

Abstract
Sulfated fucans from sea urchin egg jelly express well-defined chemical structures that vary with species. This species specificity regulates the sperm acrosome reaction, a critical step to assure intra-specific fertilization. In addition, these polysaccharides are involved in other biological activities such as anticoagulation. Although sulfation patterns are relevant to the levels of response in both activities, conformation and dynamics of these glycans are also contributing factors. However, data about these features of sulfated fucans are very rare. To address this, we have employed nuclear magnetic resonance experiments combined with molecular dynamics on structurally defined oligosaccharides derived from two sulfated fucans. The results have indicated that the oligosaccharides are flexible in solution. Ring conformation of their composing units displays just the 1C4 chair configuration. In a particular octasaccharide, composed of two tetrasaccharide sequences, inter-residual hydrogen bonds play a role to decrease dynamics in these repeating units. Conversely, the linking disaccharide [-3)-α-l-Fucp-2()-(1–3)-α-l-Fucp-4()-(1-] located right between the two tetrasaccharide units has amplified motions suggested to be promoted by electrostatic repulsion of sulfates on opposite sides of the central glycosidic bond. This conjunction of information about conformation and dynamics of sulfated fucan oligosaccharides provides new insights to explain how these glycans behave free in solution and influenced by sulfation patterns. It may also serve for future studies concerning structure–function relationship of sulfated fucans, especially those involving sea urchin fertilization and anticoagulation.
Keywords: conformation, MD, NMR, NOE, oligosaccharides, spin relaxation, sulfated fucan
Introduction
The jelly surrounding sea urchin eggs contain sulfated fucans with unique structures, composed of α-l-fucopyranose (Fucp) units. When compatible sperm approach the sea urchin egg, the sulfated fucan binds to sperm receptors and induces the acrosome reaction (Pomin and Mourão 2008; Vilela-Silva et al. 2008). This is a necessary event prior to the fertilization of sea urchins and involves polymerization of actin to form a finger-like acrosomal structure which protrudes from the sperm head. The sulfated fucans from the sea urchin egg jelly present well-defined chemical structures and each species possesses a polysaccharide with a particular structure (Pomin 2009). The species-specific structures guarantee intra-specific fertilization by inducing acrosome reaction only among homologous sperm (Pomin and Mourão 2008; Vilela-Silva et al. 2008). Therefore, sulfated polysaccharides play a role in the fertilization process, in addition to their better known interactions with growth factors, coagulation factors and selectins. The structural changes of egg jelly sulfated fucans may drive speciation in the sea urchin, as a result of fertility incompatibility or even evolutionary causes (Biermann et al. 2004; Mourão 2007).
The sulfation and glycosylation sites are not the only structural players involved in regulating the biological responses of the sulfated fucans. Conformations and dynamics of these polymers either free in solution or bound to their functional protein partners are also quite relevant to the biological outcome, as pointed for other sulfated polysaccharides (Mulloy and Forster 2000; Becker et al. 2007; Pomin 2014a). However, no information is available so far concerning the conformation and dynamics of the sea urchin sulfated fucans of well-defined chemical structures.
To address this, we have employed a variety of methods of solution nuclear magnetic resonance (NMR) spectroscopy combined with molecular dynamics (MD) simulations. This set of NMR+MD analyses were employed in structurally defined oligosaccharides produced by mild acid hydrolysis of two sulfated fucans (Structures 1 and 2, Figure 1) synthesized by the sea urchin Lytechinus variegatus (Mulloy et al. 1994; Cinelli et al. 2007). Both structures are polymers composed of 3-linked α-l-Fucp units. Sulfation occurs at the 2-O- and/or the 4-O-positions. Structure 1 is a sulfated fucan composed of a tetrasaccharide repeating sequence, in which the Fucp units are conventionally labeled A, B, C and D for their sulfation patterns: B for 2,4-disulfated, C for 2-sulfated, A for 2-sulfated and D for 4-sulfated units. Structure 2 is a uniformly 4-sulfated polymer.
Fig. 1.

Structural representation of the two sulfated fucans, Structures 1 and 2, both synthesized by the sea urchin L. variegatus. While Structure 1 is a tetrasaccharide repeating polysaccharide made up of the following sequence [→3)-α-l-Fucp-2,4-di()-(1 → 3)-α-l-Fucp-2()-(1 → 3)-α-l-Fucp-2()-(1 → 3)-α-l-Fucp-4()-(1→]n, Structure 2 is a monosaccharide repeating polysaccharide made up of the following unit [→3)-α-l-Fucp-4()-(1→]n. The residues of Structure 1 are labeled with letters A, B, C and D for individual assignments and recognition in the further experiments and explanations. The carbon-linked hydrogens were omitted in the cartoon in order to provide a clear illustration. Sulfated groups are highlighted by dashed ellipses for fast visualization of the sulfation pattern.
NMR and MD conformational and dynamical studies of long polysaccharides are challenging, and both sulfated fucans (Structures 1 and 2, Figure 1) from L. variegatus have naturally occurring high-molecular weights (MWs), >100 kDa (Mulloy et al. 1994; Cinelli et al. 2007; Pomin and Mourão 2014). Therefore, short- or medium-sized oligosaccharides were prepared from these fucans. We have chosen these two sulfated fucans for our current study because they can produce oligosaccharides of well-defined chemical structures when submitted to mild acid hydrolysis reaction. This method is critical since no enzymes have been reported so far to selectively cleave the invertebrate sulfated fucan. In addition, the mild acid hydrolysis reaction cleaved the sulfated fucans from L. variegatus in a very organized way, leading to uniform and homogeneous fragments in terms of structure and size.
Although producing simple products, the mild acid hydrolysis of Structure 1 is a complex event in terms of kinetics (Pomin, Pereira, et al. 2005; Pomin, Valente, et al. 2005). When this sulfated fucan is submitted to acidification, oligosaccharides of well-defined MWs and chemical structures can be produced through a series of reacting steps. First, a 2-desulfation reaction occurs at the unit A. Secondly, a cleavage at the glycosidic bond between the recently desulfated unit (now labeled as A′) and the adjacent unit (a 4-sulfated unit now labeled as D′) occurs. The hydrolysis of this specific glycosidic bond of the desulfated residue is the main factor for the production of oligosaccharides of well-defined chemical structures. The generated oligosaccharides are multiples of tetrasaccharide repeating units like D′-B-C-A′, D′-B-C-A-D-B-C-A′ and D′-[B-C-A-D]n-B-C-A′ (Pomin, Pereira, et al. 2005; Pomin, Valente, et al. 2005). Thus, all the Fucp units within the products will likely show different sulfation patterns (D, B, C, A and A′), although possessing the same monosaccharide type (Fucp), the same anomeric configuration (α), enatiomeric form (L) and glycosidic bond (C3-linkage position). The resulting oligosaccharides can then be used to correlate the sulfation pattern and residue position within the backbone to the conformational and dynamical behaviors. In our work, we have investigated the acidic depolymerization of sulfated fucan Structure 2 and have established the optimal conditions for producing oligosaccharides with equivalent MWs of the oligosaccharides obtained from hydrolysis of Structure 1. Based on NMR+MD analyses on the oligosaccharides, we were able to understand the role of sulfation patterns, residue position and chain lengths onto the solution conformational and dynamical properties of the sulfated fucan-derived oligosaccharides in solution. From this set of achievements, new explanations for the biological functions (fertilization and anticoagulation) of the studied sulfated fucans can be raised.
Results and discussion
Preparation, MW, and structure of oligosaccharides from sulfated fucans, Structures 1 and 2
Oligosaccharides derived from sulfated fucan Structure 1 were obtained by mild acid hydrolysis using the same condition employed in our previous works (Pomin, Pereira, et al. 2005; Pomin, Valente, et al. 2005). After proper neutralization, 80 mg of hydrolyzed material were applied to a 200 × 1.0 cm Bio-Gel P10 column for fractionation by size-exclusion chromatography (SEC) (Figure 2). The peaks corresponding to the oligosaccharides were labeled by roman numerals after Lv. In order to identify the MWs of these fractionated oligosaccharides, we submitted Lv I, Lv II and Lv III to an SEC analysis using the columns TSK G3000sw+G2000sw coupled to an ultra-fast liquid chromatography (UFLC) system (Figure 3). The two pre-packed columns combined give a fractionation range up to ∼20 kDa. The retention times of these oligosaccharides, 60.92, 57.98 and 55.43 min, respectively, (Figure 3B) were compared with those obtained from the oligosaccharides derived from a low-molecular-weight heparin (LMWH) molecular-weight standard (Figure 3A). Since heparin-based structures are heavier than sulfated fucan oligosaccharides because of the presence of amino sugars and higher sulfation content, we can therefore deduce from the comparative retention times in the UFLC-SEC chromatogram that Lv I, Lv II and Lv III are, respectively, octa-, dodeca- and hexadecasaccharides. Assuming their structures as D′-B-C-A-D-B-C-A′, D′-[B-C-A-D]2-B-C-A′ and D′-[B-C-A-D]3-B-C-A′, their MWs are, respectively, 1897, 2797 and 3776 Da. In order to check the structural integrity of these and the other Lv oligosaccharides, we collected 1D 1H NMR spectra for the majority of the fractions (Supplementary data, Figures S1A) obtained from the Bio-Gel P10 column (Figure 2). Based on these spectra, we confirmed that the tetrasaccharide repeating units is preserved among the Lv oligosaccharide series (Supplementary data, Figure S1B).
