

Abstract
Duchenne muscular dystophy (DMD) is a severe muscle wasting disease caused by mutations in the dystrophin gene. By utilizing antisense oligonucleotides, splicing of the dystrophin transcript can be altered so that exons harbouring a mutation are excluded from the mature mRNA. Although this approach has been shown to be effective to restore partially functional dystrophin protein, the level of dystrophin protein that is necessary to rescue a severe muscle pathology has not been addressed. As zebrafish dystrophin mutants (dmd) resemble the severe muscle pathology of human patients, we have utilized this model to evaluate exon skipping. Novel dmd mutations were identified to enable the design of phenotype rescue studies via morpholino administration. Correlation of induced exon-skipping efficiency and the level of phenotype rescue suggest that relatively robust levels of exon skipping are required to achieve significant therapeutic ameliorations and that pre-screening analysis of exon-skipping drugs in zebrafish may help to more accurately predict clinical trials for therapies of DMD.
Keywords: dystrophin, DMD, Duchenne muscular dystrophy, zebrafish, muscle, exon skipping
Introduction
Mutations in the dystrophin gene can lead to two forms of muscular dystrophy. Null mutations that cause complete loss of dystrophin function typically result in Duchenne muscular dystophy (DMD), a lethal disease characterized by progressive muscle wasting. In-frame deletions that lead to internally deleted dystrophin protein generally result in Becker muscular dystrophy (BMD), which shows milder symptoms than DMD with a large variation in clinical severity.
The finding of patients with a mild form of BMD that have in-frame deletions of often large regions of the dystrophin gene, together with the existence of revertant, dystrophin positive fibres in many DMD patients [1] suggests that exclusion of exons harbouring mutations can result in largely functional dystrophin protein. Similarly, the functionality of an artificial mini-dystrophin harbouring deletion of exons 18 to 58 has been reported suggesting much of the dystrophin protein may be dispensable for its function [2]. Currently, clinical trials are underway that are based on administration of exon-skipping antisense oligonucleotides (AO) [3, 4]. In exon skipping, AOs are employed to sterically block exon recognition splice motifs located in the pre-mature transcript. Effective blocking leads to faulty transcript processing that does not splice targeted exons correctly, so that they are excluded from the mature mRNA, resulting in a shorter, but partly to largely functional dystrophin. Although this approach will only be useful for the treatment of a subset of patients, it is estimated that 83% of all DMD patients would theoretically benefit from single or double exon skipping [5]. The exon-skipping approach has shown much promise in recently finished clinical trials [3, 4]. However, these clinical trials were limited, as the AO was only administered locally into a single muscle and healthy volunteers could not be tested due to the possibility of adverse affects on dystrophin function. Exon skipping has also been performed in the mdx mouse and in the golden retriever muscular dystrophy (GRMD) dog [6, 7], and the efficacy of exon-skipping protocols in generating Dystrophin protein has been demonstrated in both these models. However, the mdx mouse does not resemble the severity of the human condition and a systematic and statistical evaluation of exon skipping in dogs is hampered by their inefficient breeding and phenotypic variety [8]. Therefore, the level of dystrophin function necessary to effect full functional rescue in the context of a severe dystrophin-deficient pathology remains unclear.
In contrast to the most commonly used model, the mdx mouse [9], dystrophin-deficient zebrafish, named dmdta222a [10], closely resemble the human condition in severity, onset and specific symptoms [11]. The progressive increase in expression of dystrophin protein at non-junctional sites in zebrafish correlates well with a number of pathological similarities between dystrophin-deficient zebrafish and patients suffering from DMD. Dystrophin-deficient zebrafish larvae show extensive muscle degeneration accompanied by fibrosis, inflammatory response, activation of the muscle stem cell compartment and greater variation in myofibre cross-sectional areas [11]. The detection of dystrophic muscle by reduction in birefringence [12], a light effect of the fish muscle visible under polarized light, enables efficient usage of dmd mutants in statistically evaluated experiments. In addition, zebrafish as a model system provide valuable advantages as its embryos develop ex utero, are translucent, amenable for drug screening [13], highly manipulable and genetically tractable [14], suggesting that dystrophin-deficient zebrafish could be a clinically relevant context in which to evaluate efficacy of therapeutic strategies.
In this study, novel dystrophin-deficient zebrafish mutants were isolated and used to evaluate efficacy of phenotype rescue by administration of morpholinos. Exon-skipping protocols were established that deliver a graded restoration of dystrophin levels demonstrating that only robust levels of induced exon skipping lead to sufficient dystrophin function capable of rescuing the dmd pathology. This analysis therefore provides a benchmark for the level of exon skipping that is required to effect clinically relevant pathological amelioration in the face of a fully penetrant dystrophic phenotype.
Materials and methods
Zebrafish lines and maintenance
The dmdtm90c and dmdta222a mutant alleles were obtained from the Tübingen Stock Collection [12]. All mutant lines were maintained in the TU (Tübingen) zebrafish strain. Genotyping was accomplished via PCR followed by restriction digestion (Fig. S1). All animal experiments were approved by MAS/2009/05.
