

Key Points
Viral tropism is the ability of a given virus to productively infect a particular cell (cellular tropism), tissue (tissue tropism) or host species (host tropism). Various host innate immune antiviral cytokines, in particular the interferons (IFNs) and tumour necrosis factor (TNF), have a role in mediating viral tropism at these different levels.
Type I IFNs have a key role in determining the tropism of various viruses. These IFNs probably mediate their effects through the induction of interferon-stimulated genes; however, the exact genes that determine tropism for each virus have not been fully characterized.
Type II IFN has a more limited role in determining viral tropism, contributing mainly in the central nervous system.
The ability of type III IFNs to dictate viral tropism is largely determined by the tissue-specific expression of the type III IFN receptor. Type III IFNs probably have a major role in determining viral tropism in tissues and cells of epithelial origin.
TNF influences viral tropism through altering the expression of cell surface receptors required for viral infection. TNF can alter viral tropism in both a positive and negative manner.
Pro-inflammatory cytokines, particularly the IFNs, might be good therapeutic agents against various viruses that are capable of causing zoonotic infections. However, a better understanding of the mechanisms involved in these treatments is needed.
Defects in the IFN and TNF responsiveness of cancer cells can be exploited to create tumour-specific viral infections in an approach known as viral oncolysis. The synergistic responses of multiple cytokines might have a key role in this phenomenon.
Grant McFadden and colleagues discuss how the interferons and tumour necrosis factor can establish an intracellular antiviral state that determines the specificity of a particular virus for a specific cell type, tissue or species. Our knowledge of these antiviral cytokines can be exploited to enhance immunity against zoonotic infections and to improve the therapeutic specificity of tumour-targeted viruses.
Abstract
The specificity of a given virus for a cell type, tissue or species — collectively known as viral tropism — is an important factor in determining the outcome of viral infection in any particular host. Owing to the increased prevalence of zoonotic infections and the threat of emerging and re-emerging pathogens, gaining a better understanding of the factors that determine viral tropism has become particularly important. In this Review, we summarize our current understanding of the central role of antiviral and pro-inflammatory cytokines, particularly the interferons and tumour necrosis factor, in dictating viral tropism and how these cytokine pathways can be exploited therapeutically for cancer treatment and to better counter future threats from emerging zoonotic pathogens.
Main
Viral infection is a profound challenge to host survival, in which the ability of the virus to replicate and spread is countered by the antiviral defence mechanisms that are mounted by the host. Based on the outcome of these sequential interactions, which begin almost immediately after virus challenge, a viral infection can be either productive (that is, progeny viruses are produced, new cells are infected and the virus transmission continues) or abortive (that is, progeny viruses are not produced or virus dissemination is blocked). For most viruses, the results of the initial infection can vary widely depending on the site of entry, the cell types infected, the responses of local sentinel immune cells, the architecture and vasculature of the tissues involved, and the host species being infected1,2. The summation of these variable elements during viral infection has led to the concept of viral tropism. In a general sense, viral tropism refers to the ability of a given virus to productively infect a particular cell (cellular tropism), tissue (tissue tropism) or host species (host tropism) (Fig. 1). For example, if a particular virus can productively infect rabbits but not other vertebrate hosts such as humans, that virus would be said to have a host tropism limited to rabbits. Similarly, if the same virus could productively infect rabbit macrophages but not any other rabbit cells, that virus would be said to have a cellular tropism limited to macrophages. Importantly, this definition of tropism can only be applied to viruses with lytic replication cycles that produce progeny viruses. Viruses that establish latent infections can successfully enter cells and maintain the ability to produce infectious virus, but while they remain latent they never actually undergo productive replication. It is therefore debatable whether such latent infections are 'productive' or not and in this Review we focus only on the tropism of viruses that actively replicate and disseminate through multiple cells, tissues and hosts. We consider the roles of various host innate immune antiviral cytokines, in particular the interferons (IFNs) and tumour necrosis factor (TNF), in mediating viral tropism at these various levels.
Figure 1. Levels of viral tropism.
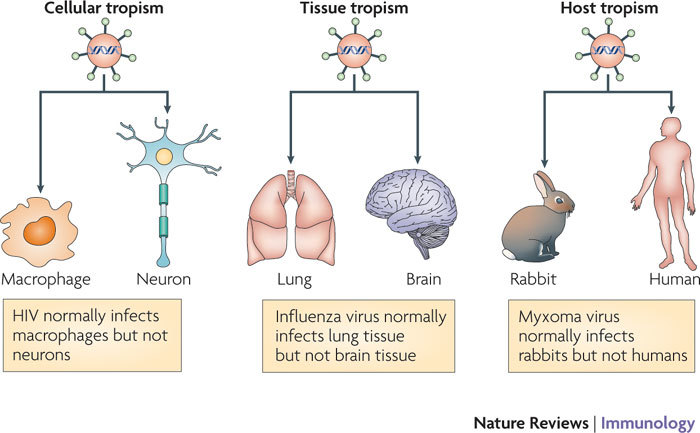
Viral tropism can be divided into three distinct categories depending on the physiological level at which it is measured. Tropism in which the virus replicates in one cell type but not another is known as cellular tropism , tropism in which the virus replicates in a particular tissue or organ but not another is known as tissue tropism, and tropism in which the virus replicates in one host species but not another is known as host tropism.
Infection of vertebrate hosts by most viruses begins with the breaching of the natural barriers that are designed to protect the host from challenge, such as the skin, mucus, saliva, stomach acid and tears. Once the virus finds its first sensitive cellular target, often in the local tissue or through the respiratory or gastrointestinal tracts, the infection itself is usually initiated by binding of the virion to the cell surface, often with the aid of host cell surface receptors that are hijacked from their normal cellular functions. For example, HIV-1 requires the presence of the T cell co-receptor CD4, CC-chemokine receptor 5 (CCR5) and CXC-chemokine receptor 4 (CXCR4) to bind and enter cells3,4, whereas human rhinovirus requires intercellular adhesion molecule 1 (ICAM1)5,6. As these receptors are often expressed in a cell type-specific manner — for example, CD4, CCR5 and CXCR4 are mainly expressed by T cells — their availability for virus binding is considered to be a primary determinant of cellular and tissue tropism. However, following identification of the binding and/or entry receptors for many viruses, it has become apparent that the distribution of receptors for a particular virus is frequently much wider than the distribution of functionally susceptible cells for that virus7. Moreover, some viruses, such as poxviruses, can bind and enter cells and initiate infection without making use of any obvious cell surface receptor8. However, poxviruses cannot productively replicate in all of the cell types that they successfully enter8. This indicates that viral tropism at the cellular level can be affected by downstream cellular factors that are distinct from the availability of any cell surface receptor used by the virus9,10, such as the cell lineage, the activation state of the cell, the route of infection and the ability of the virus to reach certain tissues, the availability of host factors and various intrinsic or induced antiviral signalling pathways8 (Table 1).
Table 1.
Host factors determining viral tropism
| Determinants of viral tropism | Example | Factor(s) involved | Type of tropism | Refs |
|---|---|---|---|---|
| Cell surface binding or entry receptors | HIV-1 | Presence of CD4, CCR5 and CXCR4 receptors on T cells required for cell entry | Cellular | 3,4 |
| Antiviral signalling by cytokines | Myxoma virus | ERK-dependent type I IFN signalling restricts productive infection | Host and cellular | 39 |
| Availability of host factors | Hepatitis B virus | The nuclear hormone receptors hepatocyte nuclear factor 4 and retinoid X receptor-α as well as peroxisome proliferator-activated receptor-α, which positively regulate viral RNA synthesis, are enriched in the liver | Tissue and cellular | 118 |
| Route of entrance | Influenza A virus | Trachea and bronchi | Cellular | 119 |
| Cell lineage | Influenza A virus | Lung cells | Cellular | 69 |
| Activation state of the cell | Parvovirus H-1 | Constitutive production of higher levels of nitric oxide and superoxide anion resticts virus infection | Cellular | 120 |
| CCR5, CC-chemokine receptor 5; CXCR4, CXC-chemokine receptor 4; ERK, extracellular signal-regulated kinase; IFN, interferon. | ||||
One mechanism by which a host can rapidly alter the intracellular milieu to inhibit viral replication is the induction and secretion of self-protective cytokines11. Cytokines, such as the IFNs, TNF and many immunoregulatory interleukins, are among the earliest host defence factors to be induced following various viral infections. Each induced cytokine binds to unique cognate cellular receptors, which are frequently ubiquitously expressed by most somatic cells, and thereby mediates distinct intracellular signalling events that help to inhibit viral infection. This inhibition can occur either directly, by inducing a specific antiviral state in which virus replication is blocked in responding cells (Table 2), or through the regulation of the adaptive immune response (summarized elsewhere12,13,14,15). TNF, IFNs and the interleukins can all inhibit viral replication through both of these mechanisms; however, TNF and the IFNs more frequently inhibit viral replication through the direct induction of an intracellular antiviral state, whereas the interleukins tend to exert their effects more indirectly through modulation of the adaptive immune response. Here, we summarize our current understanding of the direct roles of antiviral cytokines, in particular type I, II and III IFNs and TNF, in dictating viral tropism and the mechanisms that they are known to use. We also discuss the prospects of exploiting these cytokines to enhance innate immune responses against zoonotic infections and to achieve improved specificity of tissue-targeted oncolytic virotherapy.
Table 2.
