) with 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 0.3% Triton X-100 and 3% bovine serum albumine (BSA). Slides were incubated with primary rabbit anti-Hsp72 antibody, mouse monoclonal anti-Hsp27 antibody or mouse anti-active Bax (6A7) antibody at 4°C, over night. Subsequently, slides were incubated with secondary Alexa Fluor 594-conjugated antibody (room temperature, 1 hr). Nuclei were counterstained with Hoechst (1 μg/ml), slides mounted in Vectashield H-1000 (Vector Laboratories, Inc., Orton Southgate, Peterborough, UK) and analysed under Zeiss 510 Meta confocal laser scanning microscopy equipped with an inverted Zeiss Axiovert 200M microscope (Carl Zeiss MicroImaging, Inc., Thornwood, NY, USA). Mix dyes were acquired by sequential multiple channel fluorescence scanning to avoid bleed through.
Detection of apoptosis
H82 cells were co-transfected with EGFP and plasmids encoding p73 isoforms, Hsp72, asHsp72 and/or empty (control) vector. One day after transfections, cells were treated with 5 μM VP16. At 24 hrs after treatment, cells were stained with Hoechst and scored in a fluorescence microscope as percentage of EGFP expressing cells displaying apoptotic nuclei. For each sample, at least 300 cells sorted for green (EGFP) fluorescence were assessed for nuclear morphology by two independent investigators.
Assessment of mitochondrial depolarization
H82 cells were co-transfected with EGFP and plasmids encoding p73 isoforms, Hsp72, asHsp72 and/or empty (control) vector. One day after transfections, cells were treated with 5 μM VP16. At 24 hrs after treatment, cells were stained with TMRE, a fluorochrome dye that is only taken up by mitochondria having an intact electrochemical gradient, and Hoechst and scored in a fluorescence microscope as percentage of EGFP expressing cells displaying loss of mitochondrial membrane potential, as depicted by a loss of TMRE staining. For each sample, at least 300 cells sorted for green (EGFP) fluorescence were assessed for TMRE staining by two independent investigators.
Assessment of lysosomal stability
To determine the effect of TAp73 and Hsp72 on lysosomal stability, H82 cells were co-transfected with EGFP and plasmids encoding p73 isoforms, Hsp72, asHsp72 and/or empty (control) vector, and treated with 5 μM VP16. At 24 hrs after treatment, cells were incubated with 50 nM LysoTracker Red (Invitrogen Corporation) for 30 min. at 37°C to label the lysosomes. Cells were visualized by fluorescence microscopy and scored as percentage of EGFP expressing cells displaying loss of lysosomal membrane potential, as depicted by a loss of LysoTracker Red staining.
Flow cytometric analysis of Bax activation
One day after co-transfection of EGFP and plasmids encoding p73 isoforms, Hsp72, asHsp72 and/or empty (control) vector, cells were treated with 5 μM VP16 for 24 hrs. H82 cells were subsequently harvested, fixed in 1% PFA and blocked/permeabilized in PBS with 10 mM HEPES, 0.3% Triton X-100 and 3% BSA. Cells were incubated with mouse anti-Bax (6A7) antibody (4°C over night), followed by secondary Alexa Fluor 594-conjugated antibody (room temperature for 30 min.). Cells were resuspended in PBS and samples were analysed using a FACSCalibur flow cytometer using Cell Quest software (BD Biosciences). For each sample, at least 10,000 cells sorted for green (EGFP) fluorescence were assessed for intensity of Bax staining.
Flow cytometric analysis of mitochondrial depolarization
H82 cells were co-transfected with EGFP and plasmids encoding p73 isoforms, Hsp72, asHsp72, and/or empty (control) vector, and treated with 5 μM VP16. At 24 hrs after treatment, cells were stained with TMRE for 30 min. at 37°C and then analysed on a FACSCalibur flow cytometer equipped with Cell Quest software (BD Biosciences). For each sample, at least 10,000 cells sorted for green (EGFP) fluorescence were assessed for TMRE staining.
Chromatin immunoprecipitation
H82 cells were transfected with empty (control) vector or plasmids encoding p73α, and p73β and immunoprecipitation were carried out using p73 antibody (Ab-4) and ChIP assay kit (Upstate; MilliPore Corporate Headquarters, Billerica, MA, USA) according to the manufacturer’s protocol. PCRs were performed with following primers: Hsp72 promoter (forward primer −935bp 5′-CAC CGC ACA TTC CTA GGC CGC-3′ and reverse primer −571bp 5′-GGA GCC CCC ACC TAC TCG CT-3′), G3PDH (forward primer 5′-ATG GCC TTC CGT GTC CCC ACT G-3′ and reverse primer 5′-TGA GTG TGG CAG GGA CTC CCC A-3′).
Statistical analysis
Statistical analyses were performed with two-tailed, paired Student’s t-test, where ***P < 0.001, **P < 0.01 and *P < 0.05.
Results
TAp73α induces Hsp72 expression
We have shown that full-length TAp73α represses drug-induced apoptosis in SCLC cells, whereas full-length TAp73β strengthens drug-induced apoptosis in the same settings [16]. In addition, the anti-apoptotic actions of TAp73α upon drug-induced apoptosis was shown to be exerted upstream of the mitochondria, on the level of Bax [16]. Previously, the capability of the ΔN forms of p73 and p63 (ΔNp73α and ΔNp63α) to regulate the expression of Hsp72 has been demonstrated [23, 28]. Moreover, similar to TAp73α, Hsp72 has been shown to exert many of its pro-survival functions upstream of the mitochondria [17]. To test whether the anti-apoptotic effect of TAp73α in SCLC cells could be due to a direct regulation of Hsp72 protein expression, we performed luciferase gene reporter assay on the HSP72 promoter in SCLC H82 and HEK-293 cells. Full-length TAp73α was able to transactivate the HSP72 promoter in both H82 and HEK-293 cells (Fig. 1A), as well as in HeLa, HCT116 p53+/+ and HCT116 p53−/– cells (data not shown). However, no transcriptional activity of the full-length TAp73β isoform on the HSP72 promoter could be seen in any of the cell lines tested. In contrast, the transcriptional activity of ΔNp73α on the HSP72 promoter demonstrated a cell-type specificity, being significantly active in HEK-293, HeLa, HCT116 p53+/+ and HCT116 p53–/– cells, but not in H82 cells (Fig. 1A, and data not shown). An additional set of experiments was performed to confirm the induction of Hsp72 by TAp73α on both mRNA and protein levels. Indeed, a significant induction in Hsp72 mRNA levels could be seen after TAp73α overexpression, but not upon expression of TAp73β (Fig. 1B). Moreover, transient expression of TAp73α, but not TAp73β, was able to significantly induce Hsp72 protein level, as shown in single cells by immunofluorescent staining of H82 cells (Fig. 1C) and by immunoblot on HEK-293 cell extracts (Fig. 1D). However, no effect of either TAp73α or TAp73β expression could be detected on the levels of the Hsp72-related proteins HSF1, Hsp90/HSPC1, Hsc70/HSPA8 or Hsp27/HSPB1 (Fig. 1E). As control, Hsp72 protein expression during VP16 treatment and up to 24 hrs is also depicted in Figure S1A. Thus, based on these data we conclude that TAp73α is a selective and potent inducer of Hsp72 expression.