Non-complementation screen and allele identification
Forty-eight males were mutagenized with N-ethyl-N-nitrosourea (ENU) as described [15] with the alteration of using 0.004% Tricaine (Sigma-Aldrich, St. Louis, MO, USA) during treatment. Surviving fish were out-crossed to females. The resulting F1 generation was crossed to heterozygous dmdta222a/+ fish and the offspring analysed for their birefringence at 3 days after fertilization (dpf). This non-complementation approach resulted in the identification of two novel dystrophin alleles after a survey of 243 genomes. To stabilize their genetic background, founder animals were back-crossed to TU over six generations before further experiments were performed. To determine the mutation rate, DNA was isolated from all F1 males and 1704 kbp at 25 randomly chosen loci were sequenced. Identified mutations were confirmed in independent PCR, resulting in a mutation rate of 1 mutation every 107 kbp. Cryostat cross-sections were stained with Harris haematoxylin and eosin or immunostained as described [11].
Morpholino oligomer injections
Morpholino oligomers were designed and named as described [16] and ordered from Gene Tools LLC (Philomath, OR, USA). Sequences were: Z32A(–18 + 7): 5′-gtttatccttaaacagacacatttg-3′, Z32E(+83+107): 5′-ttcactttcacctcctccagcactc-3′ and Z32E(+133+157): 5′-gtgactgcaccacttcctgctccac-3′, targeting exon 32 as indicated by the number in brackets. Zebrafish embryos were injected at the 1- to 2-cell stage into the yolk with 1.4 nl of morpholino solutions of different concentrations. Isogenic, uninjected embryos from the same clutches were analysed as controls.
RNA extraction and semi-quantitative RT-PCR
Total RNA was extracted from larvae at different stages with TriReagent (Sigma) and dystrophin cDNA was prepared with the SuperScript III First-Strand-Synthesis System (Invitrogen, Carlsbad, CA, USA) using the primer 34cDNA: 5′-ccaaatcacttggcataccctcc-3′. The cDNA was then subjected to PCR performed with Advantage-HF 2 Polymerase Mix (Clontech, Mountain View, CA, USA) using the primers 30For: 5′-ctgaacaccactgaccgacatc-3′ and 34Rev: 5′-cttctccagttcctgctttcctt-3′. Resulting amplicons were confirmed by sequencing. To determine the relative abundance of different dystrophin transcripts, we performed semi-quantitative RT-PCR using wild-type (WT) dystrophin transcript (549 bp) as internal control. For each morpholino concentration described, the PCR was optimized to keep the number of PCR cycles to a minimum. Proportions of different amplified dystrophin transcripts were quantified by measuring the band intensities using ImageJ (densitometry).
For statistical analysis of the correlation of morpholino efficiency and the ratio of dystrophic larvae, three clutches of 80–100 eggs from independent breeding pairs were injected with the described morpholino concentrations and 3 dpf hatched larvae were analysed for their muscle birefringence. Two larvae per clutch, with a dystrophic phenotype when applicable, were pooled and used for RT-PCR based analysis of dystrophin transcripts. Data are represented as means ± S.E.M.
Results
Screen for novel dystrophin alleles
In order to examine the effectiveness of the zebrafish dmd mutant for evaluating exon-skipping protocols of DMD patients, we determined its conservation in comparison to the human dystrophin gene. Alignment of the human (muscle specific Dp427m isoform) and zebrafish dystrophin shows that 57% of the human protein sequence is identical (84% conserved) to zebrafish dystrophin. Additionally, the exon–intron structure is highly conserved: with the exception of the first and the last exon, each human dystrophin exon has a corresponding exon in the zebrafish sequence and skipping of any exon induces the same frame shift in the zebrafish and the human dystrophin transcript (Fig. S2). Thus, the zebrafish model has the potential to examine the functional consequence of exon skipping in the context of a highly penetrant in vivo model of DMD.
In order to study the feasibility and characteristics of phenotype rescue by exon skipping, the targeted exon should not encode for a region essential for dystrophin function. Unfortunately, the original dmdta222a allele that we initially characterized harbours a premature stop codon in an exon encoding for a N-terminal calponin homology domain that is believed to be essential for actin binding [10, 17] and the only other identified allele, sapcl100, carries a splice site mutation, which itself is unsuitable for testing exon-skipping protocols [18]. Thus, we sought to generate new dystrophin alleles that would fall within exons appropriate for the evaluation of exon skipping.
To identify novel dystrophin alleles, a non-complementation screen based on the dmdta222a fish was performed. Adult male zebrafish were treated with ENU and subsequently crossed to WT female to generate F1 founders. The F1 founders were subsequently crossed to heterozygous dmdta222a/+ fish. Resulting offspring were screened for their levels of birefringence at 3 dpf. Non-complementation of the dmdta222a allele, indicated by a reduction in birefringence, identified 2 novel dmd mutants. In addition, the zebrafish mutant dmdtm90c (synonym: sapje, saptm90c), previously identified in a large scale screen but yet to be molecularly characterized [12], was also confirmed not to complement the dmdta222a allele (Fig. 1A–D). Whole mount immunohistochemistry with antibody against dystrophin indicated loss of dystrophin in all three mutants and each of the mutants possessed a similar level of phenotypic severity compared to the original (Fig. 1A’–D’). Subsequent sequencing of the genomic dystrophin coding region in the mutants and comparison with the WT sequence led to the discovery of premature stop codons within the exons 21, 32 and 53 for the alleles named dmdpc1, dmdpc2 and dmdtm90c, respectively (Fig. 1B”–D”). The identified mutations are widely distributed across the repetitive spectrin repeat region of dystrophin, which is often deleted within BMD patients and is also largely dispensable for the functional rescue of the mdx mouse by mini-dystrophin [2]. Thus, the mutations evident in the novel dystrophin alleles dmdpc1, dmdpc2 and dmdtm90c, lie within regions suitable for exon-skipping protocols (Fig. 1E).