Cytokine-determined viral tropism
| Cytokine | Virus | Demonstrated tropism | Antiviral mechanism | Refs |
|---|---|---|---|---|
| Type I IFNs | Myxoma virus | Host (mice) | Inhibition of viral mRNA translation through EIF2α phosphorylation by a kinase other than PKR | 39 |
| Daniels strain of Theiler's virus | Host (mice) | Unknown | 38 | |
| Coxsackievirus B3 | Tissue (cardiac tissue) | Unknown | 44 | |
| Influenza A/WSN/33 virus | Tissue (lung) | Unknown | 42 | |
| Polio virus | Tissue (CNS) | Expression of ISGs such as PKR and OAS proteins | 10 | |
| Sindbis virus | Tissue (CNS) and cellular (macrophage and dendritic cell lineage) | Inhibition of viral mRNA translation initiation | 43,50 | |
| Edmonston measles virus | Tissue (lung) | Unknown | 40 | |
| Vesicular stomatitis virus | Tissue (CNS) | Unknown | 41 | |
| Neurotropic coronavirus | Tissue (CNS) | Unknown | 46 | |
| West Nile virus | Tissue (CNS) | Expression of ISGs such as PKR and OAS proteins | 45 | |
| Type II IFN | Vaccinia virus | Tissue (CNS) and cellular (mouse fibroblasts) | Activation of IRF1 signalling by IFNγ in mouse fibroblasts | 58,59 |
| Sindbis virus | Tissue (CNS) and cellular (macrophage and dendritic cell lineage) | Activation of Jak–Stat signalling pathway in neurons | 60,61 | |
| Mouse hepatitis virus | Tissue (CNS) | Unknown | 62 | |
| Murine γ-herpesvirus (γMHV68) | Cellular (B cells, macrophages and dendritic cells) | Unknown | 121 | |
| Type III IFNs | Influenza A virus | Tissue (lung) and cellular (alveolar type II epithelial cells) | Induction of antiviral genes such as those encoding myxovirus resistance proteins, OAS proteins and ISG56 | 67,69 |
| Vaccinia virus | Tissue and cellular (epithelial cells) | Unknown | 68 | |
| TNF | Adenovirus and coxsackievirus B3 | Cellular (HUVECs) | Downregulation of the expression of virus-specific cell surface receptor (CAR) | 76,78 |
| HIV-1 | Cellular (macrophages) | Downregulation of the expression of virus-specific cell surface receptor (CD4, CCR5 and CXCR4) | 79 | |
| Type I IFNs and TNF | Myxoma virus | Cellular (macrophages) | Unknown | 66,75 |
| CAR, coxsackievirus and adenovirus receptor; CCR5, CC-chemokine receptor 5; CNS, central nervous system; CXCR4, CXC-chemokine receptor 4; EIF2α, eukaryotic initiation factor 2α; HUVEC, human umbilical vein endothelial cell; IFN, interferon; IRF1, IFN response factor 1; ISG, IFN-stimulated gene; JAK, Janus family kinase; OAS, 2′-5′ oligoadenylate synthase; PKR, dsRNA-dependent protein kinase; STAT, signal transducer and activator of transcription; TNF, tumour necrosis factor. | ||||
Interferons
The IFNs are a group of inducible cytokines that have a central role in innate antiviral immune responses because they establish an intracellular antiviral state that prevents virus replication and restricts the spread of virus between neighbouring cells16,17. They are grouped into three classes, known as type I, II and III IFNs, according to their amino acid sequence, chromosomal location and receptor specificity18. Binding of the three classes of IFN to their known cellular receptors induces similar, but not identical, signalling events. These events include the phosphorylation of Janus family kinase 1 (JAK1) and non-receptor tyrosine-protein kinase 2 (TYK2), and the activation of signal transducer and activator of transcription 1 (STAT1), STAT2 and, to a lesser extent, STAT3, STAT4 and STAT5 (Refs 18, 19, 20, 21). Activated STATs then induce the transcription of specific sets of interferon-stimulated genes (ISGs), including those encoding important mediators of the antiviral response22,23,24. The best-characterized ISGs encode the dsRNA-dependent protein kinase (PKR), the 2′-5′ oligoadenylate synthase (OAS) proteins and the myxovirus resistance proteins25. Some of these ISGs have broad antiviral effects. For example, the OAS proteins are activated by viral dsRNA and produce 2′-5′ oligoadenylates, which in turn activate the latent nuclease RNase L, resulting in the degradation of both viral and host RNA transcripts25,26. Similarly, activation of PKR, a member of the eukaryotic initiation factor 2α (EIF2α) kinase family, by viral dsRNA leads to EIF2α phosphorylation with a consequent blockade of translation of most cellular and viral mRNAs25,27. In contrast to the relatively broad antiviral activities of PKR and the OAS–RNase L system, certain ISGs such as myxovirus resistance proteins and guanylate binding proteins of the dynamin family can inhibit specific families of viruses, such as influenza and other negative-strand RNA viruses25,28,29, possibly through interference with virus assembly and trafficking in the cell; however, the detailed mechanisms of their actions have yet to be determined30.
Despite the similarities between the effects of the three types of IFN, analyses of various knockout mice have shown that each class of IFN is essential for antiviral defence and that none of the three classes is functionally redundant31. Not surprisingly, many viruses have evolved potent defence strategies against specific IFNs, and the consequences of the many levels of interplay between viruses and the IFN system can vary widely16,32.
Type I interferons. Type I IFNs trigger potent innate defence mechanisms against diverse viruses, and they are key players in the earliest stages of most host–virus encounters33. Most somatic cells can induce and respond to type I IFNs, but certain specialized cells such as plasmacytoid dendritic cells can rapidly produce large quantities of type I IFNs in response to diverse virus infections34,35,36. Type I IFNs activate a common cell signalling pathway leading to the transcription of a set of several hundred inducible genes, controlled by the IFN-stimulated gene factor 3 (ISGF3) complex, which together establish the antiviral state25,37 (Fig. 2). Although we still have only a basic understanding of this antiviral state, it clearly targets and inhibits every stage of the viral life cycle.
Figure 2. Cytokine-mediated regulation of viral tropism.
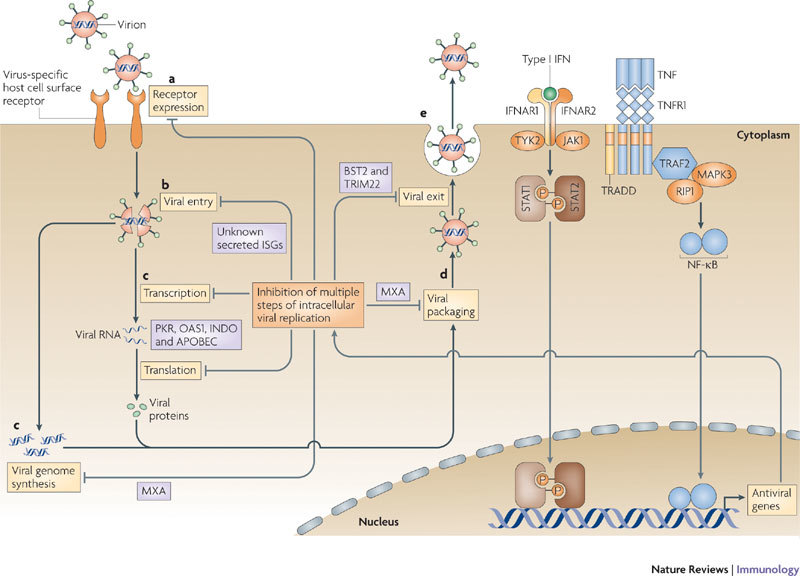
a |All viral replication cycles begin with the binding of an infectious virion to the cell surface. Frequently this step is mediated by a specific host cell surface receptor that the virus hijacks for attachment and/or entry. b | After binding, the virion is internalized into the cell and disassembles into its genome and associated proteins. c | The virus then uses a combination of viral and host proteins to transcribe and translate its own genes and replicate its genome. d | After replication, the newly synthesized genomes are packaged into nascent virus particles which then mature and traffic to the cell surface. e | Finally, the virus particles are released as infectious virus. It is important to note that this simplified life cycle is extremely general and that many viruses will deviate from this outline to some extent. Tumour necrosis factor (TNF) and interferons (IFNs) can inhibit this replication cycle by inducing the expression of proteins with antiviral properties. The important points in this replication cycle at which TNF and the IFNs can manipulate viral tropism, and the antiviral proteins that are involved, are indicated. APOBEC, apolipoprotein B mRNA editing enzyme, catalytic polypeptide; BST2, bone marrow stromal cell antigen 2; IFNAR, interferon-α/β receptor; INDO, indoleamine 2,3-dioxygenase; ISG: interferon-stimulated gene; JAK1, Janus family kinase 1; MAPK3, mitogen-activated protein kinase 3; MXA, myxovirus resistance protein A; NF-κB, nuclear factor-κB; OAS1, 2′-5′ oligoadenylate synthase 1; PKR, dsRNA-dependent protein kinase; RIP1, receptor-interacting protein 1; STAT, signal transducer and activator of transcription; TNFR1, tumour necrosis factor receptor 1; TRADD, TNFR1-associated via death domain; TRAF2, TNFR-associated factor 2; TRIM22, tripartite motif-containing 22; TYK2, tyrosine-protein kinase 2.