Fig 1.
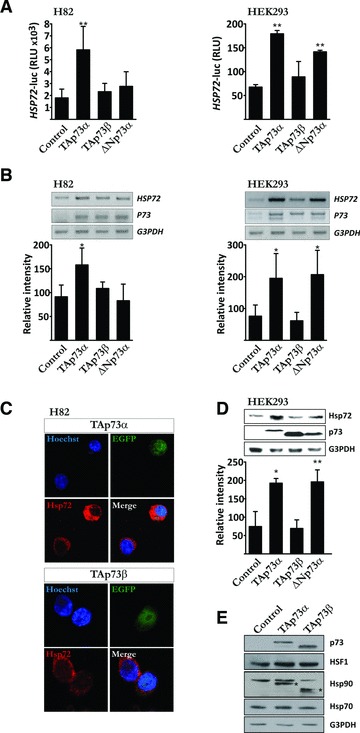
TAp73α induces Hsp72 expression. (A) H82 and HEK-293 cells were co-transfected with a HSP72 promoter-luciferase vector, a β-galactosidase reporter vector and empty vector or p73 expression vectors (TAp73α, TAp73β, ΔNp73α), as indicated. Cells were harvested after 24 hrs, and cell extracts assayed for luciferase and β-galactosidase activity. Relative luciferase units were compared after normalization to β-galactosidase activities. H82 (B) and HEK-293 (B, D) were transfected with expression vectors encoding TAp73α, TAp73β, ΔNp73α. (B) Total RNA was extracted from cells, followed by cDNA synthesis and PCR amplification of the indicated genes. Quantification of DNA band-intensity was made using ImageJ software. (C) H82 cells were co-transfected with EGFP and p73 expression vectors (TAp73α, TAp73β) (green). Samples were stained with anti-Hsp72 antibody (red) and nuclei counterstained with Hoechst (blue). Images are representatives of three independent experiments. (D) Total protein cell extracts were analysed by immunoblotting for the presence of Hsp72 and p73. G3PDH was used as protein loading control. (E) Total protein cell extracts were analysed by immunoblotting for the expression of p73, HSF1, Hsc70 and Hsp90 (bands marked * is due to previous staining with antibody against p73 and hence depict TAp73 protein). Data are represented as mean ± S.D. of at least three independent experiments, where *P < 0.05 and **P < 0.01.
Hsp72 is required for TAp73α anti-apoptotic effect
We suggested that since TAp73α is a potent inducer of Hsp72 expression, the anti-apoptotic effect of TAp73α might depend on the induction of Hsp72 protein. To investigate whether depletion of Hsp72 affects the anti-apoptotic activity of TAp73α we took advantage of an antisense Hsp72 vector (asHsp72). Transfection of cells with asHsp72 lead to a reduction in Hsp72 protein levels as shown both by Western blot (Fig. 2A) and confocal imaging (Fig. 2B). However, expression of the asHsp72 vector did not affect levels of Hsp72-related proteins HSF1, Hsp90, Hsc70 (as shown by Western blot, Fig. 2C) or Hsp27 (as shown by immunofluorescent staining, Fig. 2C). Subsequently, H82 cells were co-transfected with EGFP, p73 isoforms and asHsp72 vector, and treated with VP16 for 24 hrs. TAp73α and ΔNp73α are able to repress drug-induced apoptosis, whereas TAp73β enhances it (Fig. 2D, and as previously reported [16]). Upon co-transfection of ΔNp73α with asHsp72, ΔNp73α still exhibits an anti-apoptotic effect upon VP16 treatment (Fig. 2D, ΔNp73α black and grey bars). These data were further confirmed using a siRNA targeting HSP72/HSPA1A mRNA (Fig. 2E and F). This indicates the anti-apoptotic effect of ΔNp73α being independent on induction of Hsp72, consistent with the data described above (Fig. 1) where, in H82 cells, ΔNp73α do not show any induction of Hsp72, neither on the level of the promoter nor on mRNA and protein levels. The pro-apoptotic activity of TAp73β was unaffected by the co-expression of asHsp72. However, co-transfection of TAp73α together with asHsp72 significantly reduced the anti-apoptotic effect of TAp73α (Fig. 2D, TAp73α black and grey bars). Hence, the anti-apoptotic activity of TAp73α in SCLC H82 cells treated with chemotherapeutic drugs seems to depend on the induction of Hsp72.
Fig 2.