Fig. 2.

SEC profile from Bio-Gel P10 column (200 × 1.0 cm) of the oligosaccharides obtained from the 6 h-mild acid hydrolysis of the sulfated fucan Structure 1. The resultant peaks are labeled by roman numerals. The profile was detected by metachromatic property using the dye DMB and absorbance at 525 nm.
Fig. 3.
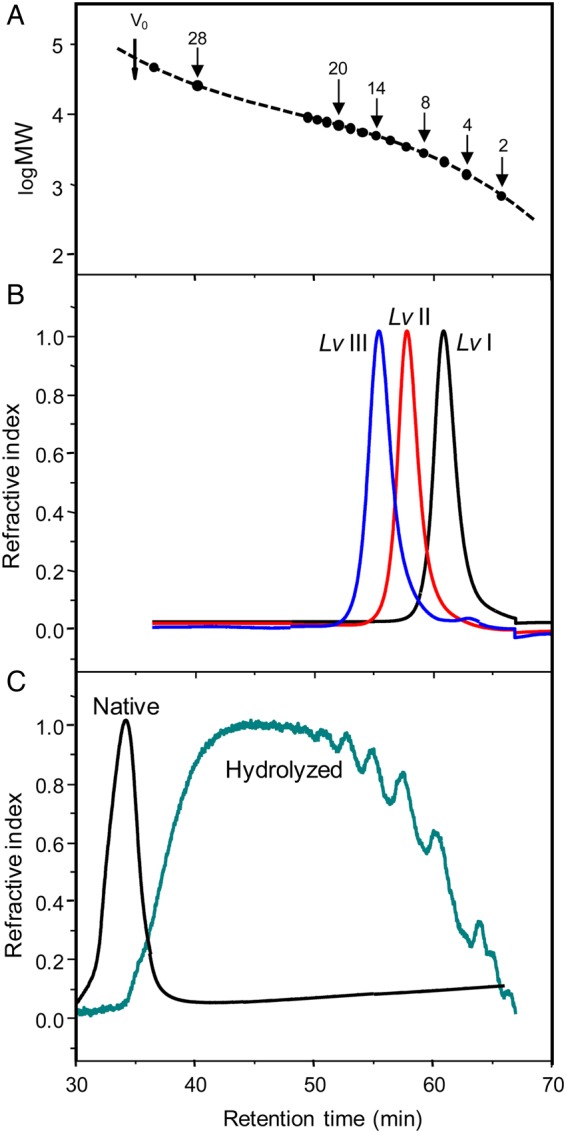
MW determination by SEC-UFLC analyses using a TSK gel G3000sw and TSK gel G2000sw pre-packed columns linked together. (A) Chromatographic profile of oligosaccharides from LMWH standard showing the respective degrees of polymerization of each fraction and the correspondent retention time in the system. Vo was determined applying a native sulfated fucan sample. (B) Chromatographic profiles of the three oligosaccharides derived from hydrolyzed sulfated fucan Structure 1, Lv I (in black), Lv II (in red) and Lv III (in blue). The retention time of these oligosaccharides was 60.92, 57.98 and 55.43 min, respectively. (C) Chromatographic profiles of the native sulfated fucan Structure 2 (black curve) and the 2 h-hydrolyzed sample (green curve). Note the similarity between the oligosaccharide distributions of the hydrolyzed sulfated fucan Structure 2 with the one of the LMWH standard. For color notation, refer to the online version of this figure.
Figure 4 shows a 1H–13C heteronuclear single quantum coherence (HSQC) spectrum of the anomeric 1H–13C pairs of Lv I, the octasaccharide derived from sulfated fucan Structure 1, which will be taken here as a representative oligosaccharide for the NMR and MD conformational and dynamical studies. In the 1H–13C HSQC spectrum and the 1D 1H spectrum of this oligosaccharide (top spectrum of Supplementary data, Figure S1A), as well as in the spectra of the other oligosaccharides (lower spectra of Supplementary data, Figure S1A), there is an extra signal in the anomeric region, assigned as A (δH/C = 5.38/94.1 ppm). This indicates that the 2-desulfation reaction has occurred just partially in unit A of the octasaccharide. The appearance of this 2-desulfation resistant unit is expected based on the sequential events of the mild acid hydrolysis of sulfated fucan Structure 1 (Pomin, Pereira, et al., 2005; Pomin, Valente, et al. 2005). This 2-sulfated unit (A) in the Lv I octasaccharide (D′-B-C-A-D-B-C-A′) hinders further cleavage into two D′-B-C-A′ tetrasaccharides. Note that in the 1H–13C HSQC spectrum of this octasaccharide, there is a downfield displacement of the D1 cross-peak from its original 13C-chemical shift at 97.3 ppm toward a new position of δC at 98.1 ppm (resonance now assigned as D′). This shift of 13C, but not 1H, is certainly not due to desulfation, and more likely due to the terminal position of the unit D′. The 1H signal of D′ overlaps with the one from the D residue and explains why the former resonance is not seen in the 1D 1H NMR spectra of the oligosaccharides (Supplementary data, Figure S1B), and was unassigned in our previous works (Pomin, Pereira, et al. 2005; Pomin, Valente, et al. 2005). Figure 4 also depicts the resonances utilized in the further 13C-based T1 relaxation analysis.
Fig. 4.

1H–13C HSQC spectrum (expansion δH/δC 5.54–4.84/100.0–90.2 ppm, which covers all anomeric cross-peaks) of octasaccharide Lv I obtained from the SEC fractionation displayed at Figure 2. The Fucp units with different sulfation patterns are labeled as the following: A for 2-sulfation, B for 2,4-disulfation, C for 2-sulfation, D for 4-sulfation, A′ for 2-desulfated (non-sulfation) and D′ for the non-reducing end 4-sulfated unit which misses the linked adjacent A unit. The spectrum was recorded at a Varian 600 MHz instrument at 298 K.
After testing various conditions of mild acid hydrolysis on the sulfated fucan Structure 2 (data not shown), the production of oligosaccharides with approximately equivalent MWs of the oligosaccharide distributions of LMWH (Figure 3C versus A), and of the hydrolyzed sulfated fucan Structure 1, was achieved only at a sugar concentration of 5.0 mg/mL submitted to 0.04 M HCl at 60°C during 2 h of reaction. Supplementary data, Figure S2A shows a polyacrylamide gel electrophoresis (PAGE) analysis of the hydrolyzed material compared with the native form, and two other sulfated polysaccharides of known MWs, which were used as molecular markers. The 1D 1H NMR spectrum of the hydrolyzed sulfated fucan Structure 2 (Supplementary data, Figure S2B) matched the spectrum of the native polysaccharide (Cinelli et al. 2007). Since no chemical shift changes were noted in the hydrolyzed sample, the structure [-3)-α-l-Fucp-4()-(1-]n was shown to be preserved in the resulting oligosaccharides.