It has long been known that type I IFNs have an important role in dictating viral host tropism, in part owing to the availability of targeted gene knockout mouse models lacking various elements of the type I IFN-induced signalling pathway. Most of these observations were made using knockout mice lacking a functional type I IFN receptor, which had a marked increase in susceptibility to a wide variety of viruses31. For example, wild-type 129Sv mice were shown to be resistant to infection with the Daniels strain of Theiler's virus, whereas type I IFN receptor-deficient 129Sv mice died from the infection38. Similarly, in vitro data show that wild-type mice are completely resistant to myxoma virus (MYXV) infection owing to the virus-induced upregulation and secretion of IFNβ39. By contrast, STAT1-deficient mice, which lack the ability to transmit a functional type I IFN signalling response, are highly susceptible to infection with MYXV39. Hence, both Daniels strain of Theiler's virus and MYXV are considered to be atropic in mice owing to the presence of an active type I IFN response.
The type I IFN response can also dictate viral tissue tropism. For example, infection of wild-type mice with Edmonston measles virus 40, vesicular stomatitis virus (VSV)41, influenza A/WSN/33 virus42, Sindbis virus (SBV)43, coxsackievirus B3 (CVB3)44, poliovirus10, West Nile virus (WNV)45 or neurotropic coronavirus46 results in tissue-specific infections that are frequently sub-clinical. However, infection of mice lacking a functional type I IFN response with any of these viruses resulted in a highly disseminated systemic infection, which indicates an increased cellular and/or tissue tropism for these viruses in the absence of a type I IFN response. For example, in the case of CVB3 infection, type I IFN-mediated signalling limited the productive virus infection to cardiac tissues, which prevented an otherwise lethal systemic infection. It is reasonable to postulate that this is an example of an evolutionarily tuned defence mechanism and not just a random tissue tropism effect. After disruption of the type I IFN response pathway, the limited cardiotropic nature of the infection could be relieved, resulting in lethal viral replication in the liver44. Similarly, in Stat1−/− mice, influenza A/WSN/33 virus infection, the tissue tropism of which is normally restricted to the lungs, progressed to a lethal systemic infection42. Finally, infection of wild-type mice with WNV led to limited replication in the central nervous system (CNS), whereas infection of mice lacking the type I IFN receptor with WNV led to a highly disseminated lethal infection45. In many of these cases, it is not presently known whether the restriction by type I IFNs is mediated by limiting the tropism of these viruses directly to specific cells of that tissue or by limiting total viral load in the specific tissue. In some cases, it is possible that type I IFNs function by severely restricting viral replication in all of the cells of the tissue, leading to insufficient virus progeny to mediate continued dissemination. More work on the relative IFN responses of different cell types in individual tissues is therefore needed to clarify the exact mechanism(s) by which type I IFNs functionally determine the tissue tropism of specific viruses.
Several studies have attempted to address the potential mechanisms underlying the ability of type I IFNs to restrict viral tropism. However, it is important to realize that there is no consensus mechanism that applies to all cells, as the JAKs and STATs and many of the ISGs have different roles in different cells25. This raises interesting questions as to how the responses to IFNs intersect with more general aspects of cellular physiology and how the specificity of cytokine responses is maintained, but these questions have yet to be fully addressed. For poliovirus, the type I IFN system has an important role in determining tissue tropism by protecting atropic tissues from productive infection through the maintenance of expression of ISG products, such as PKR and OAS proteins, even in the absence of infection10. Conversely, in tissues that could be productively infected, ISG expression was low in the uninfected state and a significant increase in the ISG response was not observed even after poliovirus infection10. Another study47 showed that the ability of type I IFNs to protect mice against lethal WNV infection was largely based on the IFN-induced PKR and OAS–RNase L pathways. By contrast, studies of MYXV restriction in primary mouse embryo fibroblasts showed that MYXV-elicited phosphorylation of EIF2α on Ser51, with a consequent blockade of translation of most cellular and viral mRNAs, can occur without PKR activation39. In this case, it is presumed that an IFN-induced EIF2α kinase other than PKR is responsible for the observed Ser51 phosphorylation. Similarly, the type I IFN-determined tropism of SBV for cells of the dendritic cell lineage was shown to be independent of both the PKR and the OAS–RNase L pathways48,49. Instead, at the cellular level, type I IFNs inhibited SBV mRNA translation; this occurred after initial translation factor cap binding but before formation of the mature 80S ribosome50, a point in translation initiation that had not previously been identified as being regulated by IFN. Taken together, these data indicate that type I IFN responses determine viral tropism at the cellular level through several distinct mechanisms that can differ from one virus–host pairing to another. Furthermore, it is reasonable to propose that the different responses by hosts of different species, and even different tissues in a single host, to the same type I IFNs have been (and continue to be) fine-tuned by the evolutionary pressures exerted by multiple pathogens over time.
Despite having crucial roles in vertebrate innate immunity by slowing down initial viral infections and severely limiting virus spread (and therefore downregulating viral tropism), IFN treatment (in particular with type I IFNs) has, in some cases, been shown to enhance virus replication, resulting in increased viral tropism. For example, in vivo, porcine reproductive and respiratory syndrome virus (PRRSV) has a clear tropism for a subset of differentiated macrophages that express porcine sialoadhesin (PoSn) but not for peripheral blood monocytes51, which have no or extremely low levels of PoSn expression, indicating that PoSn levels have an important role in determining susceptibility to PRRSV infection. In vitro treatment of cells with IFNα markedly increased PoSn expression by monocytes to levels similar to those expressed by macrophages, resulting in increased susceptibility of monocytes to PRRSV infection; this effect could be blocked with an IFNα-specific neutralizing monoclonal antibody52. However, as the outcome varies depending on the time of IFN treatment (before, during or after virus infection), it is important to appreciate that the effect of IFNα on the susceptibility of monocytes to PRRSV infection is determined by the balance between enhancement of infection due to induction of PoSn expression and reduction of infection as a result of the antiviral actions of IFNα52.
A similar stimulatory effect of IFN was also described for porcine circovirus type 2 (PCV2), for which both IFNα and the type II IFN IFNγ increased virus infection in continuous cell lines53. Recently, it was shown that virus-induced IFNα expression can also increase the susceptibility of animals to bacterial superinfections54, which indicates that IFN-dependent enhancement of infection might be a new strategy, not only of viruses but also of other pathogens, to evade the innate immune response.
Type II interferon. The only known member of the type II IFN family is IFNγ, which is secreted by activated immune cells — mainly T cells and natural killer (NK) cells and to a lesser extent B cells, NKT cells and professional antigen-presenting cells. IFNγ mediates its biological function through a heterodimeric receptor known as the IFNγ receptor25. Mice lacking either IFNγ or its functional receptor have increased susceptibility to both viral and bacterial infections55, which indicates that IFNγ has an important role in antiviral and antibacterial responses56,57. An early study found that IFNγ mediated the clearance of vaccinia virus (VACV) from the CNS but had no effect on viral replication in peripheral solid organs such as the ovaries or testes, showing that IFNγ could determine the tissue tropism of VACV58. A more recent study showed that IFNγ induced an IFN regulatory factor 1 (IRF1)-dependent antiviral state in mouse fibroblasts and blocked VACV replication in these cells; however, IFNγ had no antiviral role against VACV in human fibroblasts, which indicates that IFNγ could also regulate the host tropism of VACV59.
Of particular interest is the role of IFNγ in determining viral tropism in brain and spinal cord neurons. In vivo studies with SBV, which infects neurons of the CNS and causes persistent infection in immunodeficient mice, indicate that IFNγ can result in virus clearance from neurons without causing cell death60. A recent in vitro study has shown that activation of the JAK–STAT pathway is the main mechanism for IFNγ-mediated noncytolytic clearance of SBV from neurons61. Detailed analysis of brain tissues, however, indicated that this IFNγ-mediated viral clearance is tissue dependent, as virus was cleared from the cerebellum but not from the cortex, hippocampus or brainstem60. In addition, in another neurotropic virus model of mouse hepatitis virus infection, it was shown that IFNγ can selectively clear virus from CNS oligodendrocytes62.
Type III interferons. In 2003, two reports were published describing previously undiscovered antiviral cytokines, termed IFNλ1 and IFNλ2 by one group and interleukin-29 and interleukin-28 by the other20,63. These IFNs are structurally and genetically distinct from the other types of IFN, act through a distinct receptor system and were later termed type III IFNs. However, type I and type III IFNs induce similar sets of genes, including many that encode important mediators of the antiviral response22,23. Consequently, type I and type III IFNs can induce a similar cellular antiviral state21,64, but they probably have distinct roles in the determination of viral tropism in specific cells and tissues, mainly as a result of signalling through distinct receptors and because their expression patterns vary greatly in different cells and tissues65.
One basis for a role of IFNλ in determining viral tropism has been shown by several studies65,66 that indicate that the main cell responders to IFNλ are of epithelial origin. For example, a recent in vivo study showed that mouse epithelial cells in the kidney and CNS respond to IFNλ, but that endothelial cells do not. In addition, tissues that contain large numbers of epithelial cells, such as stomach, intestine, skin and lung, respond well to IFNλ, whereas tissues that contain few epithelial cells, such as liver, spleen, brain and spinal cord, do not65. This responsiveness correlates with the level of IFNλ receptor expressed by each tissue, which indicates that IFNλ responses are determined by the cellular levels of receptor expression. However, the expression of IFNλ itself in response to virus infection is also highly tissue specific. For example, alveolar type II (ATII) epithelial cells secrete higher levels of IFNλ1 than do alveolar macrophages in response to influenza A virus infection67. When ATII epithelial cells were treated with IFNλ1 they induced the expression of antiviral genes, such as those encoding myxovirus resistance proteins, OAS proteins and ISG56, and decreased their viral load in a dose-dependent manner67. IFNλ secretion is high in the liver but low in the brain after intraperitoneal challenge with viruses65. So, based on the expression pattern and response in selective tissues, IFNλ might restrict viral replication in epithelial cells but not in other cell types, and therefore be most effective against viruses that mainly infect epithelial cells such as poxviruses, herpesviruses and influenza virus.