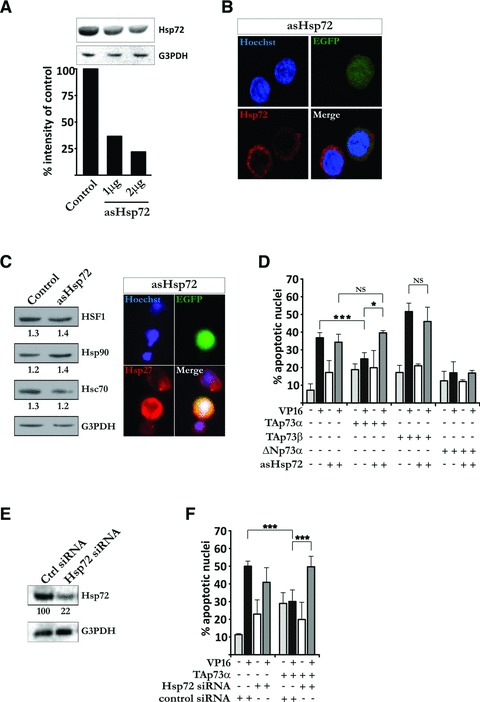
Hsp72 is required for TAp73α anti-apoptotic effect. (A) H82 cells were transfected with asHsp72 vector and total protein cell extracts were analysed by immunoblotting for the presence of Hsp72. G3PDH was used as protein loading control. Quantification of protein band-intensity was made using ImageJ software. (B) H82 cells were co-transfected with EGFP and asHsp72 vector (green). Samples were stained with anti-Hsp72 antibody (red) and nuclei counterstained with Hoechst (blue). Images are representatives of three independent experiments. (C) H82 cells were transfected with asHsp72 vector and total protein cell extracts were analysed by immunoblotting for the expression of HSF1, Hsp90 and Hsc70. G3PDH was used as protein loading control and numbers indicate the ratio between protein (HSF1, Hsp90 or Hsc70) and G3PDH band intensity. H82 cells were co-transfected with EGFP and asHsp72 vector (green) and the effect of asHsp72 expression on Hsp27 protein levels were detected using immunofluorescent staining for Hsp27 (red) and counterstaining of nuclei with Hoechst (blue). (D) H82 cells were co-transfected with EGFP, asHsp72 vector and empty vector or p73 expression vectors (TAp73α, TAp73β, ΔNp73α). Cells were treated with VP16 for 24 hrs, nuclei counterstained with Hoechst and apoptosis was assayed by scoring of EGFP transfected cells presenting condensed or fragmented nuclei. (E) H82 cells were transfected with siRNAs pool designed to interfere specifically with the expression of human Hsp72 mRNA and total protein cell extracts were analysed by immunoblotting for the expression of Hsp72. G3PDH was used as protein loading control. Quantification of protein band-intensity was made using ImageJ software. (F) H82 cells were co-transfected with EGFP, Hsp72 siRNAs pool and empty or TAp73α expression vectors. Cells were treated with VP16 for 24 hrs, nuclei counterstained with Hoechst and apoptosis was assayed by scoring of EGFP transfected cells presenting condensed or fragmented nuclei. Figures are mean ± S.D. of three independent experiments, where *P < 0.05, **P < 0.01 and ***P < 0.001.
Hsp72 represses TAp73β pro-apoptotic effect
Given that TAp73α is able to induce Hsp72 expression and that the anti-apoptotic activity of TAp73α seems to be dependent on this induction, and since it is known from before that the p73 isoforms can interact and regulate the activity of each other [16, 29], we sought to investigate whether the overexpression of Hsp72 could counteract the pro-apoptotic function of TAp73β upon drug-induced apoptosis. Transfection of H82 cells with Hsp72 vector lead to an enhanced expression of Hsp72 protein, as shown by immunoblot (Fig. 3A) and confocal imaging (Fig. 3B). Subsequent transfection of H82 cells with Hsp72 reduced the level of VP16-induced apoptosis (Fig. 3C, black and grey bars). Moreover, co-transfection of TAp73β together with Hsp72 abolished TAp73β pro-apoptotic effect upon VP16 treatment. Hence, TAp73β augmentation of drug-induced apoptosis can be repressed by simultaneous co-expression of Hsp72.
Fig 3.
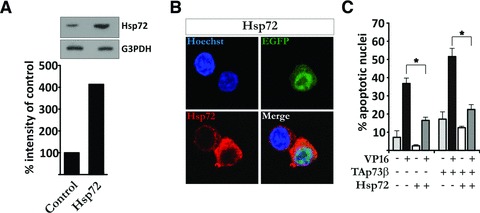
Hsp72 represses TAp73β pro-apoptotic effect. (A) H82 cells were transfected with Hsp72 expression vector and extracts were analysed as described in Figure 2A. (B) H82 cells were co-transfected with EGFP and Hsp72 vector (green), and samples assayed as described in Figure 2B. (C) H82 cells were co-transfected with EGFP, Hsp72 vector and empty vector or TAp73β. Samples were treated, stained and assayed as described in Figure 2D. Figures are mean ± S.D. of three independent experiments, where *P < 0.05, **P < 0.01 and ***P < 0.001.
Hsp72 depletion favours Bax activation and loss of ΔΨm upon VP16 treatment in the presence of TAp73α
Significant activation of Bax in H82 cells upon VP16 treatment is observed by FACS analysis at 18–24 hrs after exposure (Fig. S1B). We have previously shown that the anti-apoptotic effect of TAp73α in SCLC cells is exerted upstream of mitochondria, at the level of Bax activation [16]. In addition, Hsp72 has been shown to exert some of its protective effects at the same level, inhibiting apoptosis via the prevention of Bax activation and its translocation to the mitochondria [17]. To investigate whether the simultaneous expression of TAp73α and depletion of Hsp72 could affect the apoptotic process at the level or upstream of mitochondria, H82 cells were co-transfected with TAp73α and asHsp72, and treated with VP16. Indeed, TAp73α was able to prevent VP16-induced disruption of mitochondrial membrane potential (ΔΨm) as shown both by microscopic count of TMRE– cells (Fig. 4A, black bars) and FACS analysis (Fig. 4B, black bars). However, upon co-transfection of TAp73α with asHsp72 the protective effect disappeared leading to a drop in ΔΨm upon VP16 treatment (Fig. 4A and B, TAp73α black and dark grey bars). Moreover, TAp73α-mediated inhibition of Bax activation was eliminated by co-transfection with asHsp72, as depicted by immunofluorescent staining (Fig. 4C) and FACS analysis on percentage of cells staining positive for active Bax (Fig. 4D, black and dark grey bars). Thus, depletion of Hsp72 leads to a loss of TAp73α anti-apoptotic effect upon drug-treatment and favours Bax activation and mitochondrial dysfunctions.
Fig 4.