Ring conformational analyses via NMR- and MD-derived spin–spin coupling constants
Table I compares the NMR experimental, the averaged quantum mechanics (QM)-calculated and the Karplus-derived three-bond proton–proton scalar coupling constant (3JH,H) values obtained for the individual Fucp units of the octasaccharide Lv I. The NMR spin–spin coupling constants were obtained from the double quantum filtered-correlation spectroscopy (DQF-COSY) spectrum of this oligosaccharide (Supplementary data, Figure S3). The DQF-COSY spectrum of Lv I has sufficient resolution of the 3JH1,H2 and 3JH2,H3 multiplets to allow accurate measurements of their coupling constants, whereas the multiplet peaks related to 3JH3,H4 and 3JH4,H5 have high-order chemical shift degeneracy. 3JH1,H2 and 3JH2,H3 of α-l-Fucp are the most reliable vicinal proton pairs for predicting the ring conformation because they generate significantly different coupling constant values when the α-l-Fucp ring adopts different chair conformations. Based on Becker et al. 2007 for 3JH1,H2 and 3JH2,H3 values of 1C4 and 4C1 chair configurations of α-l-Fucp, the following set of values can be assumed: 3JH1,H2 = ∼3.0 Hz and 3JH2,H3 = ∼8.0 Hz for 1C4; and 3JH1,H2 = ∼1.0 Hz and 3JH2,H3 = ∼4.0 Hz for 4C1. All the measured 3JH2,H3 coupling constants for different residues in Lv I were >10 Hz (Table I), indicating that each α-l-Fucp in Lv I maintained a predominantly 1C4 chair conformation in solution. Larger coupling constant values measured directly from DQF-COSY experiments are normal when linewidths are somewhat broad because of cancellation of overlapping parts of the antiphase doublets.
Table I.
NMR, averaged QM-calculated and Karplus-derived 3JH,H coupling constants
| Unita |
3JH1,H2 (Hz) |
3JH2,H3 (Hz) |
|||||
|---|---|---|---|---|---|---|---|
| NMRb | QM calculatedc | Karplus derivedd | NMR | QM calculated | Karplus derived | ||
| α-l-Fucp | 4.4 ± 0.9 | 4.9 ± 0.8 | 2.9 ± 0.5 | 11.6 ± 2.3 | 10.0 ± 0.5 | 7.1 ± 0.3 | |
| Cr | 2-Sulfated α-l-Fucp | 4.5 ± 0.9 | 4.5 ± 1.2 | 3.1 ± 0.5 | 10.2 ± 2.0 | 9.8 ± 0.9 | 7.0 ± 0.4 |
| Br | 2,4-Disulfated α-l-Fucp | 4.9 ± 1.0 | 3.9 ± 0.6 | 3.0 ± 0.5 | 12.1 ± 2.4 | 9.1 ± 0.3 | 7.1 ± 0.4 |
| Dr | 4-Sulfated α-l-Fucp | 4.0 ± 0.8 | 4.4 ± 0.5 | 3.1 ± 0.5 | 11.7 ± 2.3 | 9.6 ± 0.7 | 7.1 ± 0.4 |
| Anr | 2-Sulfated α-l-Fucp | 4.7 ± 0.9 | 3.6 ± 0.6 | 2.8 ± 0.5 | 10.3 ± 2.0 | 10.0 ± 0.4 | 7.1 ± 0.3 |
| Cnr | 2-Sulfated α-l-Fucp | 4.5 ± 0.9 | 4.1 ± 0.9 | 3.0 ± 0.5 | 10.2 ± 2.0 | 10.0 ± 0.6 | 7.0 ± 0.4 |
| Bnr | 2,4-Disulfated α-l-Fucp | 4.9 ± 1.0 | 3.7 ± 0.4 | 3.0 ± 0.5 | 12.1 ± 2.4 | 9.5 ± 0.6 | 7.1 ± 0.4 |
| 4-Sulfated α-l-Fucp | 4.0 ± 0.8 | 5.1 ± 0.3 | 3.1 ± 0.5 | 11.7 ± 2.3 | 9.6 ± 0.4 | 7.0 ± 0.4 | |
aUnits labeled with subscript “nr” belong to the tetrasaccharide repeating units placed at the non-reducing end side, while those units labeled with subscript “r” belong to the tetrasaccharide repeating unit of the reducing end side.
bValues obtained from DQF-COSY spectrum (Supplementary data, Figure S3). Five DQF-COSY spectra were collected under the same conditions for generating average and error values.
cAll quantum calculations were performed at B3LYP/HIII-SU3//HF/6-31++G(2d,2p) level of theory, with all of the fucose ring torsions constrained at the average solution conformation.
dParameterization of the Karplus equation followed (Haasnoot et al. 1980).
QM-calculated scalar coupling constants were measured at B3LYP/HIII-SU3//HF/6-31++G(2d,2p) level of theory, assuming all of Fucp ring torsions were constrained in the solution conformation. Karplus-derived equations according to Haasnoot et al. were used for scalar coupling calculations. The QM-calculated 3JH2,H3 from the representative structures of the MD simulation agreed with the NMR results (Table I). Although all the Karplus equation derived 3JH2,H3 coupling constants were a bit smaller than the experimental both support anti-positions for 3JH2,H3 in all residues. In addition, the QM-calculated 3JH1H2 and 3JH2H3 values also matched the corresponding ones from NMR results. During the course of the simulation, all of the residues in Lv I maintained a 1C4 chair conformation and no ring flips were observed.
Conformational analyses via experimental NOE and theoretical interproton distances
Since the studied oligosaccharides may fall into the MW range that can result in weak NOEs, both nuclear Overhauser effect spectroscopy (NOESY) and rotational frame Overhauser effect spectroscopy (ROESY) data were collected for Lv I and the mixture derived from fucan 2. The NOESY spectra (Figure 5) were very similar to the ROESY spectra (Supplementary data, Figure S4) and so the NOESY was chosen for analysis, since ROE data can be complicated with total correlation spectroscopy artifacts. Table II displays the chemical shifts, the NOE connectivities assigned for both Structures (Figure 5A and B), and the interproton distances derived from both NMR and MD. The correct 1H–1H NOE assignments were supported by the previous assignments using the 1H–1H DQF-COSY (Supplementary data, Figure S3) data. As expected for Fucp units in the 1C4 chair conformation in solution, intra-residue NOE signals of the H1–H2 pairs have distances at ∼2.4 Å (Table II). The inter-reside NOE resonances are mostly those observed between protons across the glycosidic bonds (H1 and H3 of the adjacent unit).
Fig. 5.

1H–1H NOESY spectra of oligosaccharides from sulfated fucans Structures 1 (A) and 2 (B), recorded with mixing time at 250 ms and temperature at 298 K. (A) Octasaccharide Lv I obtained from SEC fractionation (Figure 2) after the 6 h-mild acid hydrolysis with 0.01 M HCl at 60°C. The fucosyl units with different sulfation patterns are labeled as follows: A for 2-sulfation, B for 2,4-disulfation, C for 2-sulfation, D for 4-sulfation, A′ for 2-desulfation (non-sulfation) and D′ for the non-reducing end 4-sulfated unit. The numbers after these letters indicate the respective hydrogens within the sugar chain. Inter-residual NOE signals are always assigned between the anomeric 1H1 and the 1H3 of the subsequent unit. They necessarily correspond to the glycosidic bonds. Intra-residual NOE are always between the anomeric 1H1 and 1H2, or 1H3 for the single case of the disulfated unit labeled as B. (B) Oligosaccharides obtained by the 2 h-mild acid hydrolysis with 0.04 M HCl at 60°C (Supplementary data, Figure S3). Note that there are just two types of NOE connectivities for the oligosaccharides from sulfated fucan Structure 2: the inter-residual NOE between the anomeric 1H1 and 1H3, which represents the glycosidic bonds, and the intra-residual NOE between the anomeric 1H1 and the 1H2. The spectra were recorded at a Bruker 600 MHz instrument.
Table II.