The first description of an in vivo antiviral role for IFNλ was reported from a study in which the authors expressed mouse IFNλ using a recombinant VACV model68. Infection with VACV expressing either IFNλ2 or IFNλ3 was attenuated in mice when the virus was administered intranasally but not when it was injected intradermally, which indictaes that IFNλ can directly shape VACV tissue tropism. In addition, when different routes of viral infection were compared, IFNλ protected against a lethal influenza A virus lung infection but did not protect against hepatotropic thogotovirus infection, or encephalomyocarditis virus or lymphocytic choriomeningitis virus infections in the heart and spleen69. Mice that lacked expression of IFNλ receptors had decreased resistance to influenza A virus in the lungs, indicating that IFNλ might have an antiviral role in this tissue. These observations suggest that IFNλ contributes to innate resistance against viral pathogens that infect the lungs but not those that infect other tissues.
In general, it is clear that intact IFN signalling is a key factor in determining the outcome of a viral infection and in some cases dictating viral host and tissue tropism. This action is mostly mediated through the induction of sets of cell-specific ISGs that ultimately block virus replication and dissemination at the tissue and organism levels.
Tumour necrosis factor
TNF has been shown to induce inflammation and apoptosis and thereby inhibit various viral and bacterial infections70,71. TNF signals by binding to its cognate receptors, TNFR1 and TNFR2. This binding induces a complex series of signalling events, including receptor phosphorylation and the recruitment of various signal transduction molecules, including TNFR1-associated via death domain (TRADD), TNFR-associated factor 2 (TRAF2) and receptor-interacting protein 1 (RIP1). These signal transducers then activate downstream signals, including the mitogen-activated protein kinase kinase–extracellular signal-regulated kinase (MEK–ERK) signalling pathway, leading to activation of the pro-inflammatory transcription factor nuclear factor-κB (NF-κB) and of the pro-apoptotic procaspases 8 and 10 (Ref. 72). Interestingly, the ability of TNF to induce an antiviral state analogous to that induced by the IFNs was first reported in the 1980s, but this has not been studied in as much detail as the IFN-induced antiviral state73,74. It is known that the induced set of cellular genes that mediate the antiviral state in response to TNF is unique and includes many genes that are not induced by the IFNs. However, some antiviral genes, such as those that encode OAS proteins and myxovirus resistance proteins, are induced at varying levels by both IFNs and TNF, although the antiviral function of the low-level induction of these genes by TNF has yet to be determined75. Further investigation is needed to determine the complete repertoire of antiviral genes that are induced by TNF.
Owing to its highly pleiotropic nature, TNF can exert antiviral effects through a wide variety of mechanisms. Few studies, however, have addressed the direct role of TNF in viral tropism. TNF can have a direct effect on viral tropism by altering the expression levels of cell surface receptors used by viruses (Fig. 2). For example, infection with adenovirus or CVB3 requires the expression of coxsackievirus and adenovirus receptor (CAR)76. Primary human umbilical vein endothelial cells (HUVECs) normally express CAR and are highly susceptible to these virus infections77. Treatment of HUVECs with TNF, however, downregulates expression of CAR and prevents adenovirus infection78. Importantly, TNF also downregulates expression of CAR by primary human dermal microvascular endothelial cells, thereby inhibiting adenovirus infection of these cells, but not by bronchial epithelial cells or A549 cancer cells, which indicates that TNF can directly regulate the cellular and tissue tropism of adenovirus78. Similarly, infection with HIV-1 requires the expression of various cell surface receptors, including CD4, CCR5 and CXCR4 (Refs 3,4). Treatment of primary human macrophages with TNF has been shown to downregulate expression of all three of these receptors, leading to a reduction in HIV-1 infection79. However, it is not yet known whether the downregulation of these receptors by TNF is directly intended to inhibit the binding and entry of these viruses or is a by-product of another cellular function of TNF. Interestingly, TNF can also upregulate the expression of certain cell surface receptors for viruses, leading to a gain of viral tropism. For example, treatment with TNF leads to increased expression of feline aminopeptidase N, the receptor for feline infectious peritonitis virus, by macrophages. This increased receptor expression results in increased virus infection and new virus production80. This gain of viral tropism shows that not all of the effects of TNF are directly antiviral.
TNF also affects the expression level of a wide variety of other known cell surface receptors used by viruses, such as CD55 (also known as DAF)81, which is required for Hantavirus infection82; ICAM1 (6), which is the main receptor for human rhinovirus5; and several integrins83,84, which are thought to be receptors for human cytomegalovirus85. To our knowledge, however, a direct link between TNF-mediated modulation of these cell surface receptors and viral tropism has not yet been shown.
So, TNF has been shown to alter viral tropism in both positive and negative manners by altering the expression level of cell surface receptors. Although this effect might be much more widespread than currently appreciated, the in vivo relevance of these observations largely remains to be determined.
Clinical implications
Zoonotic infections, emerging pathogens and bioterrorist threats. Occasionally, probably owing to virus mutation or recombination, a virus crosses the species barrier from its current or long-term evolutionary host to a new host species (new host tropism), a phenomenon that is referred to as a zoonotic infection when it occurs in humans. Zoonotic infections introduce previously unencountered viral pathogens into the human population and are thought to have caused some of the most lethal viral pandemics in history, such as the 1918 influenza virus pandemic that originated from chickens or pigs86, the severe acute respiratory syndrome (SARS) outbreak in 2002 that originated from a bat coronavirus87, HIV, which probably originated from African monkeys88, and various haemorrhagic fever outbreaks. In the past few years there has been an increase in the number of reported zoonotic disease transmissions involving animal viruses, such as cowpox and monkeypox viruses89,90,91,92 as well as swine and avian influenza viruses93, probably as a result of improved detection and monitoring techniques. Although many of these viruses did not necessarily 'jump' species owing to species-specific alterations in the host responses to innate cytokines, the prevalence and severity of these zoonotic infections highlight the growing need to better understand the barriers in humans that are designed to restrict the host tropism of the viruses, particularly with regards to how these cytokine defences might be exploited to inhibit new viruses from crossing species boundaries or to treat viruses that can jump species.
Particularly troubling is the idea that some of these zoonotic viruses, in particular the haemorrhagic fever viruses, could be used as biological weapons or agents of bioterrorism. As a result of this concern, there has been a concerted effort to develop new vaccines and/or therapies for zoonotic viruses that are still poorly characterized, particularly in humans. Various recombinant cytokines, such as the IFNs, have been shown by several groups to be an effective treatment for an array of viruses with the potential to cause zoonotic infections in vivo. For example, injection of recombinant IFNα protects mice against lethal challenge with the haemorrhagic fever virus Punta Toro virus94, and injection of IFNα or IFNγ protects rhesus monkeys against challenge with another haemorrhagic fever virus, Rift Valley virus, which infects mainly domestic livestock95,96. In addition, recombinant IFNα or IFNβ fused to albumin, for increased stability and pharmacological properties, has been shown to inhibit infection by various potentially zoonotic viruses in vitro, including Punta Toro virus, Venezuelan equine encephalitis (VEE) virus, Ebola virus and the SARS coronavirus97.
Although these data seem promising, several other studies have shown that some viruses can counteract the effects of IFN, and therefore treatment with recombinant IFNs is not effective against all zoonotic infections98,99. One study showed that IFNα effectively protects mice against challenge with VEE and Banzi viruses whereas IFNγ protects only against VEE virus99. A later study tested the effectiveness of IFNα, IFNβ and IFNγ against herpes simplex virus type 2, Banzi virus, Semliki forest virus and Caraparu virus98. In both studies, the authors concluded that the different IFNs had varying efficacies against each virus and that the dose and timing of the treatment were crucial. Also, many of these cytokines had to be administered before infection to have their optimal antiviral effects. So, although cytokines such as the IFNs might be one potential treatment for serious zoonotic infections, they seem unlikely to be a 'magic bullet' that will effectively eliminate the virus. Additional studies focusing on the mechanisms by which IFNs regulate viral tropism are therefore needed to develop a better understanding of how tropism-modifying cytokines, both alone and in combination (Box 1), might be used to effectively prevent or treat such trans-species infections.
Oncolytic virotherapy. Recently, the possibility of using live viruses as discriminating therapeutic agents against cancer has been investigated100,101,102. This concept, known as oncolytic virotherapy, is based on the observation that certain viruses replicate in tumour cells but not in untransformed primary cells. Such viruses could be said to have tumour-specific tropism, which can be thought of as a specific variation of cellular or tissue tropism. One mechanistic explanation for tumour tropism is linked to the inability of many cancer cells to respond properly to pro-inflammatory or antiviral cytokines (Fig. 3). For example, in normal primary mouse cells, the replication of VSV is strongly inhibited by IFNα103. In many cancer cells, however, VSV replicates even in the presence of IFNα owing to defects in the IFN responses of these cells103. Similarly, IFNβ and TNF strongly inhibit MYXV replication in normal primary human fibroblasts66,75, but MYXV can productively replicate in various human cancer cells even in the presence of these cytokines104. The injection of either VSV or MYXV into solid tumours in mice, even in the presence of an active IFN-mediated response, causes regression of the tumour, through an unknown mechanism, without significant viral pathology in normal tissues105,106.
Figure 3. Cytokine-mediated viral tropism in tumour tissues.