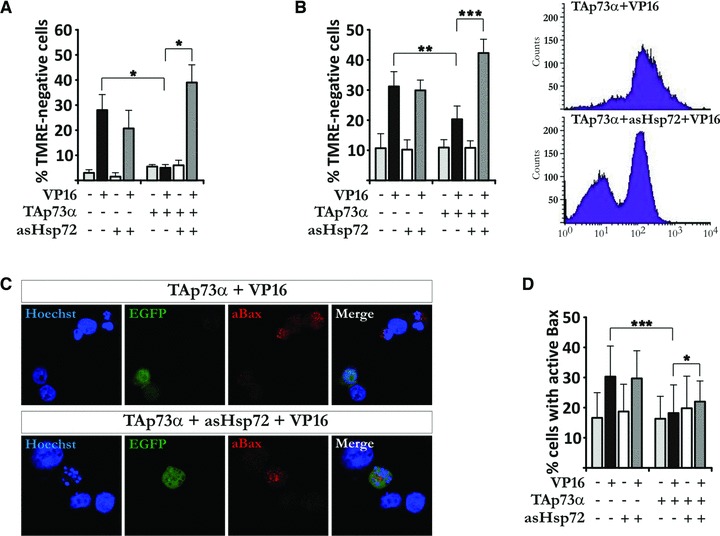
Hsp72 depletion favours Bax activation and loss of ΔΨm upon VP16 treatment in the presence of TAp73α. (A, B) H82 cells were co-transfected with EGFP, asHsp72 vector and empty vector or TAp73α. Cells were treated with VP16 for 24 hrs, stained with TMRE and nuclei counterstained with Hoechst. Loss of ΔΨm was assayed by scoring of EGFP transfected cells without TMRE staining using microscopy (A) and FACS analysis (B). (C) H82 cells were transfected and treated as described (A). Samples were stained with anti-active Bax antibody (red) and nuclei counterstained with Hoechst (blue). (D) Cells were transfected and treated as in (A), stained with anti-active Bax antibody and analysed by FACS.
Hsp72 expression prevents TAp73β-induced mitochondrial dysfunction and Bax activation
TAp73 isoforms have been shown to induce apoptosis via Puma-mediated Bax activation [30]. To investigate whether the protective effect of Hsp72 overexpression upon TAp73β-enhanced drug-induced apoptosis is exerted upstream of apoptosis-related mitochondrial events, H82 cells were co-transfected with TAp73β and Hsp72 expression vectors. Introduction of Hsp72 greatly diminished TAp73β-induced ΔΨm disruption upon VP16 treatment, as shown by both cell counting in microscope (Fig. 5A) and FACS analysis (Fig. 5B) of TMRE– cells. In addition, TAp73β-induced Bax activation was reduced upon introduction of Hsp72, as depicted by confocal imaging (Fig. 5C) and FACS analysis (Fig. 5D). Hence, TAp73β-mediated Bax activation and disruption of ΔΨm in response to VP16 can be prevented by the simultaneous expression of Hsp72.
Fig 5.
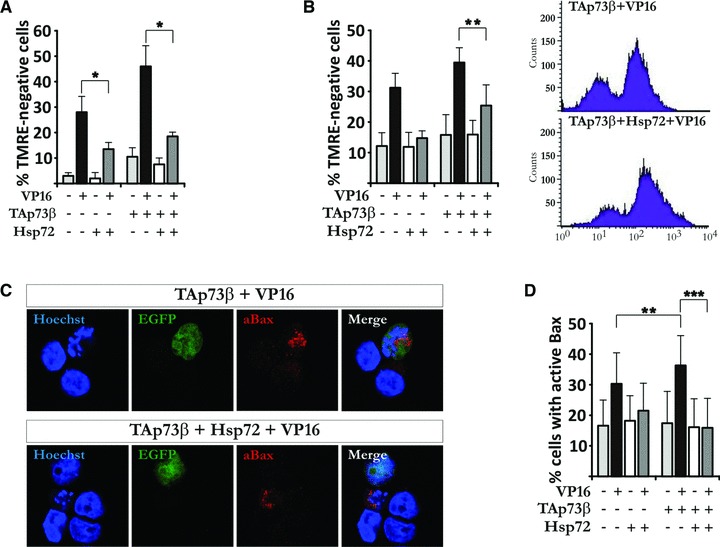
Hsp72 expression prevents TAp73β-induced mitochondrial dysfunction and Bax activation. (A, B) H82 cells were co-transfected with EGFP, Hsp72 vector and empty vector or TAp73β. Treatment and staining were performed as described in Figure 4A and B, and analysis was carried out using microscopy (A) and FACS analysis (B). (C, D) H82 cells were transfected, treated and stained as described in Figure 4C and D. Images are representatives of three independent experiments. Figures are mean ± S.D. of three independent experiments, where *P < 0.05, **P < 0.01 and ***P < 0.001.
TAp73α repress Etoposide-induced loss of lysosomal membrane potential in a Hsp72-dependent manner
Etoposide was shown to induce destabilization of lysosomes [18], an event which in turn can trigger Bax activation in a cathepsin B dependent manner [31]. On the other hand, lysosomal permeabilization has been shown to depend on Bax [32, 33]. Both ways, the destabilization of and subsequent leakage from lysosomal compartments can be prevented by simultaneous co-expression of Hsp72 [34]. Significant LMP in H82 cells upon VP16 treatment is observed by FACS analysis at 24 hrs after exposure (Fig. S1C). Hence, we set out to determine whether TAp73α can prevent VP16-induced LMP in H82 cells, and further, if this event is mediated by Hsp72. Hence, we made use of LysoTracker dye which accumulates inside acidic organelles, such as lysosomes and late endosomes [35]. A decreased LysoTracker fluorescence may then reflect LMP and/or an increase in lysosomal pH, which then would lead to less cytosolic puncta [35], as seen using fluorescence microscopy (Fig. 6C, compare white arrows in merge). Quantification of cells with a reduced punctiform staining, or no LysoTracker staining at all (here denoted as LMP), showed that VP16 treatment induced LMP (Fig. 6A). TAp73α was able to prevent VP16-induced LMP; however, co-transfection of TAp73α with asHsp72 and VP16 treatment resulted in LMP comparable to VP16 alone. On the other hand, TAp73β promoted VP16-induced LMP (Fig. 6B), whereas simultaneous co-transfection with Hsp72 resulted in a very little LMP, comparable to control levels. Given together, these results suggests that TAp73α induced Hsp72 expression protects the cell from drug-induced mitochondrial dysfunctions as well as LMP.