Theoretical and calculated NOEs (intra- and inter-residues), chemical shifts, interproton distances and intensity ranges measured for the Lv I (octasaccharide, Structure 1), and the mixture of oligosaccharides from Structure 2
| Type | Unita and 1H–1H NOEb | F1 (ppm) | F2 (ppm) | Internuclear distance (Å) |
Intensityd | |
|---|---|---|---|---|---|---|
| Experimentalc | MD | |||||
| Lv I (octasaccharide, Structure 1) | ||||||
| Intra | H1–H2 | 3.84 | 5.10 | 2.4 ± 0.00 | 2.4 ± 0.1 | Strong |
| Anr H1–H2 | 4.50 | 5.35 | 2.4 ± 0.03 | 2.4 ± 0.1 | Strong | |
| Br H1–H2 | 4.52 | 5.33 | 2.4 ± 0.03 | 2.4 ± 0.1 | Strong | |
| Bnr H1–H2 | 2.4 ± 0.1 | |||||
| Cr H1–H2 | 4.52 | 5.31 | 2.6 ± 0.06 | 2.4 ± 0.1 | Medium | |
| Cnr H1–H2 | 2.4 ± 0.1 | |||||
| Anr H1–H3 | 4.12 | 5.35 | 3.4 ± 0.1 | 3.8 ± 0.1 | Medium | |
| Br H1–H3 | 3.90 | 5.33 | 3.5 ± 0.09 | 3.8 ± 0.1 | Medium | |
| Bnr H1–H3 | 3.8 ± 0.1 | |||||
| H1–H3 | 3.98 | 5.05 | 3.4 ± 0.08 | 3.8 ± 0.1 | Medium | |
| H1–H2 | 3.82 | 5.05 | 2.3 ± 0.03 | 2.4 ± 0.1 | Strong | |
| Dr H1–H2 | 3.84 | 5.10 | 2.5 ± 0.05 | 2.4 ± 0.1 | Strong | |
| Inter | Anr H1–Dr H3 | 4.07 | 5.35 | 2.6 ± 0.06 | 2.6 ± 0.4 | Medium |
| Cnr H1–Anr H3 | 3.99 | 5.31 | 2.6 ± 0.04 | 2.6 ± 0.2 | Medium | |
| Cnr H1–Anr H4 | 4.06 | 5.31 | 2. 9 ± 0.05 | 2.3 ± 0.3 | Medium | |
| Br H1–Cr H3 | 3.99 | 5.33 | 2.5 ± 0.04 | 2.5 ± 0.3 | Strong | |
| Bnr H1–Cr H3 | 2.6 ± 0.3 | |||||
| Dr H1–Br H3 | 4.23 | 5.10 | 2.1 ± 0.02 | 2.5 ± 0.3 | Strong | |
| Oligosaccharide mixture (Structure 2) | ||||||
| Intra | H1–H2 | 3.90 | 5.14 | 2.4 | – | Strong |
| Inter | H1–H3 | 4.07 | 5.14 | 3.5 | – | Medium |
aUnits labeled with subscript “nr” belong to the tetrasaccharide repeating units placed at the non-reducing end side, while those units labeled with subscript “r” belong to the tetrasaccharide repeating unit of the reducing end side.
bConnectivities are from NOESY spectra of Figure 5, supported by assignments from DQF-COSY spectrum (Supplementary data, Figure S3).
cThe internuclear distances were obtained using the formula NOE = NOEref (rref/r)1/6, in which r denotes the distances between the proton pairs, NOE is the intensity of the peaks and ref is the referential values used for normalization. Since the studied units are Fucp at the exclusive 1C4 ring configuration, the H1–H2 intra-residual NOE-based interproton distance must be 2.4 Å. The D′nr H1–H2 distance of 2.4 Å was used for normalization. Ten NOESY spectra were collected under the same conditions for generating average and error values.
dNOE intensities were set as strong (≤2.5 Å) and medium (>2.5 and ≤3.7 Å).
NOE build-up curves were also generated for the octasaccharide Lv I, hexadecasaccharide Lv III and the oligosaccharide mixture from the sulfated fucan Structure 2 (Figure 6). As expected, the different build-up rates as a function of MW can be seen from the plots of Figure 6A versus B, where the maximum point of the hexadecasaccharide Lv III is 500 ms while the maximum point for the octasaccharide Lv I, is 750 ms. Based on these data, the NOE-based interproton distances can be measured based on the spectra displayed in Figure 5, which were collected at mixing times of 250 ms. This point is near the initial linear region of both curves (Figure 6A and C), and so allows application of the isolated spin-pair approximation for calculating interproton distances.
Fig. 6.

NOE build-up curves of all NOE resonances (inset panels) seen in the series of NOESY spectra of the oligosaccharides from sulfated fucans, Structures 1 (A and B) and 2 (C), recorded at temperature of 298 K. (A) Oligosaccharides Lv I (octasaccharide) and (B) Lv III (hexadecasaccharide) obtained from SEC fractionation (Figure 2) after the 6 h-mild acid hydrolysis with 0.01 M HCl, at 60°C of Structure 1. (C) Oligosaccharides obtained by the 2 h-mild acid hydrolysis with 0.04 M HCl at 60°C of Structure 2 (Supplementary data, Figure S3). Note that the horizontal scale of mixing times is different in (A). The spectra were recorded at Bruker 500 MHz instrument at 298 K.
Since each α-l-Fucp in Lv I maintained a dominant 1C4 chair conformation in solution, the intra-residual proton–proton distances in Lv I should be the same among all residues. Data from the MD simulation showed consistent intra-residue H1–H2 and H1–H3 proton–proton distances among different residues in Lv I (Table II). The average distances of H1–H2 and H1–H3 from the MD simulation (Table II) agreed with the average distances from the Fucp units observed in crystal structures, regardless of ring substitutions or location of the Fucp unit within the oligosaccharide chain (Supplementary data, Table SI). The inter-residual proton–proton distances, H1–H3 and H1–H4 obtained from the MD simulation also agreed quite well with the experimental values (Table II).
Dynamical analyses via NMR spin relaxation: the influence of sulfation patterns
As expected, the longitudinal (T1) spin relaxation times of the 1H anomeric signals from the oligosaccharides of sulfated fucan Structure 1 (Lv I, Lv II and Lv III) increased with temperature and decreased with chain length (Table III). Within each compound, the relaxation times for units A′ and D are longer. The terminal residue A′ would be expected to have a greater degree of motion based on its terminal position, although the lack of sulfation and strong interaction with solvent may also contribute to dynamics. The internal 4-sulfated residue D appears more dynamic than the 2- and 2,4-disulfated units, which suggests that the 2-sulfation might limit motions of the internal units of the Lv oligosaccharides. This is consistent with possible steric hindrance effects caused by the proximity of the 2-sulfation to the (1–3) glycosidic linkage.
Table III.
Longitudinal spin relaxation (T1) (in s) of the anomeric 1H signals of Lv I, Lv II and Lv III (Structure 1); and all 1H signals from the oligosaccharide mixture from Structure 2
|
1H T1 (s)a |
|||
|---|---|---|---|
| 298 K | 310 K | 323 K | |
| Anomeric unit (sulfation type) | Lv I (octasaccharide, Structure 1) | ||
| A H1 (2-sulfated) | 1.13 | 1.15 | 1.31 |
| B H1 (2,4-disulfated) | 1.06 | 1.07 | 1.30 |
| C H1 (2-sulfated) | 1.17 | 1.23 | 1.31 |
| D H1 (4-sulfated) | 1.26 | 1.30 | 1.36 |
| A′ H1 (unsulfated) | 1.30 | 1.36 | 1.37 |
| Anomeric unit (sulfation type) | Lv II (dodecasaccharide, Structure 1) | ||
| A H1 (2-sulfated) | 0.92 | 0.93 | 0.98 |
| B H1 (2,4-disulfated) | 0.81 | 0.87 | 0.91 |
| C H1 (2-sulfated) | 0.94 | 0.94 | 1.02 |
| D H1 (4-sulfated) | 1.17 | 1.16 | 1.21 |
| A′ H1 (unsulfated) | 1.25 | 1.34 | 1.38 |
| Anomeric unit (sulfation type) | Lv III (hexadecasaccharide, Structure 1) | ||
| A H1 (2-sulfated) | 0.74 | 0.76 | 0.94 |
| B H1 (2,4-disulfated) | 0.74 | 0.76 | 0.93 |
| C H1 (2-sulfated) | 0.75 | 0.75 | 0.94 |
| D H1 (4-sulfated) | 0.96 | 1.09 | 1.16 |
| A′ H1 (unsulfated) | 0.96 | 1.09 | 1.18 |
| Hydrogen position | Oligosaccharide mixture (Structure 2) | ||
| H1 | 1.35 | – | – |
| H2 | 0.98 | – | – |
| H3 | 1.44 | – | – |
| H4 | ndb | – | – |
| H5 | 0.85 | – | – |
| H6 | 0.86 | – | – |
aThe T1 values were measured by 1H 1D NMR inversion-recovery experiment at three temperatures using a Varian 500 MHz.
bNot determined due to its close resonance with HOD, this signal was also suppressed by water suppression step of the NMR experiment.
Since spin relaxation of 1H can be influenced by hydrogen bonds or interatomic contacts of the oligosaccharide structures in solution, we also measured 13C-related spin relaxation for the Lv I, Lv II and Lv III (Table IV) using an 1H–13C inversion-recovery filtered HSQC experiment. The 13C-related T1 relaxation times are distributed in a much broader range of values than those seen for 1H nuclei which improves reliability in analyses and interpretation. Table IV shows that T1 relaxation times of the 2-sulfated units are, in general, slightly shorter than those of the terminal non- (A′) and 4-sulfated (D′) units. The differences become as more evident as the oligosaccharide length decreases. This set of results supports the conception that 2-sulfation, within a residue or in the nearby residue, is a restricting factor to the solution dynamics of the oligosaccharides, especially the octasaccharide derived from sulfated fucan Structure 1. Based on Tables III and IV, we can observe that the 4- and non-sulfated units located at the non- and reducing ends of the oligosaccharides are experiencing more dynamics in solution than the units placed at the internal positions.