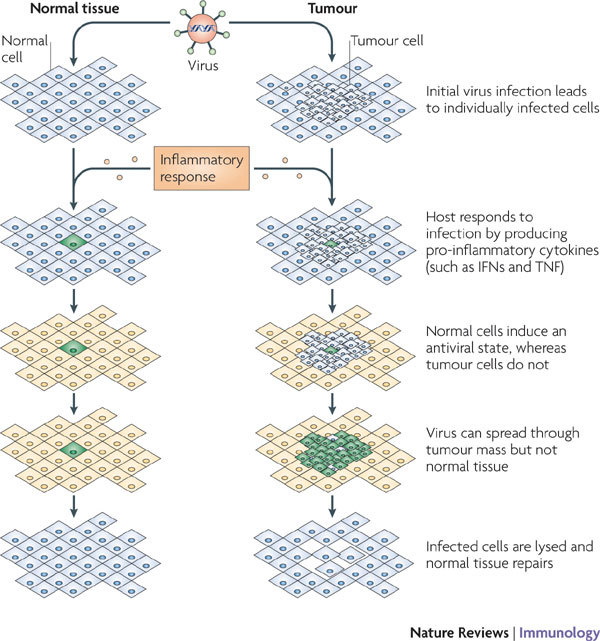
In a normal tissue, the host innate immune defences respond to infection of a single cell (shown in green) by releasing pro-inflammatory and antiviral cytokines (such as interferons (IFNs) and tumour necrosis factor (TNF)). These cytokines not only affect immune responses but also induce an antiviral state (shown in yellow) in normal tissue, such that the virus cannot productively infect neighbouring cells and the spread of infection is stopped or impeded. However, in a tumour, although the immune sentinel cells in the host might still initiate a potent innate immune response, including the release of the same antiviral cytokines, the tumour cells are frequently unable to respond to these secreted cytokines and so fail to establish an antiviral state. This can favour virus spread within the tumour tissue (and lysis of tumour cells) but not into neighbouring normal tissues, in which the antiviral state has been established.
Whereas wild-type VSV and MYXV have intrinsic tumour tropism owing to their unique IFN and/or TNF sensitivities in untransformed cells, certain other potential oncolytic viruses, including VACV, adenovirus, influenza virus, measles virus and poliovirus, can be modified to increase tumour tropism through specific gene mutations and deletions107. One common method of genetically modifying an oncolytic virus candidate to increase tumour tropism is to delete or modify the viral genes responsible for countering the IFN-mediated immune response. For example, influenza virus can normally productively infect humans; however, deletion of the NS1 gene, which counters the host IFN response, results in a virus that can replicate only in the absence of an IFN-mediated response108. The mutant virus can preferentially replicate in cancer cells109 and in Stat1−/− mice110, and causes tumour regression108, but is unable to replicate or cause disease in wild-type mice109. Similar results have been observed with E1B gene-deleted adenovirus111,112 and B18R gene-deleted VACV113, which have also had cellular genes that encode IFNs inserted into the viral genome113,114. These recombinant viruses have stricter tumour tropism and higher oncolytic potential than their single-gene-deleted predecessors113,114,115. Together, these data indicate that the regulation of tumour tropism by innate pro-inflammatory and antiviral cytokines, such as TNF and the IFNs, might have a key role in the potential use of viruses as oncolytic therapeutics.
Concluding remarks
Cytokine responses are crucial for effective host defences against invading pathogens. Indeed, the ability of any pathogen to emerge, re-emerge or persist quietly in a host can be linked to its ability to subvert or evade protective antiviral cytokine responses. The outcomes of such encounters can determine viral tropism at all three levels (cellular, tissue and host). Indeed, several lines of evidence have shown a major role for antiviral cytokines, in particular the IFNs and TNF, in directly dictating both tissue and host specificity of viruses. It is unclear at this point whether the dictation of viral tropism by the IFNs and TNF is an evolved cellular response or an unintended consequence of the innate immune response in general. It seems unlikely that the host would evolve a defence mechanism aimed at allowing viral replication in one tissue but not others. For several reasons, including differential gene expression or splicing, the differential activation or expression of transcription factors such as STATs116 and altered signalling pathways, the response to innate cytokines can differ significantly from cell type to cell type or tissue to tissue. This could be because the innate immune system has evolved to counter all pathogen infections in a largely nonspecific manner. Although the innate immune system is highly efficient, it has not evolved to perfectly counter every infection under every circumstance, merely to counter most infections under most circumstances. So, the innate immune response will always be suboptimal for any single infecting pathogen. Each virus has probably evolved specifically to take advantage of one aspect of this suboptimal response in at least one host species. Exactly which aspect a virus has evolved to take advantage of is likely to dictate the tropism of that particular virus. So, the balance between the requirement of the innate immune system to be specific enough to counter one invading pathogen infection while still being nonspecific enough to counter a totally unrelated pathogen infection might be the evolutionary explanation for how inflammatory cytokines, such as TNF and the IFNs, dictate viral tropism.
Further studies of how cytokines regulate viral tropism might result in practical outcomes, such as the improved development of more selective tumour-restricted oncolytic viruses. We can also anticipate new insights into the fundamental mechanisms by which some viruses can occasionally cross from a long-term evolutionary host species to cause zoonotic infections in humans. By focusing on the essential interactive nature of the innate antiviral cytokine response pathways (Box 1), we also predict the development of innovative therapeutic strategies to better respond to emerging infections in general, even before the next new viral pathogen has appeared in the human population.
Box 1 | Synergistic antiviral cytokine interactions.
Individual cytokines such as the interferons (IFNs) and tumour necrosis factor (TNF) have similar but distinct abilities to regulate viral tropism. Several studies, however, have shown that certain viruses that are normally resistant to inhibition by individual cytokines are nevertheless susceptible to the effects of cytokine combinations (reviewed elsewhere117). However, so far there are few specific documented examples showing that combinations of cytokines actually regulate viral tropism at the host level. One example, in contrast to the complete restriction of myxoma virus (MYXV) replication by type I IFNs in mouse fibroblasts39, is that the complete restriction of MYXV infection in primary human fibroblasts requires both type I IFNs and TNF66,75. Moreover, MYXV infection of primary human macrophages was shown to induce the simultaneous production of TNF and type I IFNs, through activation of the cytoplasmic RNA sensor retinoic acid inducible gene-I (RIG-I; also known as DDX58)66. Although all of the observations about the synergistic nature of antiviral cytokines have been made in vitro, and have yet to be rigorously validated in vivo, it is probable that cytokine synergism makes a significant contribution to viral tropism in complex tissues in situ. This knowledge could be particularly useful in terms of its implications for the modulation of viral tropism in the clinic to combat natural zoonotic infections or threats from potential viral pandemics that have not yet emerged in the human population.
Acknowledgements
This work was supported by the University of Florida College of Medicine, USA.
Glossary
- Antiviral state
An intracellular state in which virus replication is blocked, restricting the spread of virus to neighbouring cells. By signalling through the type I IFN receptor, IFNs activate the inducible expression of hundreds of genes that together establish the antiviral state.
- Zoonotic infection
The ability of a given virus to cross the host species barrier from its current or long-term evolutionary host to humans, causing disease.
- Oncolytic virotherapy
The treatment of cancer by using a virus specifically tailored to infect cancer cells while leaving normal cells unharmed. The engineering of such viruses involves ensuring that the viruses can replicate only inside cancer cells, lysing the cells when they exit, and ensuring a high dosage at the site of the tumour.
- Interferon-stimulated genes
(ISGs). Genes that are induced or expressed as a result of IFN action and encode proteins such as: PKR, a dsRNA-activated kinase that phosphorylates eIF2α with a consequent blockade of the translation of most cellular and viral mRNAs; oligoadenylate synthases, which produce 2'-5' oligoadenylates, which in turn activate the latent nuclease RNase L, resulting in the degradation of both viral and host RNA transcripts; and myxovirus resistance proteins, which possibly interfere with viral assembly and trafficking in the cell.
- Plasmacytoid dendritic cell
An immature dendritic cell with a morphology that resembles that of a plasma cell. Plasmacytoid dendritic cells produce type I IFNs (that is, IFNα and IFNβ) in response to viral infection.
- IFN-stimulated gene factor 3 (ISGF3) complex
An IFN-induced signal transduction and transcription activation complex. ISGF3 is assembled from three proteins, STAT1, STAT2 and interferon-regulatory factor 9. Of these components, STAT2 provides a fundamental and essential transcription activation function.
- Oligodendrocyte
A type of glial cell that creates the myelin sheath that insulates axons and improves the speed and reliability of signal transmission by neurons.
- Alveolar type II (ATII) epithelial cell
ATII cells are cuboidal in shape, with short microvilli along their apical surface. They secrete a pulmonary surfactant that decreases the surface tension of the alveolar surface, allowing the alveoli to expand during inspiration and preventing their collapse during expiration.
Biographies
Grant McFadden received his Ph.D. from McGill University in Montreal, Canada, in 1975. He held several academic positions in Canada until 2006, when he relocated to the University of Florida, USA. His scientific expertise is on how viral pathogens interact with the host immune system. His laboratory studies the mechanisms that determine the tropism and host range of poxviruses and the molecular basis of how certain poxviruses can sometimes cause zoonotic infections in humans.
Mohamed R. Mohamed received his Ph.D. from the State University of New York (SUNY) at Buffalo, USA, in 2001. After initial postdoctoral studies at SUNY, he joined the University of Florida in 2007. His research focuses on how poxviruses interfere with and manipulate the diverse host immune responses, in particular the NF-κB pathway.