Fig 6.
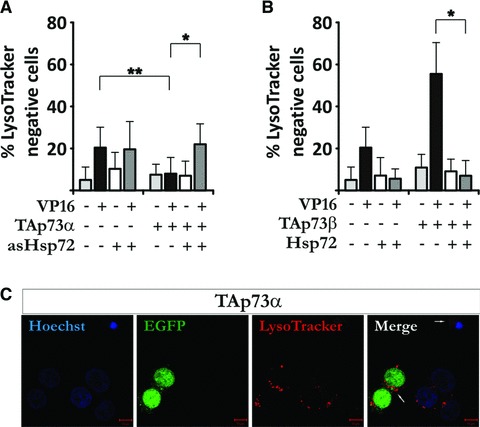
TAp73α repress Etoposide-induced LMP in a Hsp72-dependent manner. H82 cells were co-transfected with EGFP, asHsp72, TAp73α, Hsp72 and/or TAp73β, treated with VP16 for 24 hrs and stained with Lysotracker Red. Cells were quantified live using fluorescence microscopy (A, B), or fixed and stained with Hoechst for confocal analysis (C). (A) and (B) are mean ± S.D. of at least three independent experiments, where *P < 0.05, **P < 0.01 and ***P < 0.001. (C) is a representative image.
The Hsp72-mediated anti-apoptotic effect of TAp73α can be counteracted by TAp73β
It is known since before that the p73 isoforms can interact and regulate the activity of each other [16, 29]. Given that TAp73α is able to transactivate the HSP72 promoter (Fig. 1), we decided to investigate whether TAp73β could affect the activity of TAp73α with regards to the HSP72 promoter. Subsequently, H82 cells were transfected with HSP72-luciferase construct, TAp73α and increasing amounts of TAp73β. In a dose-dependent manner, TAp73β was able to significantly reduce the level of transactivation of TAp73α on the HSP72 promoter (Fig. 7A). Using chromatin immunoprecipitation assay and primers covering a region of the promoter including a p53 responsive element, we show that TAp73α directly binds to the HSP72 promoter, whereas TAp73β do not (Fig. 7B). Consequently, to investigate if this effect was reflected on the outcome of apoptosis, H82 cells were transfected with TAp73α and increasing amounts of TAp73β, and treated with VP16. Indeed, in a dose-dependent manner TAp73β was able to reverse the anti-apoptotic effect of TAp73α on drug-induced apoptosis (Fig. 7C). Thus, the Hsp72-dependent anti-apoptotic effect of TAp73α can be counteracted by simultaneous co-expression of TAp73β.
Fig 7.
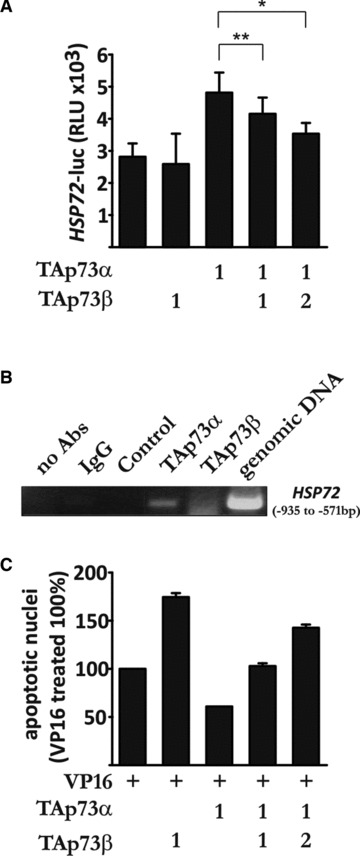
The Hsp72-mediated anti-apoptotic effect of TAp73α can be counteracted by TAp73β. (A) H82 cells were transfected and assayed as described in Figure 1A. (B) H82 cells were transfected with TAp73α, TAp73β, ΔNp73α, TAp73α and TAp73β, or empty vector. Samples were immunoprecipitated using p73 antibody, and binding to the HSP72 promoter was detected with PCR. (C) H82 cells were transfected, treated and assayed by scoring of EGFP transfected cells presenting condensed or fragmented nuclei (as described in Fig. 2D). Figures are mean ± S.D. of three independent experiments, where *P < 0.05, **P < 0.01 and ***P < 0.001.
Discussion
In this study we show that the TAp73α isoform transcriptionally regulates the expression of Hsp72. The protective effect of TAp73α upon treatment of SCLC cells with chemotherapeutic agents is shown to be dependent on Hsp72. Furthermore, we discovered an additional level of complexity where TAp73β is able to counteract TAp73α anti-apoptotic effects (also previously reported by us [16]) and transcriptional activation of and binding to the HSP72 promoter.
We show that TAp73α can regulate expression of the HSP72 gene, an activity not shared by TAp73β, which provide additional evidence that there indeed exists an isoform-specific regulation of target genes. The diverse TAp73 carboxy-terminal isoforms differs in regard to relative efficiency in transactivation of promoters of target genes, post-translational modifications and interaction partners. The final outcome of these variations among the TAp73 carboxy-terminal isoforms are ultimately displayed as different potencies in the induction of apoptosis, cell cycle arrest and/or differentiation [7, 36, 37]. TAp73β has been shown a potent transactivator of the Aquaporin 3 [38] and P57Kip2[39] genes, whereas TAp73α is inefficient in this aspect. Of note, p57Kip2 can promote drug-induced cell death [40], providing one possible explanation to why TAp73β in many cases is a more potent inducer of apoptosis than TAp73α[8]. On the other hand, TAp73α, but not TAp73β, can suppress myogenic differentiation due to an attenuation of MyoD transcriptional activity [41]. Due to its extended carboxy-terminus, TAp73α, but not TAp73β, can be covalently modified by SUMO-1. Sumoylation at Lysine 627 leads to a more rapid degradation of TAp73α by the proteasome [42]. Interaction of TAp73α with different proteins, most likely mediated by the SAM domain and the extreme carboxy-terminus, might provide one explanation to some of the functional differences between TAp73α and TAp73β. For instance, receptor for activated kinase 1 interacts with TAp73α carboxy-terminus ultimately resulting in reduced transcriptional activity and inhibition of TAp73α-induced apoptosis [43].