Fig 1.
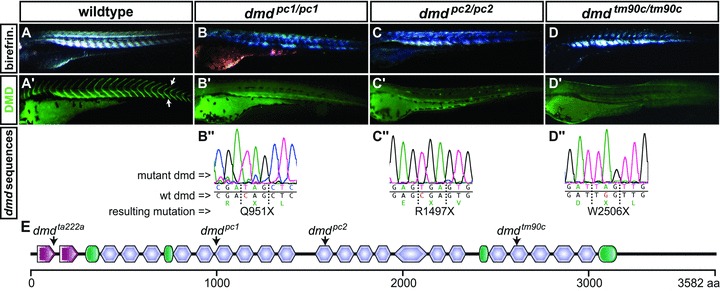
Novel dmd mutants identified in a non-complementation screen. (A) Birefringence of the muscle of a 3 dpf WT larva, evoked by polarized light, indicates an ordered array of myofilaments. (B–D) All novel dmd mutants display reduced muscle birefringence. (A’) Immunofluorescence revealed that dystrophin (green) is present at the somite borders (arrows) in siblings, (B’–D’) but absent in the homozygous dmd mutants. (B”–D”) Genomic sequencing of the dmd mutants revealed premature stop codons within exon 21 of dmdpc1/pc1, exon 32 of dmdpc2/pc2 and exon 53 of dmdtm90c/tm90c. (E) Arrows in the schematic diagram of zebrafish dystrophin point to the location of the premature stop codon encoded in each indicated mutant. Amino acid positions are displayed in the scale bar below; calponin-homology domains are in red, hinge domains in green and spectrin-like repeats in blue.
Dystrophin restoration in dmd homozygotes via exon skipping
AO, designed to sterically hinder specific splicing signals, can be employed to alter the splicing of the dystrophin pre-mRNA so that the targeted exon is excluded from the mature mRNA leading to an internally deleted, but partially to largely functional dystrophin protein. In such an exon-skipping approach, removal of the exon may neither disrupt the open reading frame nor remove essential regions of the protein. The skipping of exon 32, which is mutated in dmdpc2, satisfies these two criteria.
An phosphordiamidate morpholino oligomer, Z32A(–18+7), was designed to target the 5′-end of exon 32. To avoid nonspecific morpholino effects at high concentrations, various concentrations of Z32A(–18 + 7) were administered. By comparison of abnormal embryo rates versus skipping efficiency, the optimal concentration of Z32A(–18 + 7) for injections was established to be 500 μM (data not shown), a concentration which induced a slight curve in the angle of the body axis and a small delay in development. Two days after delivery of Z32A(–18 + 7) into WT embryos, RNA was isolated and RT-PCR across exons 30–34 performed. Although RT-PCR on uninjected embryos led to detection of only WT dystrophin transcript, RT-PCR after injection of Z32A(–18 + 7) resulted in two additional transcripts: one corresponding to a larger transcript arising from insertion of intron 31 upstream and one corresponding to a shorter amplicon originating from simultaneous skipping of the two exons 32 and 33 (Fig. 2A and B). The latter amplicon emanates from a dystrophin transcript with a preserved open reading frame encoding for a shorter dystrophin protein missing 121 amino acids encoded by exons 32 and 33. Interestingly, the proportion of the two Z32A(–18 + 7)-induced transcripts in relation to all detected dystrophin transcripts is significantly higher in dmdpc2/pc2 homozygotes (82%± 0.6%) than in WT embryos (45%± 0.5%; P < 0.01, n = 3), suggesting that the stop mutation-containing transcript could well undergo nonsense mediated decay (Fig. 2F). In addition, even though the amount of the two additional transcripts was declining, they could still be detected 7 days after Z32A(–18 + 7) delivery (Fig. 2F).
Fig 2.