Table IV.
Longitudinal spin relaxation (T1) (in s) of the anomeric 1H–13C cross-peaks of 1H–13C HSQC spectra of Lv I, Lv II and Lv III (Structure 1); and all 1H–13C cross-peaks from 1H–13C HSQC spectra of the oligosaccharide mixture from Structure 2, measured at 298 K
| NMR resonance | 1H–13C T1 (s)a |
|---|---|
| Anomeric unit (sulfation type) | Lv I (octasaccharide, Structure 1) |
| A H1 (2-sulfated) | 0.69 |
| B H1 (2,4-disulfated) | 0.83 |
| C H1 (2-sulfated) | 0.25 |
| D H1 (4-sulfated) | 1.07 |
| D′ H1(4-sulfated, non-reducing end) | 1.08 |
| A′H1 (unsulfated) | 1.13 |
| Lv II (dodecasaccharide, Structure 1) | |
| A H1 (2-sulfated) | 0.51 |
| B H1 (2,4-disulfated) | 0.51 |
| C H1 (2-sulfated) | n.d.b |
| D H1 (4-sulfated) | 0.22 |
| D′ H1(4-sulfated, non-reducing end) | 0.64 |
| A′ H1(unsulfated) | 0.71 |
| Lv III (hexadecasaccharide, Structure 1) | |
| A H1 (2-sulfated) | 0.26 |
| B H1 (2,4-disulfated) | 0.41 |
| C H1 (2-sulfated) | 0.14 |
| D H1 (4-sulfated) | 0.22 |
| D′ H1(4-sulfated, non-reducing end) | 0.49 |
| A′ H1(unsulfated) | 0.34 |
| Cross-peak within the ring chain | Oligosaccharide mixture (Structure 2) |
| 1H1–13C1 | 0.35 |
| 1H2–13C2 | 0.38 |
| 1H3–13C3 | 0.44 |
| 1H4–13C4 | 0.43 |
| 1H5–13C5 | 0.35 |
| 1H6–13C6 | 0.66 |
aThe T1 values were measured by 2D NMR 1H–13C inversion-recovery filtered HSQC experiment using a Varian 600 MHz.
bNot determined due to resonance overlap.
Relaxation data T1 were also collected for the oligosaccharide mixture derived from sulfated fucan Structure 2 (Tables III and IV). Although longer 1H relaxation values could be seen for 1H1 and 1H3 compared with 1H2, 1H5 and 1H6 (Table III), indicating more mobility close to the glycosylation site, the 13C T1 relaxation values did not show a similar pattern (Table IV), except for the exocyclic C6. It is expected that the methyl 1H–13C pairs should have higher mobility in relationship to ring-belonged 1H–13C pairs, implying thus longer 13C-related T1 values for the former (Table IV). Overall, the dynamic nature of the oligosaccharide mixture from sulfated fucan Structure 2 looks more complex than the series of Lv oligosaccharides and due to structural simplicity a clear influence of sulfation pattern, chain length and unit position on dynamics cannot be stated.
Nonetheless, the relaxation times of the oligosaccharide mixture from Structure 2, in comparison with the series of oligosaccharides derived from Structure 1, showed overall higher dynamical behavior if we consider molecular size in the analyses. Considering the fact that the oligosaccharide mixture from Structure 2 has MW distribution very similar to LMWH (Figure 3), relaxation rates of the oligosaccharide mixture derived from the sulfated fucan Structure 2 should be lower than those observed for the individual hexadecasaccharide Lv III of Structure 1 based on molecular size. But this is not the case. The data are generally indicating that the uniformly 4-sulfated oligosaccharides are more dynamic than the oligosaccharides from Structure 1. This conclusion is in agreement with the fact that 2-sulfation may be participating in reduced dynamics on the oligosaccharides derived from sulfated fucan Structure 1.
Dynamical analyses via MD simulations: how sulfation patterns influence dynamics of fucan oligosaccharides?
In order to verify the influence of sulfate groups on the dynamical properties of Lv I, molecular models of difucoses with different sulfation patterns were built and submitted to MD simulation. Disaccharide composed of 2-sulfated units (difucose A, Figure 7A) showed a dominant glycosidic linkage conformation (state A in Figure 7B), while a disaccharide composed of a 2-sulfated followed by a 4-sulfated unit (difucose B, Figure 7C) adopted two additional major conformations (states B and C in Figure 7D). Although the simulation of a non-sulfated disaccharide (difucose C) explored five glycosidic linkage conformations (states A–E in Figure 7G), the total occupancy of three of the states (states B, D and E) was <5%. As can be seen from the structural differences among difucoses A, B and C, the presence of the sulfate groups and their positions within the ring both influenced the interglycosidic dihedral angle distributions, leading thus to the safe conclusion that the major conformational populations of the difucose units are different.
Fig. 7.

Chemical structures of difucose A (A), difucose B (C) and difucose C (F); dihedral angle (ψ/φ) distributions of difucose A (B), difucose B (D), difucose C (G) in the MD simulation and difucose B (E) in the MD simulation with atomic charges of sulfate groups turned off. Each dot in panels represents a trajectory frame in the MD simulation.
The anomeric and exo-anomeric effects provide significant resistance to torsions about the glycosidic linkage (Foley et al. 2012; Nivedha et al. 2014), which prefers a tight φ/ψ angle distribution. Comparison of structures within the φ/ψ angle distributions for difucoses A and C showed that hydrogen bond interactions between the sulfate group and hydroxyl group across the glycosidic linkage in difucose A may contribute to the tight φ/ψ angle distribution. As a result, difucose A adopted a single glycosidic linkage conformation for most of the simulation, which matches a low-energy conformation of the carbohydrate intrinsic energy functions (Nivedha et al. 2014).
In difucose B, the axial 4-hydroxyl group was replaced by a sulfate group at the reducing end residue of difucose A, which forces the glycosidic linkage to adopt two additional conformations during the course of MD simulations. To understand the origins of these additional conformations, a simulation of difucose B without atomic charges on the sulfate groups was carried out (Figure 7E). During this simulation, the glycosidic linkage of difucose B obtained the same states found in the simulation in which the atomic charges of sulfate groups were on. However, state A gained a marked increase of occupancy from state B, while state C only decreased by 5% (Figure 7E). This information suggests that electrostatic repulsion (Zsiska and Meyer 1993) and steric hindrance between 2- and 4-sulfate groups flanking the glycosidic bond are likely forcing the glycosidic linkage to deviate from the previous dominant conformation (state A in Figure 7B and G), and adopt the two additional conformations (states B and C seen in Figure 7D).
Based on the above observations, it is seen that the glycosidic linkage between two adjacent α-l-Fucp units maintains a dominant low-energy conformation when the sulfation pattern allows inter-residual hydrogen bonds between the sulfate and the hydroxyl groups. In contrast, when the sulfate groups from adjacent residues repel each other, the glycosidic linkage adopts additional conformations, thus enhancing the dynamics of the molecule. A similar influence of sulfation pattern on the dynamics of the difucoses was also observed in Lv I (Figure 8). The sulfation patterns in the first and last three disaccharides (D′nr-Bnr, Bnr-Cnr, Cnr-Anr and Dr-Br, Br-Cr, Cr-A′r, respectively) of Lv I allow the inter-residual hydrogen bonds between the adjacent Fucp. The atomic distances involving potential hydrogens bonds were measured in the simulation of Lv I (Table V; Supplementary data, Table SII). The sulfation pattern between Anr and Dr residues produces the repulsions between the equatorial 2-sulfate and axial 4-sulfate groups. The inter-residual hydrogen bonds play a role to decrease dynamics on the two tetrasaccharide repeating units of Lv I as opposed to the amplified motions produced by the repulsions between the sulfate groups in Anr and Dr residues placed right between the two tetrasaccharide units (the middle) of the octasaccharide Lv I. This proposition is summarized in Figure 9. Dynamic analyses via averaged root-mean-square deviation (RMSD) of the whole octasaccharide compared with their composing tetrasaccharides supports the conclusion that dynamics of the Lv I octasaccharide is strongly influenced by the sulfation-dependent repulsive forces in the central glycosidic bond and is not intrinsically a property of the tetrasaccharide unit (Supplementary data, Figure S5). It is worth noting that the glycosidic linkage in Cr-A′r- was more dynamic than the glycosidic linkages of other disaccharides with inter-residual hydrogen bonds (Figure 8A). This is in good agreement with the longer NMR spin relaxation rates (Tables III and IV).