Although both TAp73α and TAp73β can induce cell cycle arrest and apoptosis upon drug treatment, TAp73α expression is increased in a range of tumours and has been shown to confer resistance of some tumour cells towards drug treatment [15, 16]. Previously, we have shown that TAp73α can repress drug-induced apoptosis in a cell-type specific manner. We demonstrated this effect to be exerted upstream of the mitochondria, at the level of Bax activation [16]. Here, we provide a mechanism in support of the anti-apoptotic effect of TAp73α. We show that the anti-apoptotic effect of TAp73α is lost upon depletion of Hsp72. Moreover, the repressive effect of TAp73α on Bax activation, drop in mitochondrial membrane potential and LMP in drug-induced apoptosis was lost upon introduction of antisense Hsp72. The anti-apoptotic, inducible Hsp72 has been shown to act on different levels in the apoptotic signalling pathways – stabilizing lysosomal membranes [18] and preventing loss of mitochondrial membrane potential [19], release of pro-apoptotic mitochondrial proteins, apoptosome formation [19–21], caspase activation [19, 22] and delay drug-induced cell death via physical interaction with pro-apoptotic molecules such as Bax, AIF and Apaf-1 [44]. Here we show that TAp73β-enhanced drug-induced Bax activation, drop in mitochondrial membrane potential, LMP and subsequent cell death is reduced upon introduction of Hsp72 expression vector. Indeed, different studies have shown LMP to occur either before Bax activation [31], or to be dependent on active Bax [32, 33]. However, in SCLC cells Bax activation seems to occur prior to LMP (Fig. S1B and C), thus indicating that the effects of Hsp72 downstream of p73 is mainly exerted via Bax. Indeed, our data demonstrate that the transcriptional regulation of Hsp72 expression by TAp73α mediates its pro-survival effect. To notice, Hsp72 overexpression was shown to increase transformation in a range of different cell types [45–47]. Also, an elevated expression of Hsp72 has been reported in tumour cell lines [48–50] and in several tumour cases [51]. Interestingly, selective down-regulation of Hsp72 by small interfering RNA’s or antisense constructs promotes drug-induced cell death in a range of different tumour cell lines, e.g. cervical carcinoma (HeLa) [52], colon carcinoma, hepatocellular carcinoma [53] and breast cancer [49]. Hence, the possibility of a tumour cell to induce expression of Hsp72 seems to provide it with an advantage towards drug-induced cell death.
Consequently, with these data we reinforce the idea that there is indeed a balance between the two main TAp73 isoforms, TAp73α and TAp73β. We have previously shown that TAp73α can counteract the pro-apoptotic effect of TAp73β, and vice versa [16]. Here, we demonstrate that TAp73β can repress TAp73α binding to and transactivation of the HSP72 promoter, and that this might be a part of the explanation on how TAp73β counteracts TAp73α anti-apoptotic effect. In addition to the previously reported potential oncogenic properties of the TAp73α isoform this study gives a deeper insight into how TAp73α expression in tumours could contribute to drug resistance. Importantly, these data reveal the importance in investigating the relative expression of different p73 isoforms in human tumours. It is now clear that not only does the relation between the amino-terminal isoforms, ΔNp73 and TAp73, affect the final outcome of a cell upon drug treatment, but it can also be affected by the relative expression of individual carboxy-terminal p73 isoforms, e.g. TAp73α and TAp73β.
Acknowledgments
We thank Dr. T. Perlmann for permanent support. This work was supported by grants from the Swedish Research Council, the Swedish Cancer Society (B.Z., B.J.), Swedish Medical Society, the Åke Wiberg Foundation, Karolinska Institutet Foundations (KI Cancer) (B.J.), Stockholm Cancer Society and the European Commission (B.Z.).
Conflict of interest
The authors confirm that there are no conflicts of interest.
Supporting Information
(A-C) H82 cells treated with 5 μM VP16for the indicated time points. (A) Total protein cellextracts were analyzed by immunoblotting for the presence of Hsp72.G3PDH was used as protein loading control. Quantification ofprotein band-intesity was made using ImageJ software. (B)Cells were stained with Lysotracker Red. Cells were quantified liveusing fluoroscence microscopy. Figures (B) and (C)are mean ± SD of at least three independent experiments,where *p < 0.05, **p < 0.01 and ***p < 0.001.
References
- 1.Kaghad M, Bonnet H, Yang A, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997;90:809–19. doi: 10.1016/s0092-8674(00)80540-1. [DOI] [PubMed] [Google Scholar]
- 2.Jost CA, Marin MC, Kaelin WG., Jr p73 is a simian [correction of human] p53-related protein that can induce apoptosis. Nature. 1997;389:191–4. doi: 10.1038/38298. [DOI] [PubMed] [Google Scholar]
- 3.De Laurenzi V, Raschella G, Barcaroli D, et al. Induction of neuronal differentiation by p73 in a neuroblastoma cell line. J Biol Chem. 2000;275:15226–31. doi: 10.1074/jbc.275.20.15226. [DOI] [PubMed] [Google Scholar]
- 4.Grob TJ, Novak U, Maisse C, et al. Human delta Np73 regulates a dominant negative feedback loop for TAp73 and p53. Cell Death Differ. 2001;8:1213–23. doi: 10.1038/sj.cdd.4400962. [DOI] [PubMed] [Google Scholar]
- 5.Chi SG, Chang SG, Lee SJ, et al. Elevated and biallelic expression of p73 is associated withprogression of human bladder cancer. Cancer Res. 1999;59:2791–3. [PubMed] [Google Scholar]