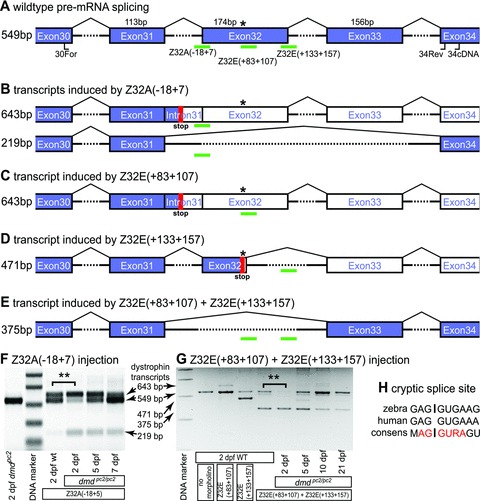
Evaluation of the efficacy of exon skipping for phenotypic rescue of dystrophin-deficient zebrafish. (A) Schematic diagram of the pre-mature dystrophin transcript with exons represented by boxes and introns (not to scale) marked by arches. The positions of the primers (34cDNA, 30For and 34Rev) used for RT-PCR are indicated and green bars mark the position of the morpholino targets. The sizes of exons as well as RT-PCR amplicons are shown in bp and the position of the dmdpc2 mutation is indicated by an asterisk. Blue exons represent translated coding sequence, whereas white exons are not translated with the first stop codon represented by a red block. (B–E) Schematic diagrams of transcripts induced by the indicated morpholinos with the size of the RT-PCR amplicons indicated. (F) Injection of Z32A(–18 + 7) induces a 643 bp and a 219 bp amplicon representing dystrophin transcripts schematically depicted in (B). Interestingly, the proportion of induced exon-skipped transcripts is, with high significance, increased in 2-dpf-old dmdpc2/pc2 homozygotes compared to WT control embryos (82%± 0.6% versus 45%± 0.5%, P < 0.01, n = 3) suggesting that nonsense-mediated decay proportionally reduced the levels of un-skipped stop codon containing transcript. (G) Administration of Z32E(+133+157) leads to exclusion of 78 bp from the 3′-end of exon 32 and administration of Z32E(+83+107) induces inclusion of the intron located upstream of exon 32. Combination of these two morpholinos, however, leads to a robust skipping of exon 32, which is still detected 21 days after administration. Again, the proportion of morpholino-induced transcript is significantly higher in 2-dpf-old dmdpc2/pc2 homozygotes than WT embryos (93%± 2% versus 66%± 3%, P < 0.01, n = 3). (H) Comparison of the zebrafish cryptic splice site elicited by Z32E(+133+157) with the corresponding human dystrophin sequence and the human 5′ splice consensus [24]. ** indicates P < 0.01.
To determine, if administration of Z32A(–18 + 7) leads to expression of dystrophin protein, whole mount immunohistochemistry with antibodies against dystrophin was performed. Three days after administration of Z32A(–18 + 7) the proportion of the exon 31–32 deletion transcript is 4.7%± 0.5% of all dystrophin transcripts (n = 8; data are means ± S.E.M.) and, in contrast to uninjected dmdpc2/pc2 homozygotes, faint dystrophin expression could be detected in Z32A(–18 + 7)-injected homozygous dmdpc2/pc2 at the same stage (Fig. 2B and C).
Under polarized light the zebrafish muscle shows a birefringence effect that is caused by parallel thread-like myofibrils within muscle fibres (Fig. 3A’). This birefringence is reduced in dystrophin-deficient zebrafish, as the fibre organization is lost due to detachment and retraction of fibres [10]. Therefore, the birefringence assay was employed to survey if the restored levels of dystrophin are sufficient to rescue the dmdpc2 phenotype. Z32A(–18 + 7)-injected homozygotes, however, demonstrate a loss of birefringence that is comparable to dmdpc2/pc2 homozygotes, indicating muscle loss and lack of phenotypic rescue (Fig. 3B’ and C’). In order to survey the phenotype with an independent method, cross-sections of uninjected and Z32A(–18 + 7)-injected dmdpc2/pc2 homozygotes larvae were stained for haematoxylin and eosin at 7 dpf. As indicated by the depicted muscle fibrosis and myofibre loss, the muscle of dmdpc2/pc2 homozygotes injected with Z32A(–18 + 7) demonstrates a level of muscle loss comparable to that detected in untreated dmdpc2/pc2 homozygotes (Fig. 3B” and C”). Taken together, these results reveal that administration of the morpholino Z32A(–18 + 7) leads to a low level of exon skipping that restores a detectable level of dystrophin protein in dmdpc2/pc2 homozygotes, but does not rescue the dmdpc2 phenotype.
Fig 3.
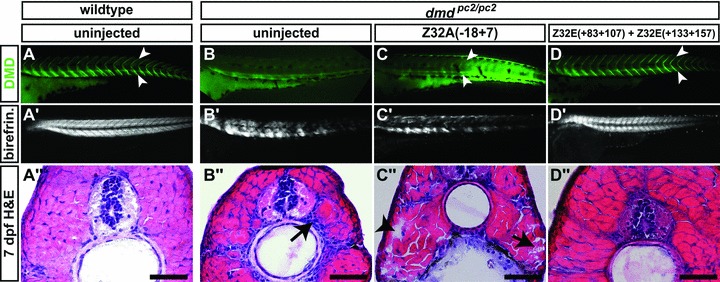
Phenotype analysis of morpholino injected dmdpc2/pc2 homozygotes. (A) Although in 3-dpf-old WT larvae antibody against dystrophin locates dystrophin expression at the somite borders (arrowheads), (B) in dmdpc2/pc2 homozygous larvae dystrophin is not detected. (C) In contrast, homozygotes dmdpc2/pc2 treated with Z32A(–18 + 7) show weak dystrophin expression and (D) homozygotes administered with a combination of Z32E(+133+157) and Z32E(+83+107) display robust restoration of dystrophin protein at the somite borders (arrowheads). Although birefringence is evident in WT larvae at 3 dpf, it is reduced in (B’) untreated and (C’) Z32A(–18 + 7)-injected dmdpc2/pc2 homozygotes. (D’) dmdpc2/pc2 homozygotes injected with Z32E(+133+157) and Z32E(+83+107) demonstrate restoration of birefringence, indicating full phenotype rescue. Accordingly, haematoxylin and eosin staining performed on cross-sections of 3-dpf-old larvae shows no phenotypic muscle in (A”) WT larvae, whereas the muscle of (B”) uninjected and (C”) Z32A(–18 + 7)-treated dmdpc2/pc2 homozygous larvae is dystrophic and the phenotype is rescued in larvae (D”) administered with Z32E(+133+157) and Z32E(+83+107). The arrow in (B”) points to mono-nucleated infiltrates and in (C”) to necrotic myofibre. The arrowhead in (C”) points to a gap where a necrotic myofibre has been replaced by mucus. Scale bar: 50 μm.