Masmudur M. Rahman received his Ph.D. from the Indian Institute of Science, Karnataka, India, in 2004. After initial postdoctoral studies at the Roberts Research Institute, Ontario, Canada, he joined the University of Florida in 2006. His research focuses on poxviral modulators of the immune response, in particular the inflammasome complex.
Eric Bartee received his Ph.D. from Oregon Health and Science University, USA, in 2007. He then joined the University of Florida in 2007. His research focuses on cytokine-mediated regulation of poxvirus tropism, with a strong emphasis on the synergistic anti-poxviral action of both type I interferons and tumour necrosis factor.
Related links
FURTHER INFORMATION
Footnotes
Mohamed R. Mohamed, Masmudur M. Rahman and Eric Bartee: These authors contributed equally to this work
References
- 1.Brandenburg B, Zhuang X. Virus trafficking — learning from single-virus tracking. Nature Rev. Microbiol. 2007;5:197–208. doi: 10.1038/nrmicro1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hickman HD, Bennink JR, Yewdell JW. Caught in the act: intravital multiphoton microscopy of host–pathogen interactions. Cell Host Microbe. 2009;5:13–21. doi: 10.1016/j.chom.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berger EA, Murphy PM, Farber JM. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu. Rev. Immunol. 1999;17:657–700. doi: 10.1146/annurev.immunol.17.1.657. [DOI] [PubMed] [Google Scholar]
- 4.Lee B, Montaner LJ. Chemokine immunobiology in HIV-1 pathogenesis. J. Leukoc. Biol. 1999;65:552–565. doi: 10.1002/jlb.65.5.552. [DOI] [PubMed] [Google Scholar]
- 5.Greve JM, et al. The major human rhinovirus receptor is ICAM-1. Cell. 1989;56:839–847. doi: 10.1016/0092-8674(89)90688-0. [DOI] [PubMed] [Google Scholar]
- 6.Tosi MF, et al. Induction of ICAM-1 expression on human airway epithelial cells by inflammatory cytokines: effects on neutrophil-epithelial cell adhesion. Am. J. Respir. Cell Mol. Biol. 1992;7:214–221. doi: 10.1165/ajrcmb/7.2.214. [DOI] [PubMed] [Google Scholar]
- 7.Schneider-Schaulies J. Cellular receptors for viruses: links to tropism and pathogenesis. J. Gen. Virol. 2000;81:1413–1429. doi: 10.1099/0022-1317-81-6-1413. [DOI] [PubMed] [Google Scholar]
- 8.McFadden G. Poxvirus tropism. Nature Rev. Microbiol. 2005;3:201–213. doi: 10.1038/nrmicro1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bieniasz PD. Intrinsic immunity: a front-line defense against viral attack. Nature Immunol. 2004;5:1109–1115. doi: 10.1038/ni1125. [DOI] [PubMed] [Google Scholar]
- 10.Ida-Hosonuma M, et al. The alpha/beta interferon response controls tissue tropism and pathogenicity of poliovirus. J. Virol. 2005;79:4460–4469. doi: 10.1128/JVI.79.7.4460-4469.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O'Shea JJ, Murray PJ. Cytokine signaling modules in inflammatory responses. Immunity. 2008;28:477–487. doi: 10.1016/j.immuni.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tato CM, Cua DJ. Snapshot: cytokines IV. Cell. 2008;132:e2. doi: 10.1016/j.cell.2008.02.024. [DOI] [PubMed] [Google Scholar]
- 13.Tato CM, Cua DJ. Snapshot: cytokines III. Cell. 2008;132:900. doi: 10.1016/j.cell.2008.02.023. [DOI] [PubMed] [Google Scholar]
- 14.Tato CM, Cua DJ. Snapshot: cytokines II. Cell. 2008;132:500. doi: 10.1016/j.cell.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 15.Tato CM, Cua DJ. Snapshot: cytokines I. Cell. 2008;132:324.e1–324.e2. doi: 10.1016/j.cell.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 16.Randall RE, Goodbourn S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 2008;89:1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- 17.Stetson DB, Medzhitov R. Antiviral defense: interferons and beyond. J. Exp. Med. 2006;203:1837–1841. doi: 10.1084/jem.20061377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004;202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x. [DOI] [PubMed] [Google Scholar]
- 19.Dumoutier L, et al. Role of the interleukin (IL)-28 receptor tyrosine residues for antiviral and antiproliferative activity of IL-29/interferon-λ1: similarities with type I interferon signaling. J. Biol. Chem. 2004;279:32269–32274. doi: 10.1074/jbc.M404789200. [DOI] [PubMed] [Google Scholar]
- 20.Kotenko SV, et al. IFN-λs mediate antiviral protection through a distinct class II cytokine receptor complex. Nature Immunol. 2003;4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- 21.Kotenko SV, Langer JA. Full house: 12 receptors for 27 cytokines. Int. Immunopharmacol. 2004;4:593–608. doi: 10.1016/j.intimp.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 22.Doyle SE, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology. 2006;44:896–906. doi: 10.1002/hep.21312. [DOI] [PubMed] [Google Scholar]
- 23.Marcello T, et al. Interferons α and λ inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology. 2006;131:1887–1898. doi: 10.1053/j.gastro.2006.09.052. [DOI] [PubMed] [Google Scholar]
- 24.Sadler AJ, Latchoumanin O, Hawkes D, Mak J, Williams BR. An antiviral response directed by PKR phosphorylation of the RNA helicase A. PLoS Pathog. 2009;5:e1000311. doi: 10.1371/journal.ppat.1000311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu. Rev. Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 26.Meurs E, Krause D, Robert N, Silverman RH, Hovanessian AG. The 2–5A system in control and interferon-treated K/BALB cells infected with encephalomyocarditis virus. Prog. Clin. Biol. Res. 1985;202:307–315. [PubMed] [Google Scholar]
- 27.Meurs EF, et al. Constitutive expression of human double-stranded RNA-activated p68 kinase in murine cells mediates phosphorylation of eukaryotic initiation factor 2 and partial resistance to encephalomyocarditis virus growth. J. Virol. 1992;66:5805–5814. doi: 10.1128/jvi.66.10.5805-5814.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arnheiter H, Meier E. Mx proteins: antiviral proteins by chance or by necessity? New Biol. 1990;2:851–857. [PubMed] [Google Scholar]
- 29.Pavlovic J, Schroder A, Blank A, Pitossi F, Staeheli P. Mx proteins: GTPases involved in the interferon-induced antiviral state. Ciba Found. Symp. 1993;176:233–243. doi: 10.1002/9780470514450.ch15. [DOI] [PubMed] [Google Scholar]
- 30.Stetson DB, Medzhitov R. Type I interferons in host defense. Immunity. 2006;25:373–381. doi: 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 31.Muller U, et al. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 32.Finlay BB, McFadden G. Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell. 2006;124:767–782. doi: 10.1016/j.cell.2006.01.034. [DOI] [PubMed] [Google Scholar]
- 33.Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc. R. Soc. Lond. B Biol. Sci. 1957;147:258–267. doi: 10.1098/rspb.1957.0048. [DOI] [PubMed] [Google Scholar]
- 34.Gondois-Rey F, et al. Hepatitis C virus is a weak inducer of interferon α in plasmacytoid dendritic cells in comparison with influenza and human herpesvirus type-1. PLoS One. 2009;4:e4319. doi: 10.1371/journal.pone.0004319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krug A, et al. Identification of CpG oligonucleotide sequences with high induction of IFN-α/β in plasmacytoid dendritic cells. Eur. J. Immunol. 2001;31:2154–2163. doi: 10.1002/1521-4141(200107)31:7<2154::AID-IMMU2154>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 36.Libri NA, Barker SJ, Rosenberg WM, Semper AE. A class C CpG toll-like receptor 9 agonist successfully induces robust interferon-alpha production by plasmacytoid dendritic cells from patients chronically infected with hepatitis C. J. Viral Hepat. 2009;16:315–324. doi: 10.1111/j.1365-2893.2008.01011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Boxel-Dezaire AH, Rani MR, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity. 2006;25:361–372. doi: 10.1016/j.immuni.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 38.Fiette L, et al. Theiler's virus infection of 129Sv mice that lack the interferon alpha/beta or interferon gamma receptors. J. Exp. Med. 1995;181:2069–2076. doi: 10.1084/jem.181.6.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang F, et al. Disruption of Erk-dependent type I interferon induction breaks the myxoma virus species barrier. Nature Immunol. 2004;5:1266–1274. doi: 10.1038/ni1132. [DOI] [PubMed] [Google Scholar]
- 40.Mrkic B, et al. Measles virus spread and pathogenesis in genetically modified mice. J. Virol. 1998;72:7420–7427. doi: 10.1128/jvi.72.9.7420-7427.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steinhoff U, et al. Antiviral protection by vesicular stomatitis virus-specific antibodies in alpha/beta interferon receptor-deficient mice. J. Virol. 1995;69:2153–2158. doi: 10.1128/jvi.69.4.2153-2158.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garcia-Sastre A, et al. The role of interferon in influenza virus tissue tropism. J. Virol. 1998;72:8550–8558. doi: 10.1128/jvi.72.11.8550-8558.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ryman KD, Klimstra WB, Nguyen KB, Biron CA, Johnston RE. Alpha/beta interferon protects adult mice from fatal Sindbis virus infection and is an important determinant of cell and tissue tropism. J. Virol. 2000;74:3366–3378. doi: 10.1128/JVI.74.7.3366-3378.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wessely R, Klingel K, Knowlton KU, Kandolf R. Cardioselective infection with coxsackievirus B3 requires intact type I interferon signaling: implications for mortality and early viral replication. Circulation. 2001;103:756–761. doi: 10.1161/01.CIR.103.5.756. [DOI] [PubMed] [Google Scholar]