- 6.Ishimoto O, Kawahara C, Enjo K, et al. Possible oncogenic potential of DeltaNp73: a newly identified isoform of human p73. Cancer Res. 2002;62:636–41. [PubMed] [Google Scholar]
- 7.Moll UM, Erster S, Zaika A. p53, p63 and p73–solos, alliances and feuds among family members. Biochim Biophys Acta. 2001;1552:47–59. doi: 10.1016/s0304-419x(01)00036-1. [DOI] [PubMed] [Google Scholar]
- 8.Stiewe T, Putzer BM. Role of p73 in malignancy: tumor suppressor or oncogene. Cell Death Differ. 2002;9:237–45. doi: 10.1038/sj.cdd.4400995. [DOI] [PubMed] [Google Scholar]
- 9.Zitterbart K, Zavrelova I, Kadlecova J, et al. p73 expression in medulloblastoma: TAp73/DeltaNp73 transcript detection and possible association of p73alpha/DeltaNp73 immunoreactivity with survival. Acta Neuropathol. 2007;114:641–50. doi: 10.1007/s00401-007-0298-2. [DOI] [PubMed] [Google Scholar]
- 10.Novak U, Grob TJ, Baskaynak G, et al. Overexpression of the p73 gene is a novel finding in high-risk B-cell chronic lymphocytic leukemia. Ann Oncol. 2001;12:981–6. doi: 10.1023/a:1011153206003. [DOI] [PubMed] [Google Scholar]
- 11.Niyazi M, Ghazizadeh M, Konishi H, et al. Expression of p73 and c-Abl proteins in human ovarian carcinomas. J Nippon Med Sch. 2003;70:234–42. doi: 10.1272/jnms.70.234. [DOI] [PubMed] [Google Scholar]
- 12.Kang MJ, Park BJ, Byun DS, et al. Loss of imprinting and elevated expression of wild-type p73 in human gastric adenocarcinoma. Clin Cancer Res. 2000;6:1767–71. [PubMed] [Google Scholar]
- 13.Yokomizo A, Mai M, Tindall DJ, et al. Overexpression of the wild type p73 gene in human bladder cancer. Oncogene. 1999;18:1629–33. doi: 10.1038/sj.onc.1202474. [DOI] [PubMed] [Google Scholar]
- 14.Frasca F, Vella V, Aloisi A, et al. p73 tumor-suppressor activity is impaired in human thyroid cancer. Cancer Res. 2003;63:5829–37. [PubMed] [Google Scholar]
- 15.Vikhanskaya F, Marchini S, Marabese M, et al. P73a overexpression is associated with resistance to treatment with DNA-damaging agents in a human ovarian cancer cell line. Cancer Res. 2001;61:935–8. [PubMed] [Google Scholar]
- 16.Nyman U, Sobczak-Pluta A, Vlachos P, et al. Full-length p73alpha represses drug-induced apoptosis in small cell lung carcinoma cells. J Biol Chem. 2005;280:34159–69. doi: 10.1074/jbc.M500394200. [DOI] [PubMed] [Google Scholar]
- 17.Stankiewicz AR, Lachapelle G, Foo CP, et al. Hsp70 inhibits heat-induced apoptosis upstream of mitochondria by preventing Bax translocation. J Biol Chem. 2005;280:38729–39. doi: 10.1074/jbc.M509497200. [DOI] [PubMed] [Google Scholar]
- 18.Nylandsted J, Gyrd-Hansen M, Danielewicz A, et al. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med. 2004;200:425–35. doi: 10.1084/jem.20040531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Creagh EM, Carmody RJ, Cotter TG. Heat shock protein 70 inhibits caspase-dependent and -independent apoptosis in Jurkat T cells. Exp Cell Res. 2000;257:58–66. doi: 10.1006/excr.2000.4856. [DOI] [PubMed] [Google Scholar]
- 20.Ravagnan L, Gurbuxani S, Susin SA, et al. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat Cell Biol. 2001;3:839–43. doi: 10.1038/ncb0901-839. [DOI] [PubMed] [Google Scholar]
- 21.Mosser DD, Caron AW, Bourget L, et al. The chaperone function of hsp70 is required for protection against stress-induced apoptosis. Mol Cell Biol. 2000;20:7146–59. doi: 10.1128/mcb.20.19.7146-7159.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mosser DD, Caron AW, Bourget L, et al. Role of the human heat shock protein hsp70 in protection against stress-induced apoptosis. Mol Cell Biol. 1997;17:5317–27. doi: 10.1128/mcb.17.9.5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanaka Y, Kameoka M, Itaya A, et al. Regulation of HSF1-responsive gene expression by N-terminal truncated form of p73alpha. Biochem Biophys Res Commun. 2004;317:865–72. doi: 10.1016/j.bbrc.2004.03.124. [DOI] [PubMed] [Google Scholar]
- 24.De Laurenzi V, Costanzo A, Barcaroli D, et al. Two new p73 splice variants, gamma and delta, with different transcriptional activity. J Exp Med. 1998;188:1763–8. doi: 10.1084/jem.188.9.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He B, Meng YH, Mivechi NF. Glycogen synthase kinase 3beta and extracellular signal-regulated kinase inactivate heat shock transcription factor 1 by facilitating the disappearance of transcriptionally active granules after heat shock. Mol Cell Biol. 1998;18:6624–33. doi: 10.1128/mcb.18.11.6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jäättelä M, Wissing D, Kokholm K, et al. Hsp70 exerts its anti-apoptotic function downstream of caspase-3-like proteases. EMBO J. 1998;17:6124–34. doi: 10.1093/emboj/17.21.6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gottwald E, Muller O, Polten A. Semiquantitative reverse transcription-polymerase chain reaction with the Agilent 2100 Bioanalyzer. Electrophoresis. 2001;22:4016–22. doi: 10.1002/1522-2683(200110)22:18<4016::AID-ELPS4016>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 28.Wu G, Osada M, Guo Z, et al. DeltaNp63alpha up-regulates the Hsp70 gene in human cancer. Cancer Res. 2005;65:758–66. [PubMed] [Google Scholar]
- 29.Ueda Y, Hijikata M, Takagi S, et al. Transcriptional activities of p73 splicing variants are regulated by inter-variant association. Biochem J. 2001;356:859–66. doi: 10.1042/0264-6021:3560859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Melino G, Bernassola F, Ranalli M, et al. p73 Induces apoptosis via PUMA transactivation and Bax mitochondrial translocation. J Biol Chem. 2004;279:8076–83. doi: 10.1074/jbc.M307469200. [DOI] [PubMed] [Google Scholar]