Phenotype rescue of dmdpc2/pc2 homozygotes via exon skipping
Given the surprising complexity of the response to the blocking of the 3′ or acceptor splice site of exon 32, additional mechanisms were sought to increase the efficacy of in-frame skipping of exon 32 in dmdpc2/pc2 homozygous mutants. In addition to blocking 3′ acceptor or 5′ donor-splice sites, targeting exonic splice enhancers (ESE) located within exons has also reported to lead to exon-skipping events [19]. Therefore, the program “RESCUE-ESE” was used to predict ESE located in exon 32 of dystrophin. Based on the prediction, the morpholino Z32E(+133+157) was designed against an ESE positioned close to the 3′-end of exon 32. RT-PCR, performed in the same manner as described above, on WT embryos treated with Z32E(+133+157) led to the detection of an additional dystrophin transcript corresponding to the deletion of bp +97 to +174 of exon 32, activating a cryptic splice site at 5′-GAG gugaaa-3′ (Fig. 2D, G and H). Therefore, a second morpholino, Z32E(+83+107), was designed to target this region. Administration of Z32E(+83+107) alone promotes inclusion of the intron upstream of exon 32. However, administration of the combined morpholinos Z32E(+133+157) and Z32E(+83+107), in a concentration of 25 μM each, induced robust skipping of targeted exon 32 without non-specific morpholino effects. Similar to the result already demonstrated with Z32A(–18 + 7) injection, the proportion of the morpholino-induced transcript in relation to the WT dystrophin transcript is significantly higher in dmdpc2/pc2 homozygotes (93%± 2%) compared to WT embryos (66%± 3%; P < 0.01, n = 3) (Fig. 2G). Although 2 days after injection of dmdpc2/pc2 homozygotes with the combination of Z32E(+133+157) and Z32E(+83+107) robust skipping of exon 32 was observed, the amount of morpholino-induced dystrophin transcript declined with progressing age of injected embryos. However, skipping of exon 32 was still detected 21 days after injection, suggesting that the stability of the morpholino AO in vivo and its efficacy in inducing exon skipping 3 weeks after injection may provide an effective therapeutic window of treatment (Fig. 2G).
In order to survey the effect of Z32E(+133+157) and Z32E(+83+107) administration on dmdpc2/pc2 homozygotes, whole mount immunohistochemistry using an antibody that recognizes zebrafish dystrophin was performed. Although dystrophin was not detected in 3 dpf dmdpc2/pc2 homozygotes, robust dystrophin expression was detected in dmdpc2/pc2 homozygotes treated with Z32E(+133+157) and Z32E(+83+107) (Fig. 3B and D). Accordingly, untreated dmdpc2/pc2 homozygotes display a severely reduced muscle birefringence under polarized light, whereas 3-dpf-old homozygotes treated with the morpholino combination show restoration of muscle birefringence, indicative of a full phenotypic rescue (Fig. 3B’ and D’). In addition, the muscle histology of 7 dpf dmdpc2/pc2 homozygous larvae administered with Z32E(+133+157) and Z32E(+83+107) was analysed on cross-sections stained with haematoxylin and eosin, which confirmed that in contrast to untreated dmdpc2/pc2 homozygotes, no muscle loss or enhanced fibrosis could be found (Fig. 3B” and D”).
Collectively, administration of two morpholinos, one against a splice enhancer and the other blocking a cryptic splice site, induced a high level of exon 32 skipping leading to restoration of a robust level of dystrophin protein, sufficient to fully rescue the phenotype of dmdpc2/pc2 homozygotes.
Determination of the threshold level of exon skipping required to effect phenotypic rescue in dystrophin-deficient zebrafish
In contrast to administration of Z32E(–18 + 7) that only leads to weak skipping of exons 31 and 32 and does not result in phenotype rescue, simultaneous administration of Z32E(+133+157) and Z32E(+83+107) shows a robust skipping effect leading to a full phenotype rescue. We therefore surveyed in more detail the level of skipping efficiency necessary to achieve dmd phenotype rescue.