- 45.Samuel MA, Diamond MS. Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J. Virol. 2005;79:13350–13361. doi: 10.1128/JVI.79.21.13350-13361.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ireland DD, Stohlman SA, Hinton DR, Atkinson R, Bergmann CC. Type I interferons are essential in controlling neurotropic coronavirus infection irrespective of functional CD8 T cells. J. Virol. 2008;82:300–310. doi: 10.1128/JVI.01794-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Samuel MA, et al. PKR and RNase L contribute to protection against lethal West Nile Virus infection by controlling early viral spread in the periphery and replication in neurons. J. Virol. 2006;80:7009–7019. doi: 10.1128/JVI.00489-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ryman KD, et al. Sindbis virus translation is inhibited by a PKR/RNase L-independent effector induced by alpha/beta interferon priming of dendritic cells. J. Virol. 2005;79:1487–1499. doi: 10.1128/JVI.79.3.1487-1499.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ryman KD, White LJ, Johnston RE, Klimstra WB. Effects of PKR/RNase L-dependent and alternative antiviral pathways on alphavirus replication and pathogenesis. Viral Immunol. 2002;15:53–76. doi: 10.1089/088282402317340233. [DOI] [PubMed] [Google Scholar]
- 50.Tesfay MZ, et al. Alpha/beta interferon inhibits cap-dependent translation of viral but not cellular mRNA by a PKR-independent mechanism. J. Virol. 2008;82:2620–2630. doi: 10.1128/JVI.01784-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Duan X, Nauwynck HJ, Pensaert MB. Effects of origin and state of differentiation and activation of monocytes/macrophages on their susceptibility to porcine reproductive and respiratory syndrome virus (PRRSV) Arch. Virol. 1997;142:2483–2497. doi: 10.1007/s007050050256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Delputte PL, Van Breedam W, Barbe F, Van Reeth K, Nauwynck HJ. IFN-α treatment enhances porcine Arterivirus infection of monocytes via upregulation of the porcine Arterivirus receptor sialoadhesin. J. Interferon Cytokine Res. 2007;27:757–766. doi: 10.1089/jir.2007.0001. [DOI] [PubMed] [Google Scholar]
- 53.Meerts P, Misinzo G, Nauwynck HJ. Enhancement of porcine circovirus 2 replication in porcine cell lines by IFN-γ before and after treatment and by IFN-α after treatment. J. Interferon Cytokine Res. 2005;25:684–693. doi: 10.1089/jir.2005.25.684. [DOI] [PubMed] [Google Scholar]
- 54.Navarini AA, et al. Increased susceptibility to bacterial superinfection as a consequence of innate antiviral responses. Proc. Natl Acad. Sci. USA. 2006;103:15535–15539. doi: 10.1073/pnas.0607325103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-γ: an overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 56.van Boxel-Dezaire AH, Stark GR. Cell type-specific signaling in response to interferon-γ. Curr. Top. Microbiol. Immunol. 2007;316:119–154. doi: 10.1007/978-3-540-71329-6_7. [DOI] [PubMed] [Google Scholar]
- 57.Young HA, Bream JH. IFN-γ: recent advances in understanding regulation of expression, biological functions, and clinical applications. Curr. Top. Microbiol. Immunol. 2007;316:97–117. doi: 10.1007/978-3-540-71329-6_6. [DOI] [PubMed] [Google Scholar]
- 58.Kundig TM, Hengartner H, Zinkernagel RM. T cell-dependent IFN-γ exerts an antiviral effect in the central nervous system but not in peripheral solid organs. J. Immunol. 1993;150:2316–2321. [PubMed] [Google Scholar]
- 59.Trilling M, et al. Gamma interferon-induced interferon regulatory factor 1-dependent antiviral response inhibits vaccinia virus replication in mouse but not human fibroblasts. J. Virol. 2009;83:3684–3695. doi: 10.1128/JVI.02042-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Binder GK, Griffin DE. Interferon-gamma-mediated site-specific clearance of alphavirus from CNS neurons. Science. 2001;293:303–306. doi: 10.1126/science.1059742. [DOI] [PubMed] [Google Scholar]
- 61.Burdeinick-Kerr R, Govindarajan D, Griffin DE. Noncytolytic clearance of sindbis virus infection from neurons by gamma interferon is dependent on Jak/STAT signaling. J. Virol. 2009;83:3429–3435. doi: 10.1128/JVI.02381-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parra B, et al. IFN-γ is required for viral clearance from central nervous system oligodendroglia. J. Immunol. 1999;162:1641–1647. [PubMed] [Google Scholar]
- 63.Sheppard P, et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nature Immunol. 2003;4:63–68. doi: 10.1038/ni873. [DOI] [PubMed] [Google Scholar]
- 64.Ank N, West H, Paludan SR. IFN-λ: novel antiviral cytokines. J. Interferon Cytokine Res. 2006;26:373–379. doi: 10.1089/jir.2006.26.373. [DOI] [PubMed] [Google Scholar]
- 65.Sommereyns C, Paul S, Staeheli P, Michiels T. IFN-lambda (IFN-λ) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008;4:e1000017. doi: 10.1371/journal.ppat.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang F, et al. RIG-I mediates the co-induction of tumor necrosis factor and type I interferon elicited by myxoma virus in primary human macrophages. PLoS Pathog. 2008;4:e1000099. doi: 10.1371/journal.ppat.1000099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang J, et al. Differentiated human alveolar type II cells secrete antiviral IL-29 (IFN-λ1) in response to influenza A infection. J. Immunol. 2009;182:1296–1304. doi: 10.4049/jimmunol.182.3.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bartlett NW, Buttigieg K, Kotenko SV, Smith GL. Murine interferon lambdas (type III interferons) exhibit potent antiviral activity in vivo in a poxvirus infection model. J. Gen. Virol. 2005;86:1589–1596. doi: 10.1099/vir.0.80904-0. [DOI] [PubMed] [Google Scholar]
- 69.Mordstein M, et al. Interferon-λ contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS Pathog. 2008;4:e1000151. doi: 10.1371/journal.ppat.1000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Benedict CA, Banks TA, Ware CF. Death and survival: viral regulation of TNF signaling pathways. Curr. Opin. Immunol. 2003;15:59–65. doi: 10.1016/S0952-7915(02)00018-3. [DOI] [PubMed] [Google Scholar]
- 71.Rahman MM, McFadden G. Modulation of tumor necrosis factor by microbial pathogens. PLoS Pathog. 2006;2:e4. doi: 10.1371/journal.ppat.0020004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ihnatko R, Kubes M. TNF signaling: early events and phosphorylation. Gen. Physiol. Biophys. 2007;26:159–167. [PubMed] [Google Scholar]
- 73.Mestan J, et al. Antiviral effects of recombinant tumour necrosis factor in vitro. Nature. 1986;323:816–819. doi: 10.1038/323816a0. [DOI] [PubMed] [Google Scholar]
- 74.Wong GH, Goeddel DV. Tumour necrosis factors alpha and beta inhibit virus replication and synergize with interferons. Nature. 1986;323:819–822. doi: 10.1038/323819a0. [DOI] [PubMed] [Google Scholar]
- 75.Bartee E, Mohamed MR, Lopez MC, Baker HV, McFadden G. The addition of tumor necrosis factor plus beta interferon induces a novel synergistic antiviral state against poxviruses in primary human fibroblasts. J. Virol. 2009;83:498–511. doi: 10.1128/JVI.01376-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bergelson JM, et al. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science. 1997;275:1320–1323. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- 77.Carson SD, Hobbs JT, Tracy SM, Chapman NM. Expression of the coxsackievirus and adenovirus receptor in cultured human umbilical vein endothelial cells: regulation in response to cell density. J. Virol. 1999;73:7077–7079. doi: 10.1128/jvi.73.8.7077-7079.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vincent T, Pettersson RF, Crystal RG, Leopold PL. Cytokine-mediated downregulation of coxsackievirus-adenovirus receptor in endothelial cells. J. Virol. 2004;78:8047–8058. doi: 10.1128/JVI.78.15.8047-8058.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bailer RT, Lee B, Montaner LJ. IL-13 and TNF-α inhibit dual-tropic HIV-1 in primary macrophages by reduction of surface expression of CD4, chemokine receptors CCR5, CXCR4 and post-entry viral gene expression. Eur. J. Immunol. 2000;30:1340–1349. doi: 10.1002/(SICI)1521-4141(200005)30:5<1340::AID-IMMU1340>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 80.Takano T, Hohdatsu T, Toda A, Tanabe M, Koyama H. TNF-α, produced by feline infectious peritonitis virus (FIPV)-infected macrophages, upregulates expression of type II FIPV receptor feline aminopeptidase N in feline macrophages. Virology. 2007;364:64–72. doi: 10.1016/j.virol.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Moutabarrik A, et al. Cytokine-mediated regulation of the surface expression of complement regulatory proteins, CD46(MCP), CD55(DAF), and CD59 on human vascular endothelial cells. Lymphokine Cytokine Res. 1993;12:167–172. [PubMed] [Google Scholar]