- 31.Bidere N, Lorenzo HK, Carmona S, et al. Cathepsin D triggers Bax activation, resulting in selective apoptosis-inducing factor (AIF) relocation in T lymphocytes entering the early commitment phase to apoptosis. J Biol Chem. 2003;278:31401–11. doi: 10.1074/jbc.M301911200. [DOI] [PubMed] [Google Scholar]
- 32.Feldstein AE, Werneburg NW, Li Z, et al. Bax inhibition protects against free fatty acid-induced lysosomal permeabilization. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1339–46. doi: 10.1152/ajpgi.00509.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oberle C, Huai J, Reinheckel T, et al. Lysosomal membrane permeabilization and cathepsin release is a Bax/Bak-dependent, amplifying event of apoptosis in fibroblasts and monocytes. Cell Death Differ. 17:1167–78. doi: 10.1038/cdd.2009.214. [DOI] [PubMed] [Google Scholar]
- 34.Fehrenbacher N, Bastholm L, Kirkegaard-Sorensen T, et al. Sensitization to the lysosomal cell death pathway by oncogene-induced down-regulation of lysosome-associated membrane proteins 1 and 2. Cancer Res. 2008;68:6623–33. doi: 10.1158/0008-5472.CAN-08-0463. [DOI] [PubMed] [Google Scholar]
- 35.Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene. 2008;27:6434–51. doi: 10.1038/onc.2008.310. [DOI] [PubMed] [Google Scholar]
- 36.Zhu J, Jiang J, Zhou W, et al. The potential tumor suppressor p73 differentially regulates cellular p53 target genes. Cancer Res. 1998;58:5061–5. [PubMed] [Google Scholar]
- 37.Lee CW, La Thangue NB. Promoter specificity and stability control of the p53-related protein p73. Oncogene. 1999;18:4171–81. doi: 10.1038/sj.onc.1202793. [DOI] [PubMed] [Google Scholar]
- 38.Zheng X, Chen X. Aquaporin 3, a glycerol and water transporter, is regulated by p73 of the p53 family. FEBS Lett. 2001;489:4–7. doi: 10.1016/s0014-5793(00)02437-6. [DOI] [PubMed] [Google Scholar]
- 39.Blint E, Phillips AC, Kozlov S, et al. Induction of p57(KIP2) expression by p73beta. Proc Natl Acad Sci USA. 2002;99:3529–34. doi: 10.1073/pnas.062491899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vlachos P, Nyman U, Hajji N, et al. The cell cycle inhibitor p57(Kip2) promotes cell death via the mitochondrial apoptotic pathway. Cell Death Differ. 2007;14:1497–507. doi: 10.1038/sj.cdd.4402158. [DOI] [PubMed] [Google Scholar]
- 41.Li CY, Zhu J, Wang JY. Ectopic expression of p73alpha, but not p73beta, suppresses myogenic differentiation. J Biol Chem. 2005;280:2159–64. doi: 10.1074/jbc.M411194200. [DOI] [PubMed] [Google Scholar]
- 42.Minty A, Dumont X, Kaghad M, et al. Covalent modification of p73alpha by SUMO-1. Two-hybrid screening with p73 identifies novel SUMO-1-interacting proteins and a SUMO-1 interaction motif. J Biol Chem. 2000;275:36316–23. doi: 10.1074/jbc.M004293200. [DOI] [PubMed] [Google Scholar]
- 43.Ozaki T, Watanabe K, Nakagawa T, et al. Function of p73, not of p53, is inhibited by the physical interaction with RACK1 and its inhibitory effect is counteracted by pRB. Oncogene. 2003;22:3231–42. doi: 10.1038/sj.onc.1206382. [DOI] [PubMed] [Google Scholar]
- 44.Calvaruso G, Giuliano M, Portanova P, et al. Hsp72 controls bortezomib-induced HepG2 cell death via interaction with pro-apoptotic factors. Oncol Rep. 2007;18:447–50. [PubMed] [Google Scholar]
- 45.Jaattela M. Over-expression of hsp70 confers tumorigenicity to mouse fibrosarcoma cells. Int J Cancer. 1995;60:689–93. doi: 10.1002/ijc.2910600520. [DOI] [PubMed] [Google Scholar]
- 46.Volloch VZ, Sherman MY. Oncogenic potential of Hsp72. Oncogene. 1999;18:3648–51. doi: 10.1038/sj.onc.1202525. [DOI] [PubMed] [Google Scholar]
- 47.Samali A, Cotter TG. Heat shock proteins increase resistance to apoptosis. Exp Cell Res. 1996;223:163–70. doi: 10.1006/excr.1996.0070. [DOI] [PubMed] [Google Scholar]
- 48.Helmbrecht K, Zeise E, Rensing L. Chaperones in cell cycle regulation and mitogenic signal transduction: a review. Cell Prolif. 2000;33:341–65. doi: 10.1046/j.1365-2184.2000.00189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nylandsted J, Rohde M, Brand K, et al. Selective depletion of heat shock protein 70 (Hsp70) activates a tumor-specific death program that is independent of caspases and bypasses Bcl-2. Proc Natl Acad Sci USA. 2000;97:7871–6. doi: 10.1073/pnas.97.14.7871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ekedahl J, Joseph B, Marchetti P, et al. Heat shock protein 72 does not modulate ionizing radiation-induced apoptosis in U1810 non-small cell lung carcinoma cells. Cancer Biol Ther. 2003;2:663–9. [PubMed] [Google Scholar]
- 51.Lanneau D, Brunet M, Frisan E, et al. Heat shock proteins: essential proteins for apoptosis regulation. J Cell Mol Med. 2008;12:743–61. doi: 10.1111/j.1582-4934.2008.00273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yaglom JA, Gabai VL, Sherman MY. High levels of heat shock protein Hsp72 in cancer cells suppress default senescence pathways. Cancer Res. 2007;67:2373–81. doi: 10.1158/0008-5472.CAN-06-3796. [DOI] [PubMed] [Google Scholar]
- 53.Nylandsted J, Wick W, Hirt UA, et al. Eradication of glioblastoma, and breast and colon carcinoma xenografts by Hsp70 depletion. Cancer Res. 2002;62:7139–42. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A-C) H82 cells treated with 5 μM VP16for the indicated time points. (A) Total protein cellextracts were analyzed by immunoblotting for the presence of Hsp72.G3PDH was used as protein loading control. Quantification ofprotein band-intesity was made using ImageJ software. (B)Cells were stained with Lysotracker Red. Cells were quantified liveusing fluoroscence microscopy. Figures (B) and (C)are mean ± SD of at least three independent experiments,where *p < 0.05, **p < 0.01 and ***p < 0.001.