For this purpose, we in-crossed dmdpc2/+ carriers and injected the offspring with Z32E(+133+157) and Z32E(+83+107) combined at an equal concentration of 12 μM. First, to test the accuracy of the morpholino administration, dmdpc2/pc2 larvae were identified by PCR and subsequently analysed for their induced skipping effect individually. As shown in Figure S3, the variation in the detected skipping effects was relatively low. Therefore, in subsequent experiments with morpholino concentrations varying from 0 to 12 μM, two dmdpc2/pc2 homozygous larvae were combined and the proportion of exon 32 skipped transcript was analysed in relation to all dystrophin transcripts by RT-PCR. Also, progression of the dystrophic pathology after each morpholino administration was assessed by analysis of the muscle birefringence at 3 dpf (Fig. 4). The extremes of the injected concentrations, 0 and 12 μM, resulted in a Mendelian ratio of 24.6%± 0.5% of affected larvae and no detectable dystrophic phenotype, respectively (data in mean ± S.E.M.). Intermediate concentrations, however, resulted in partial rescue of the birefringence in a highly dose-dependent manner as manifested by intermediate phenotype ratios. Collected data suggest that a skipping efficiency of about 10% results in 10% of detected dystrophic fish, representing a reduction of the expected Mendalian ratio by about half. In contrast to this partial rescue, a skipping efficiency of about 30–40% seems to result in less than 3% of injected larvae with a detectable dystrophic pathology, revealing that a skipping efficiency of 30–40% is sufficient to evoke a near full rescue of the dystrophic phenotype. As described above, injection of Z32E(+133+157) and Z32E(+83+107) into WT embryos resulted in lower exon-skipping efficiencies. Therefore, skipping efficiencies measured in homozygous mutants were also analysed in WT embryos. Although exon skipping could not be detected at morpholino concentrations of 0 μM, 1 μM and 2 μM, injection of 12 μM of the morpholino combination resulted in 12%± 1% skipping efficiency, 6 μM in 3.5%± 0.2%, 5 μM in 2.8%± 0.3%, 4 μM in 1.5%± 0.2% and 3 μM in 0.6%± 0.1% efficiency.
Fig 4.

Determination of phenotypic rescue threshold for exon-skipping efficiency. Different concentrations of an equal mixture of Z32E(+133+157) and Z32E(+83+107) were injected into embryos of an in-cross of heterozygous dmdpc2/+ fish. At 3 days post administration, resulting levels of exon-skipping efficiency (blue line) were evaluated by RT-PCR and depicted as the percentage of skipped transcript out of total dystrophin transcript. The corresponding level of phenotypic rescue, as evaluated by the birefringence assay, is displayed as the percentage of larvae possessing a dystrophic phenotype from all injected larvae. Administration of doses of combined Z32E(+133+157) and Z32E(+83+107) below 2 μM does not change the Mendalian ratio (25%) of detected dystrophic mutants. In contrast, no larvae display a dystrophic phenotype when injected with a concentration of 12 μM and administration of intermediate concentrations lead to partial rescue as specified in the depicted diagram. These data suggest an exon-skipping efficiency of 20–30% is required to induce effective phenotypic rescue of the highly penetrant dystrophic phenotype evident in the zebrafish model. Data are means ± S.E.M.
Discussion
This report describes the efficacy of exon-skipping approaches in the dystrophin-deficient zebrafish mutant dmd that accurately resembles the severe dystrophic muscle phenotype of human DMD. Statistical and systematic evaluation of exon-skipping efficiency demonstrates that relatively robust levels of exon skipping are required to achieve a significant level of phenotype rescue in a severe dystrophic pathology in vivo.
Recently, two promising clinical trials employing exon skipping of dystrophin have successfully been completed [3, 4], but some unanswered questions remain. Amongst the problems that have been encountered is the fact that individual AO cannot generally be tested for side-effects in healthy volunteers, only small biopsies can be taken that are not statistically evaluated, and it remains unclear to which level dystrophin function needs to be restored to significantly improve the condition of DMD patients. Exon skipping can be surveyed in tissue culture, however, the canine model, as well as our survey, shows a large discrepancy in the outcome of exon-skipping experiments undertaken in vitro and in vivo [20], pointing to the importance of establishing rescue protocols in vivo.
Our comparison of the exon–intron structure of the human and the zebrafish dystrophin gene shows a remarkable degree of conservation. With the exception of the first and the last exon, this level of conservation enables modelling of skipping of human dystrophin exons in the zebrafish in vivo.
One of the novel dmd alleles that we identified in a genetic screen, dmdpc2, carries a mutation in exon 32, deletion of which does not disrupt the open reading frame nor results in deletion of an essential region of the dystrophin protein. This leaves us in the unique position of testing exon skipping in a genetically tractable model of DMD in vivo. We have established the experimental paradigm that enables us to survey exon-skipping experiments in our dmdpc2 mutant, measure the necessary skipping efficiency to rescue the dmd phenotype, and histologically survey the outcome of the rescue. Our data suggest that about 10% to 20% of normal dystrophin levels lead to only partial restoration of muscle function, whereas levels of at least 30% to 40% are needed to significantly improve muscle function in the context of the zebrafish dystrophic condition. These results are comparable to studies that correlated the level of dystrophin, quantified by antibodies, to the severity of symptoms evident within a given patient. Although patients with less than 10% of the normal level of dystrophin are reported to show a severe clinical phenotype of DMD [21], patients with 15% or less display a moderate to severe DMD phenotype [22] and BMD patients with mild symptoms are found to have dystrophin levels above 30%[22, 23].