- 82.Krautkramer E, Zeier M. Hantavirus causing hemorrhagic fever with renal syndrome enters from the apical surface and requires decay-accelerating factor (DAF/CD55) J. Virol. 2008;82:4257–4264. doi: 10.1128/JVI.02210-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Herzberg F, et al. IL-4 and TNF-α induce changes in integrin expression and adhesive properties and decrease the lung-colonizing potential of HT-29 colon carcinoma cells. Clin. Exp. Metastasis. 1996;14:165–175. doi: 10.1007/BF00121213. [DOI] [PubMed] [Google Scholar]
- 84.Nista A, Mattioni M, Gismondi A, Palmieri G, Santoni A. β1-Integrin expression and function in human bladder cancer cells: modulation by TNFα. Anticancer Res. 1996;16:581–588. [PubMed] [Google Scholar]
- 85.Feire AL, Koss H, Compton T. Cellular integrins function as entry receptors for human cytomegalovirus via a highly conserved disintegrin-like domain. Proc. Natl Acad. Sci. USA. 2004;101:15470–15475. doi: 10.1073/pnas.0406821101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vana G, Westover KM. Origin of the 1918 Spanish influenza virus: a comparative genomic analysis. Mol. Phylogenet. Evol. 2008;47:1100–1110. doi: 10.1016/j.ympev.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 87.Wang LF, et al. Review of bats and SARS. Emerg. Infect. Dis. 2006;12:1834–1840. doi: 10.3201/eid1212.060401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Holmes EC. On the origin and evolution of the human immunodeficiency virus (HIV) Biol. Rev. Camb. Philos. Soc. 2001;76:239–254. doi: 10.1017/S1464793101005668. [DOI] [PubMed] [Google Scholar]
- 89.Baxby D, Bennett M. Poxvirus zoonoses. J. Med. Microbiol. 1997;46:17–20. [PubMed] [Google Scholar]
- 90.Hawranek T, et al. Feline orthopoxvirus infection transmitted from cat to human. J. Am. Acad. Dermatol. 2003;49:513–518. doi: 10.1067/S0190-9622(03)00762-X. [DOI] [PubMed] [Google Scholar]
- 91.Lewis-Jones S. Zoonotic poxvirus infections in humans. Curr. Opin. Infect. Dis. 2004;17:81–89. doi: 10.1097/00001432-200404000-00003. [DOI] [PubMed] [Google Scholar]
- 92.Schupp P, et al. Cowpox virus in a 12-year-old boy: rapid identification by an orthopoxvirus-specific polymerase chain reaction. Br. J. Dermatol. 2001;145:146–150. doi: 10.1046/j.1365-2133.2001.04300.x. [DOI] [PubMed] [Google Scholar]
- 93.CDC. Outbreak of swine-origin influenza A (H1N1) virus infection — Mexico, March–April 2009. Morb. Mortal. Wkly Rep.58, 467–470 (2009). [PubMed]
- 94.Sidwell RW, et al. Antiviral and immunomodulating inhibitors of experimentally-induced Punta Toro virus infections. Antiviral Res. 1994;25:105–122. doi: 10.1016/0166-3542(94)90100-7. [DOI] [PubMed] [Google Scholar]
- 95.Morrill JC, Czarniecki CW, Peters CJ. Recombinant human interferon-γ modulates Rift Valley fever virus infection in the rhesus monkey. J. Interferon Res. 1991;11:297–304. doi: 10.1089/jir.1991.11.297. [DOI] [PubMed] [Google Scholar]
- 96.Morrill JC, Jennings GB, Cosgriff TM, Gibbs PH, Peters CJ. Prevention of Rift Valley fever in rhesus monkeys with interferon-α. Rev. Infect. Dis. 1989;11:S815–S825. doi: 10.1093/clinids/11.Supplement_4.S815. [DOI] [PubMed] [Google Scholar]
- 97.Subramanian GM, et al. Potent in vitro activity of the albumin fusion type 1 interferons (albumin-interferon-α and albumin-interferon-β) against RNA viral agents of bioterrorism and the severe acute respiratory syndrome (SARS) virus. Chemotherapy. 2008;54:176–180. doi: 10.1159/000140361. [DOI] [PubMed] [Google Scholar]
- 98.Pinto AJ, Morahan PS, Brinton M, Stewart D, Gavin E. Comparative therapeutic efficacy of recombinant interferons-α, -β, and -γ against alphatogavirus, bunyavirus, flavivirus, and herpesvirus infections. J. Interferon Res. 1990;10:293–298. doi: 10.1089/jir.1990.10.293. [DOI] [PubMed] [Google Scholar]
- 99.Pinto AJ, Morahan PS, Brinton MA. Comparative study of various immunomodulators for macrophage and natural killer cell activation and antiviral efficacy against exotic RNA viruses. Int. J. Immunopharmacol. 1988;10:197–209. doi: 10.1016/0192-0561(88)90050-1. [DOI] [PubMed] [Google Scholar]
- 100.Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol. Ther. 2007;15:651–659. doi: 10.1038/sj.mt.6300108. [DOI] [PubMed] [Google Scholar]
- 101.Liu TC, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nature Clin. Pract. Oncol. 2007;4:101–117. doi: 10.1038/ncponc0736. [DOI] [PubMed] [Google Scholar]
- 102.Vaha-Koskela MJ, Heikkila JE, Hinkkanen AE. Oncolytic viruses in cancer therapy. Cancer Lett. 2007;254:178–216. doi: 10.1016/j.canlet.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Barber GN. Vesicular stomatitis virus as an oncolytic vector. Viral Immunol. 2004;17:516–527. doi: 10.1089/vim.2004.17.516. [DOI] [PubMed] [Google Scholar]
- 104.Bartee Eric, McFadden Grant. Human cancer cells have specifically lost the ability to induce the synergistic state caused by tumor necrosis factor plus interferon-β. Cytokine. 2009;47(3):199–205. doi: 10.1016/j.cyto.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Stanford MM, et al. Myxoma virus oncolysis of primary and metastatic B16F10 mouse tumors in vivo. Mol. Ther. 2008;16:52–59. doi: 10.1038/sj.mt.6300348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Stojdl DF, et al. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nature Med. 2000;6:821–825. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- 107.Cattaneo R, Miest T, Shashkova EV, Barry MA. Reprogrammed viruses as cancer therapeutics: targeted, armed and shielded. Nature Rev. Microbiol. 2008;6:529–540. doi: 10.1038/nrmicro1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Muster T, et al. Interferon resistance promotes oncolysis by influenza virus NS1-deletion mutants. Int. J. Cancer. 2004;110:15–21. doi: 10.1002/ijc.20078. [DOI] [PubMed] [Google Scholar]
- 109.Egorov A, et al. Transfectant influenza A viruses with long deletions in the NS1 protein grow efficiently in Vero cells. J. Virol. 1998;72:6437–6441. doi: 10.1128/jvi.72.8.6437-6441.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Garcia-Sastre A, et al. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology. 1998;252:324–330. doi: 10.1006/viro.1998.9508. [DOI] [PubMed] [Google Scholar]
- 111.Lee B, et al. Oncolysis of human gastric cancers by an E1B 55 kDa-deleted YKL-1 adenovirus. Cancer Lett. 2002;185:225–233. doi: 10.1016/S0304-3835(02)00279-3. [DOI] [PubMed] [Google Scholar]
- 112.Nemunaitis J, et al. Selective replication and oncolysis in p53 mutant tumors with ONYX-015, an E1B-55kD gene-deleted adenovirus, in patients with advanced head and neck cancer: a phase II trial. Cancer Res. 2000;60:6359–6366. [PubMed] [Google Scholar]
- 113.Kirn DH, Wang Y, Le Boeuf F, Bell J, Thorne SH. Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PLoS Med. 2007;4:e353. doi: 10.1371/journal.pmed.0040353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shashkova EV, Spencer JF, Wold WS, Doronin K. Targeting interferon-alpha increases antitumor efficacy and reduces hepatotoxicity of E1A-mutated spread-enhanced oncolytic adenovirus. Mol. Ther. 2007;15:598–607. doi: 10.1038/sj.mt.6300064. [DOI] [PubMed] [Google Scholar]
- 115.Shashkova EV, Kuppuswamy MN, Wold WS, Doronin K. Anticancer activity of oncolytic adenovirus vector armed with IFN-α and ADP is enhanced by pharmacologically controlled expression of TRAIL. Cancer Gene Ther. 2008;15:61–72. doi: 10.1038/sj.cgt.7701107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hevehan DL, Miller WM, Papoutsakis ET. Differential expression and phosphorylation of distinct STAT3 proteins during granulocytic differentiation. Blood. 2002;99:1627–1637. doi: 10.1182/blood.V99.5.1627. [DOI] [PubMed] [Google Scholar]
- 117.Bartee E, Mohamed MR, McFadden G. Tumor necrosis factor and interferon: cytokines in harmony. Curr. Opin. Microbiol. 2008;11:378–383. doi: 10.1016/j.mib.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tang H, Banks KE, Anderson AL, McLachlan A. Hepatitis B virus transcription and replication. Drug News Perspect. 2001;14:325–334. [PubMed] [Google Scholar]
- 119.van Riel D, et al. Human and avian influenza viruses target different cells in the lower respiratory tract of humans and other mammals. Am. J. Pathol. 2007;171:1215–1223. doi: 10.2353/ajpath.2007.070248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lopez-Guerrero JA, Rayet B, Tuynder M, Rommelaere J, Dinsart C. Constitutive activation of U937 promonocytic cell clones selected for their resistance to parvovirus H-1 infection. Blood. 1997;89:1642–1653. [PubMed] [Google Scholar]
- 121.Steed A, Buch T, Waisman A, Virgin HWT. Gamma interferon blocks gammaherpesvirus reactivation from latency in a cell type-specific manner. J. Virol. 2007;81:6134–6140. doi: 10.1128/JVI.00108-07. [DOI] [PMC free article] [PubMed] [Google Scholar]