Fig. 8.

Dihedral angle (ψ/φ) distributions of the glycosidic linkages in Lv I. Each dot in every panel represents a trajectory frame in the MD simulation of a disaccharide block of Lv I.
Table V.
Atomic distances in sulfate-related groups, key inter-residual hydrogen bonds and their occupancies in Lv Ia
| Sulfate group |
Hydroxyl group |
Distanceb (occupancy)c | ||
|---|---|---|---|---|
| Residue | Atomd | Residue | Atom | |
| Cr- | O | O4 | 2.8 ± 0.3 (63) | |
| Br- | O | Cr | O4 | 2.8 ± 0.3 (50) |
| Br- | O | Dr | O1 | 2.7 ± 0.3 (88) |
| Cnr- | O | Anr | O4 | 2.8 ± 0.3 (61) |
| Bnr- | O | Cnr | O4 | 2.8 ± 0.3 (51) |
| Bnr- | O | O2 | 2.7 ± 0.3 (100) | |
| Sulfate group | Sulfate ester | Distance | ||
| Residue | Atom | Residue | Atom | |
| Anr- | O | Dr | O4 | 5.3 ± 0.7 |
aSee schematic representation at Figure 9.
bIn Å.
cOccupancy is calculated in percentages (%), based on a distance between oxygen atoms in sulfate groups and hydroxyl groups of <3.5 Å. Each of the occupancies of the interaction listed is the sum of all the individual hydrogen bonds between the oxygen atoms in sulfate group with hydroxyl group and the distance is the average of all the individual hydrogen bonds, unless it is otherwise noted.
dOxygen in the different groups.
Fig. 9.

Schematic representation for explaining the sulfation pattern-related hydrogen bonds (in blue) and repulsive effects (in red) at (A) the Lv I octasaccharide structure, and (B) atomic distances of units. (A) Note that inter-residual hydrogen bonds play a role to decrease dynamics on the two tetrasaccharide repeating units (***labelled with “nr” for non-reducing end and “r” for reducing end) as opposed to the amplified motions occasioned by the repulsive forces between the sulfate groups in Anr-Dr disaccharide located right between the two tetrasaccharide sequences. (B) The atomic distances of sulfate-related groups and key inter-residual hydrogen bonds of adjacent units in Lv I octasaccharide are reported in Table V and Supplementary data, Table SII. For color notation, refer to the online version of this figure.
Conclusions
In this work, we report experimental and theoretical conformation and dynamic analyses of oligosaccharides from two sulfated fucans of the sea urchin L. variegatus. The glycosidic bonds were shown to be the main spots of flexibility in the sulfated fucan oligosaccharides since dynamics do not originate from changes in the ring configuration as the 1C4 chair configuration dominates in the composing Fucp units (Table I). The MD-based analyses have pointed to a very selective dynamical process right at the middle of the chain in the Lv I octasaccharide (Figure 8 and Supplementary data, Figure S5) likely due to sulfation-dependent electrostatic repulsive forces and steric hindrance effects (Figure 7). These effects are suggested to be promoted by spatially proximal axial 4-sulfate and equatorial 2-sulfate groups flanking the glycosidic bond of the central disaccharide. This shows curiously that the mobility of the sulfated fucan Structure 1 is centered right between the composing tetrasaccharide repeating units. Moreover, it helps to explain, aside from more accessibility to solvent, the amplified motions at the terminal non- and 4-sulfated Fucp units as observed by spin relaxation data (Tables III and IV) and plots of dihedral angle distribution (Figure 8). The internal units (mostly 2-sulfated) exhibit dynamics strongly regulated by hydrogen bonds. A picture outlining the key hydrogen bonds and the center dynamical spot in the Lv I octasaccharide structure is shown at Figure 9A. A scheme showing the distances of atoms nearby the interglycosidic vectors of disaccharides with sulfation-caused hydrogen bonds and electrostatic repulsion forces is depicted in Figure 9B.
Naturally, one could question the impact of the current results on the biological activities of the sulfated fucans either as species-specific inducers of the acrosome reaction in sea urchin fertilization or as new anticoagulants. Regarding the fertilization process, the structural requirements of the sulfated fucans involved in this activity have been elucidated (Pomin and Mourão 2008; Vilela-Silva et al. 2008). Clearly, the activity for 3-linked α-l-sulfated fucans depends considerably on the particular sulfation patterns which 2-O- and 4-O-positions are the exclusive sites of sulfation. But, as demonstrated here, sulfation patterns influence directly conformation and dynamics of sulfated fucans. And these features can be taken as important regulators in biologically active or inactive glycans. This hypothesis is based on the fact that the sulfated fucan Structure 1, which is composed by the tetrasaccharide repeating motifs of restricted mobility but linked through dynamical bonds, is active to induce the acrosome reaction in homologous sperm—while the uniformly 4-sulfated fucan (Structure 2) is inactive, likely because of a missing reactive structural motif formed by a oligosaccharide sequence of defined sulfation pattern and reduced flexibility.
Although the anticoagulant effect of sea urchin sulfated polysaccharides is far from its natural role as acrosome reaction inducers, the structure–function relationship regarding this effect has been considerably proposed (Pomin 2012a;2012b). It has been postulated that 2-O- or 2,4-di-O-sulfations are anticoagulant motifs in sulfated fucans made up of 3-linked α-l-Fucp units, as opposed to 4-O-sulfation which contributes to lose activity (Pomin 2012b,2014b; Pomin and Mourão 2014). Based on the fact that the sulfated fucan Structure 1 is anticoagulant, whereas sulfated fucan Structure 2 is not, the tetrasaccharide repeating unit, mostly 2-sulfated, indicates a contributing motif to the anticoagulant action. This tetrasaccharide sequence may be working in anticoagulation like the heparin pentasaccharide of high affinity for antithrombin likely because of its structure dynamically reduced and capable of better interactions with the coagulation cofactors. Although future investigations are still needed to affirm these structure–function relationship hypotheses, this work has provided a novel set of information which might be useful to explain the levels of activity of certain sulfated fucan in sea urchin fertilization and anticoagulation.
Materials and methods
Materials
The sodium salt of chondroitin sulfate A from bovine trachea (MW of ∼40 kDa) and Sephadex G-15 resin (fractionation range of dextrans <1.5 kDa) were purchased from Sigma-Aldrich Co (St. Louis, MO). LMWH standard (lot 05/112) comes from National Institute for Biological Standards and Control (NIBISC, UK). A pre-packed TSK G3000sw and TSK G2000sw SEC columns (both 7.5 × 300 mm) were purchased from Tosoh Biosciences (San Francisco, CA). Bio-Gel P-10 gel resin (fractionation range of dextrans from ∼1.5 to ∼20 kDa) in fine polyacrylamide beads, and the polypropylene chromatographic columns (200 × 1.0 cm for oligosaccharide fractionation and 1.0 × 50 cm for desalting) were purchased from Bio-Rad Life Science (Hercules, CA). Three and 5 mm glass NMR tubes were purchased from Wilmad-LabGlass (Vineland, NJ). Deuterium oxide “100%” (D 99.9%) were purchased from Cambridge Isotope Laboratories, Inc. (Andover, MA). Reagents were purchased from Merck do Brasil (Rio de Janeiro, RJ).
Purification of sulfated fucans
Sea urchin specimens of L. variegatus were collected from the sea shore in Urca, Rio de Janeiro, Brasil, and brought to the laboratory. Eggs from mature females of the sea urchins were spawned into filtered sea water after intracelomic injection of 0.5 M KCl (∼5 mL per specimen). The egg jelly, which is sorted out from the male sperms due to its yellowish color, was separated by pH shock, as described previously (Vilela-Silva et al. 1999). Sulfated polysaccharides were extracted from the egg jelly by papain digestion and partially purified by ethanol precipitation (Albano and Mourão 1986). Sulfated fucans were purified by anion-exchange chromatography, and their purity was checked by agarose gel electrophoresis and NMR spectroscopy, as previously performed (Alves et al. 1997;1998; Vilela-Silva et al. 1999, 2002; Pomin, Pereira, et al., 2005; Pomin, Valente, et al. 2005).
Mild acid hydrolysis
Sulfated fucan Structure 1 (∼80 mg) was dissolved in 10.0 mL of 0.01 M and maintained at 60°C for 6 h. Sulfated fucan Structure 2 (∼80 mg) was dissolved in 16.0 mL of 0.04 M HCl and maintained at 60°C for 2 h. After this depolymerization procedure, the pH was neutralized by addition of equal volumes (10.0 or 16.00 mL) of ice-cold 0.01 or 0.04 M NaOH (Pomin, Pereira, et al. 2005; Pomin, Valente, et al. 2005). Hydrolyzed samples were then freeze-dried and ressuspended in 3.0 mL of distilled water.