Our observations show a highly significant difference between exon-skipping protocols performed on siblings and on mutants, probably due to nonsense-mediated decay of mutant transcript that harbours a premature stop-codon. This effect has to be taken into account when skipping efficiencies in dystrophin-deficient and WT zebrafish are compared.
Interestingly, administration of the AO Z32E(+133+157) activated a cryptic splice site at 5′-GAGgugaaa-3′, which is highly conserved between zebrafish and human beings. This potential cryptic splice site should be taken into account when AO are designed against exon 32 in human patients. It also reveals a surprisingly high level of conservation of the genetic structure of the dystrophin gene across a large evolutionary distance. Furthermore, to our knowledge, this is also the first time two splice switching AOs, which singly either promoted intron inclusion or activated a cryptic splice site, have been combined to induce robust and specific targeted exon skipping, and suggests a new mechanistic basis to promote exon skipping of individual exons.
The quality of life of DMD sufferers is not considered in the dmd fish model, any treatment of DMD needs to be evaluated in the context of individual human patients, as the use of exon-skipping strategies that induce less than optimal skipping efficiency may well have a significant therapeutic benefit. Nonetheless, these results highlight the importance of testing exon-skipping protocols in an in vivo model that features a comparable mutant context to patients for which a clinical trial is designed, in this way allowing a more accurate design and prediction of the clinical trial. As demonstrated here, the zebrafish as an animal model has the advantage of rapid screening techniques to generate alleles specific for exon-skipping strategies targeting specific exons.
In summary, the dmd fish can effectively be used to evaluate proposed therapeutic strategies for the treatment of DMD. We reveal that exon skipping is an effective mechanism for restoring dystrophin function in the context of a fully penetrant dystrophic phenotype and propose that the zebrafish model will be a valuable regent in identifying, designing and evaluating future therapeutic strategies for DMD.
Acknowledgments
We are grateful to R. Kettleborough and D. Stemple for technical advice. This work was supported by grants from the National Health and Medical Research Council of Australia to PDC.
Conflict of interest
The authors confirm that there are no conflicts of interest.
Supporting Information
dmd allele identification. The followingprimer pairs amplify the dystrophin locus, characteristic for eachallele, followed by restriction digestion with the designatedenzyme. Resulting DNA band patterns are as depicted.dmdpc1: pc1F 5'-ctgcagtctccatccaagt-3' and pc1R5'-accaagtcctggaggacctgagggccagggatcga-3', ClaI.dmdpc2: pc2F 5'-aatgcctgtaaacaaatgtgtctgt-3' andpc2R 5'-ccttgccatgttaacccaaa-3', BsaXI.dmdta222a: ta222aF 5'-catacccaaggtttcaaagca-3' andta222aR 5'-gttaagggagtgcactcgagtgaagccacgttttt-3', DraI.dmdtm90c: tm90cF5'-actgcagaacatgtataaggactccagcgctt-3' and tm90cR5'-aattaccttgacatctgcattttggcc-3', DdeI.
Comparison of human and zebrafish dystrophin. Numbered boxes represent exons of the human dystrophin isoform Dp427m with amino acid numbers indicated. The conservation of each exon in comparison to the zebrafish dystrophin sequence is written inside each box. Blue boxes represent exons that have the same number of amino acids in human and zebrafish and yellow ones have a different number of amino acids, as indicated below each yellow box.
Variation of exon skipping in individual larvae.At 3 dpf, the exon-skipping efficiency was analysed in individuallarvae administered with an equal mixture of Z32E(+133+157) andZ32E(+83+107) at a concentration of 12 μM each. The variation inexon-skipping efficiency is relatively low, allowing pooling of twolarvae for subsequent analysis in further experiments.