SEC fractionation and analysis
The oligosaccharides formed after 6 h (Structure 1) and 2 h (Structure 2) of mild acid hydrolysis of the sulfated fucans (80 mg each) were fractionated on a Bio-Gel P-10 column (200 × 1.0 cm), equilibrated with aqueous 10% ethanol, containing 1.0 M NaCl, as described previously (Pomin et al. 2010). The column was eluted with the same solution at a flow rate of 4 mL h−1 and fractions of 1.0 mL were collected and assayed by metachromasia using 1,9-dimethylmethylene blue (DMB) (Farndale et al. 1986). The elution volume of blue dextran and cresol red indicates the Vo and Vt of the column, respectively. The fractions containing the various oligosaccharides (as indicated by the metachromatic absorbance measured at 525 nm, Figure 2) were individually pooled, freeze-dried and dissolved in 2.0 mL distilled water. These solutions were desalted on a Sephadex G-15 column (1.0 × 50 cm). Fractions of 0.5 mL were collected and their conductivities determined. The oligosaccharides were detected by metachromasia (Farndale et al. 1986). Fractions containing the desalted oligosaccharides were individually pooled and freeze-dried for further analyses.
The retention times of Lv I, Lv II, Lv III and LMWH-derived oligosaccharides were compared by individual applications (200 μg of each sample) into a Tosoh Biosciences TSK gel G3000sw and TSK gel G2000sw pre-packed columns coupled to UFLC systems, as described previously (Glauser et al. 2012). For a better resolution and fractionation, the columns were linked together. The system was eluted with 0.1 M ammonium acetate at 298 K at a flow rate of 0.3 mL min−1. The LMWH standard was monitored simultaneously by 232 nm (insaturated 4,5 double-bond at the non-reducing end uronic acid residue) and by refractive index for unspecific detection of sugar-containing materials.
PAGE
The native and degraded derivatives of the sulfated fucan Structure 2 (∼10 µg of each) were applied to a 6% 1-mm thick polyacrylamide gel slab in 0.06 M Tris–HCl (pH 8.6) and run for ∼60 min at 100 V. After the electrophoresis, the sulfated polysaccharides were stained with 0.1% toluidine blue in 1% acetic acid and washed for ∼2 h in 1% acetic acid. The MWs were estimated by comparison with the electrophoretic mobility of standard compounds. The standards were chondroitin 4-sulfate from bovine trachea (∼40 kDa) and LMWH (∼6.2 kDa).
NMR experiments
1H- and 13C-related spectra of the sulfated fucans and their oligosaccharide derivates were recorded by a Bruker DRX 600, Bruker DRX 500 MHz, Varian 600 or Varian 500 MHz, all with triple resonance probes. When 5 mm NMR tubes were used, ∼5.0 mg of sample were dissolved in 0.6 mL 99.9% D2O. When the 3-mm NMR tubes were used, ∼2.0 mg of sample were dissolved in 160 μL 99.9% D2O. 1D 1H NMR inversion-recovery experiment was set using delay intervals of 0.0625, 0.125, 0.25, 0.5, 1, 2, 4, 8 and 16 s. 1H–1H COSY, 1H–1H NOESY and 1H–13C HSQC spectra were recorded at 298 K using states-time proportion phase incrementation for quadrature detection in the indirect dimension. NOESY and ROESY spectra were run with 4046 × 400 points with multiple mixing times: 0, 0.25, 0.5, 0.75, 1, 1.25, 1.5, 1.75, 2, 2.25 and 2.5 s. 1H–13C HSQC spectra were run with 1024 × 256 points and globally optimized alternating phase rectangular pulses for decoupling, filtering by inversion recovery. The delay times used in the 1H–13C HSQC experiments for obtaining the longitudinal 13C-related T1 relaxation values were 0, 0.05, 0.1, 0.2, 0.4, 0.6, 0.8 and 1.2 s. Chemical shifts are relative to external trimethylsilylpropionic acid at 0 ppm for 1H and to methanol for 13C. 10 NOESY and 5 DQF-COSY spectra were collected at the same experimental conditions for measurements of average and error values.
MD simulation setup
The initial structures of Lv I and sulfated difucoses were built from carbohydrate builder on GLYCAM website (http://glycam.org). The structure and atomic charges of Lv I and sulfated difucoses were described by employing the GLYCAM06 (Kirschner et al. 2008) (version h) force field parameters and then solvated with an 8 Å TIP3P water buffer in octahedral box using LEaP program in AMBER14 package (Case et al. 2014). Sodium ions were added to neutralize the solvated system.
The energy minimizations for the solvated Lv I and sulfated difucoses were performed separately under constant volume (500 steps steepest descent, followed by 24,500 steps of conjugate-gradient minimization). Each system was then heated to 300 K over a period of 50 ps and followed by an equilibration at 300 K for 0.5 ns using nPT ensemble, with the Berendsen thermostat (Berendsen et al. 1984) for temperature control in AMBER. All covalent bonds involving hydrogen atoms were constrained using SHAKE algorithm (Vangunsteren and Berendsen 1977), allowing a simulation time step of 2 fs throughout the simulations. After the equilibration, each simulation was carried out with GPU implementation (Gotz et al. 2012) from AMBER14 and trajectory frames were collected at every 1 ps for the total of 450 ns. IOne to 4 non-bonded interactions were not scaled (Kirschner and Woods 2001) and non-bonded interaction cutoff was set to 8 Å.
MD-based scalar proton–proton coupling constants and internuclear distances
Ten representative structures of Lv I extracted from the simulation were the trajectory frames with the lowest RMSD values comparing with the average structure of Lv I from the simulation. Each of the fucoses in these representative structures of Lv I was isolated, then the missing atoms for each fucose were added by GaussView03 (Dennington et al. 2003) software package. The QM spin–spin coupling constants calculations of each Fucp were computed at B3LYP/HIII-SU3//HF/6-31++G(2d,2p) level of theory in Gaussian09 software package (Frisch et al. 2009) with the fucose ring constrained at its conformation in the MD simulation.
The Karplus equation derived spin–spin coupling constants were calculated with a generic Karplus equation (Haasnoot et al. 1980). The electronegativity parameters for the generic Karplus equation were determined by the atoms attached to the carbon atoms in the ring. The torsion angle values of H1-C1-C2-H2 and H2-C2-C3-H3 for each fucose in Lv I were applied to the generic Karplus equation with appropriate electronegativity parameters and the final results were averaged over the 450,000 trajectory frames.
Supplementary data
Supplementary data for this article are available online at http://glycob.oxfordjournals.org/.
Conflict of interest
None declared.
Funding
This study was supported by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) nominated to P.A.S.M. and V.H.P. I.N.L.Q. is a doctorate candidate at Institute of Medical Biochemistry Leopoldo de Meis, UFRJ, with a scholarship from CNPq (141872/2013-6). J.H.P. was partially supported by a grant from the US National Institutes of Health (NIH), P41 GM103390. R.J.W. acknowledges the supports from NIH, grants R01 GM094919 (EUREKA) and P41 GM103390, and the Science Foundation of Ireland, 08/IN.1/B2070. The content of this work is solely the responsibility of the author and does not necessarily represent the official views of the funding agencies.
Abbreviations
DQF-COSY, double quantum filtered-correlation spectroscopy; Fucp, fucopyranose; HSQC, heteronuclear single quantum coherence; LMWH, low-molecular-weight heparin; MD, molecular dynamics; MW, molecular weight; NMR, nuclear magnetic resonance; NOE, nuclear Overhauser effect; NOESY, nuclear Overhauser effect spectroscopy; PAGE, polyacrylamide gel electrophoresis; QM, quantum mechanics; RMSD, root-mean-square deviation; ROE, rotation frame Overhauser effect; ROESY, rotational frame Overhauser effect spectroscopy; SEC, size-exclusion chromatography; UFLC, ultra-fast liquid chromatography.
Supplementary Material
Acknowledgements
We are grateful to Dr David Live (associate research scientist from CCRC, UGA) for giving us some useful advice on the 13C-related spin relaxation analyses, Adriana A. Piquet (UFRJ) for assistance with the UFLC-SEC analysis on the sulfated fucan oligosaccharides, and Prof. Ana M.F. Tovar for helping the molecular-weight determination.
References
- Albano RM, Mourão PAS. 1986. Isolation, fractionation, and preliminary characterization of a novel class of sulfated glycans from the tunic of Styela plicata (Chordata, Tunicata). J Biol Chem. 261:758–765. [PubMed] [Google Scholar]
- Alves AP, Mulloy B, Diniz JA, Mourão PAS. 1997. Sulfated polysaccharides from the egg jelly layer are species-specific inducers of acrosomal reaction in sperms of sea urchins. J Biol Chem. 272:6965–6971. [DOI] [PubMed] [Google Scholar]
- Alves AP, Mulloy B, Moy GW, Vacquier VD, Mourão PAS. 1998. Females of the sea urchin Strongylocentrotus purpuratus differ in the structures of their egg jelly sulfated fucans. Glycobiology. 8:939–946. [DOI] [PubMed] [Google Scholar]
- Becker CF, Guimarães JA, Mourão PA, Verli H. 2007. Conformation of sulfated galactan and sulfated fucan in aqueous solutions: Implications to their anticoagulant activities. J Mol Graph Model. 26:391–399. [DOI] [PubMed] [Google Scholar]
- Berendsen HJC, Postma JPM, Vangunsteren WF, Dinola A, Haak JR. 1984. Molecular-dynamics with coupling to an external bath. J Chem Phys. 81:3684–3690. [Google Scholar]
- Biermann CH, Marks JA, Vilela-Silva AC, Castro MO, Mourão PA. 2004. Carbohydrate-based species recognition in sea urchin fertilization: Another avenue for speciation? Evol Dev. 6:353–361. [DOI] [PubMed] [Google Scholar]
- Case DA, Babin V, Berryman JT, Betz RM, Cai Q, Cerutti DS, Cheatham TE, Darden TA, Duke RE, Gohlke H, et al. 2014. AMBER 14. San Francisco: University of California. [Google Scholar]
- Cinelli LP, Castro MO, Santos LL, Garcia CR, Vilela-Silva ESA-C, Mourão PA. 2007. Expression of two different sulfated fucans by females of Lytechinus variegatus may regulate the seasonal variation in the fertilization of the sea urchin. Glycobiology. 17:877–885. [DOI] [PubMed] [Google Scholar]
- Dennington RI, Keith T, Millam JM, Eppinnett K, Hovell WL. 2003. GaussView, Version 3.0. Shawnee Mission, KS: Semichem, Inc. [Google Scholar]
- Farndale RW, Buttle DJ, Barret AJ. 1986. Improved quantification and discrimination of sulphated glycosaminoglycans by use of dimethylmethylene blue. Biochim Biophys Acta. 883:173–177. [DOI] [PubMed] [Google Scholar]
- Foley BL, Tessier MB, Woods RJ. 2012. Carbohydrate force fields. Comput Mol Sci. 2:652–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, et al. 2009. Gaussian 09. Wallingford, CT: Gaussian, Inc. [Google Scholar]
- Glauser BF, Vairo BC, Oliveira SN, Cinelli LP, Pereira MS, Mourão PA. 2012. Structure and haemostatic effects of generic versions of LMWHs available for clinical use in Brazil: similarity to the original drug. Thromb Haemost. 107:302–314. [DOI] [PubMed] [Google Scholar]
- Gotz AW, Williamson MJ, Xu D, Poole D, Le Grand S, Walker RC. 2012. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized born. J Chem Theory Comput. 8:1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haasnoot CAG, de Leeuw FAAM, Altona C. 1980. The relationship between proton-proton NMR coupling constants and substituent electronegativities-I: An empirical generalization of the Karplus equation. Tetrahedron. 36:2783–2792. [Google Scholar]
- Kirschner KN, Woods RJ. 2001. Solvent interactions determine carbohydrate conformation. Proc Natl Acad Sci USA. 98:10541–10545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner KN, Yongye AB, Tschampel SM, Gonzalez-Outeirino J, Daniels CR, Foley BL, Woods RJ. 2008. GLYCAM06: A generalizable biomolecular force field. Carbohydrates. J Comput Chem. 29:622–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourão PA. 2007. A carbohydrate-based mechanism of species recognition in sea urchin fertilization. Braz J Med Biol Res. 40:5–17. [DOI] [PubMed] [Google Scholar]
- Mulloy B, Forster MJ. 2000. Conformations and dynamics of heparin and heparan sulfates. Glycobiology. 10:1147–1156. [DOI] [PubMed] [Google Scholar]
- Mulloy B, Ribeiro AC, Alves AP, Vieira RP, Mourão PA. 1994. Sulfated fucans from echinoderms have a regular tetrasaccharide repeating unit defined by specific patterns of sulfation at the O-2 and O-4 positions. J Chem Biol. 269:22113–22123. [PubMed] [Google Scholar]
- Nivedha AK, Makeneni S, Foley BL, Tessier MB, Woods RJ. 2014. Importance of ligand conformational energies in carbohydrate docking: Sorting the wheat from the chaff. J Comput Chem. 35:526–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomin VH. 2009. Review: An overview about the structure-function relationship of marine sulfated homopolysaccharides with regular chemical structures. Biopolymers. 91:601–609. [DOI] [PubMed] [Google Scholar]
- Pomin VH. 2012a. Fucanomics and galactanomics: Current status in drug discovery, mechanism of action and role of the well-defined structures. Biochim Biophys Acta. 1820:1971–1979. [DOI] [PubMed] [Google Scholar]
- Pomin VH. 2012b. Structure-function relationship of anticoagulant and antithrombotic well-defined sulfated polysaccharides from marine invertebrates. Adv Food Nutr Res. 65:195–209. [DOI] [PubMed] [Google Scholar]
- Pomin VH. 2014a. Solution NMR conformation of glycosaminoglycans. Prog Biophys Mol Biol. 114:61–68. [DOI] [PubMed] [Google Scholar]
- Pomin VH. 2014b. Anticoagulant motifs of marine sulfated glycans. Glycoconj J. 31:341–344. [DOI] [PubMed] [Google Scholar]
- Pomin VH, Mourão PA. 2008. Structure, biology, evolution, and medical importance of sulfated fucans and galactans. Glycobiology. 18:1016–1027. [DOI] [PubMed] [Google Scholar]
- Pomin VH, Mourão PA. 2014. Specific sulfation and glycosylation – A structural combination for the anticoagulation of marine carbohydrates. Front Cell Infect Microbiol. 6(3):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomin VH, Pereira MS, Valente AP, Tollefsen DM, Pavão MS, Mourão PA. 2005. Selective cleavage and anticoagulant activity of a sulfated fucan: Stereospecific removal of a 2-sulfate ester from the polysaccharide by mild acid hydrolysis, preparation of oligosaccharides, and heparin cofactor II-dependent anticoagulant activity. Glycobiology. 15:369–381. [DOI] [PubMed] [Google Scholar]
- Pomin VH, Sharp JS, Li X, Wang L, Prestegard JH. 2010. Characterization of glycosaminoglycans by 15N NMR spectroscopy and in vivo isotopic labeling. Anal Chem. 82:4078–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomin VH, Valente AP, Pereira MS, Mourão PA. 2005. Mild acid hydrolysis of sulfated fucans: A selective 2-desulfation reaction and an alternative approach for preparing tailored sulfated oligosaccharides. Glycobiology. 15:1376–1385. [DOI] [PubMed] [Google Scholar]
- Vangunsteren WF, Berendsen HJC. 1977. Algorithms for macromolecular dynamics and constraint dynamics. Mol Phys. 34:1311–1327. [Google Scholar]
- Vilela-Silva A-CES, Alves AP, Valente AP, Vacquier VD, Mourão PAS. 1999. Structure of the sulfated α-L-fucan from the egg jelly coat of the sea urchin Strongylocentrotus franciscanus: Patterns of preferential 2-O- and 4-O-sulfation determine sperm cell recognition. Glycobiology. 9:927–933. [DOI] [PubMed] [Google Scholar]
- Vilela-Silva A-CES, Castro MO, Valente AP, Biermann CH, Mourão PAS. 2002. Sulfated fucans from the egg jellies of the closely related sea urchins Strongylocentrotus droebachiensis and Strongylocentrotus pallidus ensure species-specific fertilization. J Biol Chem. 277:379–387. [DOI] [PubMed] [Google Scholar]
- Vilela-Silva A-CES, Hirohashi N, Mourão PA. 2008. The structure of sulfated polysaccharides ensures a carbohydrate-based mechanism for species recognition during sea urchin fertilization. Int J Dev Biol. 52:551–559. [DOI] [PubMed] [Google Scholar]
- Zsiska M, Meyer B. 1993. Influence of sulfate and carboxylate groups on the conformation of chondroitin sulfate related disaccharides. Carbohydr Res. 243:225–258. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.