References
- 1.Klein CJ, Coovert DD, Bulman DE, et al. Somatic reversion/suppression in Duchenne muscular dystrophy (DMD): evidence supporting a frame-restoring mechanism in rare dystrophin-positive fibers. Am J Hum Genet. 1992;50:950–9. [PMC free article] [PubMed] [Google Scholar]
- 2.Harper SQ, Hauser MA, DelloRusso C, et al. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat Med. 2002;8:253–61. doi: 10.1038/nm0302-253. [DOI] [PubMed] [Google Scholar]
- 3.van Deutekom JC, Janson AA, Ginjaar IB, et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med. 2007;357:2677–86. doi: 10.1056/NEJMoa073108. [DOI] [PubMed] [Google Scholar]
- 4.Kinali M, Arechavala-Gomeza V, Feng L, et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009;8:918–28. doi: 10.1016/S1474-4422(09)70211-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aartsma-Rus A, Fokkema I, Verschuuren J, et al. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat. 2009;30:293–9. doi: 10.1002/humu.20918. [DOI] [PubMed] [Google Scholar]
- 6.Mann CJ, Honeyman K, Cheng AJ, et al. Antisense-induced exon skipping and synthesis of dystrophin in the mdx mouse. Proc Natl Acad Sci USA. 2001;98:42–7. doi: 10.1073/pnas.011408598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McClorey G, Moulton HM, Iversen PL, et al. Antisense oligonucleotide-induced exon skipping restores dystrophin expression in vitro in a canine model of DMD. Gene Ther. 2006;13:1373–81. doi: 10.1038/sj.gt.3302800. [DOI] [PubMed] [Google Scholar]
- 8.Zucconi E, Valadares MC, Vieira NM, et al. Ringo: discordance between the molecular and clinical manifestation in a golden retriever muscular dystrophy dog. Neuromuscul Disord. 2010;20:64–70. doi: 10.1016/j.nmd.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 9.Bulfield G, Siller WG, Wight PA, et al. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci USA. 1984;81:1189–92. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bassett DI, Bryson-Richardson RJ, Daggett DF, et al. Dystrophin is required for the formation of stable muscle attachments in the zebrafish embryo. Development. 2003;130:5851–60. doi: 10.1242/dev.00799. [DOI] [PubMed] [Google Scholar]
- 11.Berger J, Berger S, Hall TE, et al. Dystrophin-deficient zebrafish feature aspects of the Duchenne muscular dystrophy pathology. Neuromuscul Disord. 2010;20:826–32. doi: 10.1016/j.nmd.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 12.Granato M, van Eeden FJ, Schach U, et al. Genes controlling and mediating locomotion behavior of the zebrafish embryo and larva. Development. 1996;123:399–413. doi: 10.1242/dev.123.1.399. [DOI] [PubMed] [Google Scholar]
- 13.Berger J, Currie P. The role of zebrafish in chemical genetics. Curr Med Chem. 2007;14:2413–20. doi: 10.2174/092986707781745532. [DOI] [PubMed] [Google Scholar]
- 14.Lieschke GJ, Currie PD. Animal models of human disease: zebrafish swim into view. Nat Rev Genet. 2007;8:353–67. doi: 10.1038/nrg2091. [DOI] [PubMed] [Google Scholar]
- 15.Pelegri F. Mutagenesis. In: Nusslein-Volhard C, Dahm R, editors. Zebrafish: a practical approach. New York: Oxford University Press; 2002. pp. 145–74. [Google Scholar]
- 16.Mitrpant C, Adams AM, Meloni PL, et al. Rational design of antisense oligomers to induce dystrophin exon skipping. Mol Ther. 2009;17:1418–26. doi: 10.1038/mt.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Corrado K, Mills PL, Chamberlain JS. Deletion analysis of the dystrophin-actin binding domain. FEBS Lett. 1994;344:255–60. doi: 10.1016/0014-5793(94)00397-1. [DOI] [PubMed] [Google Scholar]
- 18.Guyon JR, Goswami J, Jun SJ, et al. Genetic isolation and characterization of a splicing mutant of zebrafish dystrophin. Hum Mol Genet. 2009;18:202–11. doi: 10.1093/hmg/ddn337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Deutekom JC, Bremmer-Bout M, Janson AA, et al. Antisense-induced exon skipping restores dystrophin expression in DMD patient derived muscle cells. Hum Mol Genet. 2001;10:1547–54. doi: 10.1093/hmg/10.15.1547. [DOI] [PubMed] [Google Scholar]
- 20.Yokota T, Lu QL, Partridge T, et al. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann Neurol. 2009;65:667–76. doi: 10.1002/ana.21627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoffman EP, Kunkel LM, Angelini C, et al. Improved diagnosis of Becker muscular dystrophy by dystrophin testing. Neurology. 1989;39:1011–7. doi: 10.1212/wnl.39.8.1011. [DOI] [PubMed] [Google Scholar]
- 22.Angelini C, Fanin M, Pegoraro E, et al. Clinical-molecular correlation in 104 mild X-linked muscular dystrophy patients: characterization of sub-clinical phenotypes. Neuromuscul Disord. 1994;4:349–58. doi: 10.1016/0960-8966(94)90071-x. [DOI] [PubMed] [Google Scholar]
- 23.Nicholson LV, Johnson MA, Bushby KM, et al. Integrated study of 100 patients with Xp21 linked muscular dystrophy using clinical, genetic, immunochemical, and histopathological data. J Med Genet. 1993;30:728–36. doi: 10.1136/jmg.30.9.728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang MQ. Statistical features of human exons and their flanking regions. Hum Mol Genet. 1998;7:919–32. doi: 10.1093/hmg/7.5.919. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
dmd allele identification. The followingprimer pairs amplify the dystrophin locus, characteristic for eachallele, followed by restriction digestion with the designatedenzyme. Resulting DNA band patterns are as depicted.dmdpc1: pc1F 5'-ctgcagtctccatccaagt-3' and pc1R5'-accaagtcctggaggacctgagggccagggatcga-3', ClaI.dmdpc2: pc2F 5'-aatgcctgtaaacaaatgtgtctgt-3' andpc2R 5'-ccttgccatgttaacccaaa-3', BsaXI.dmdta222a: ta222aF 5'-catacccaaggtttcaaagca-3' andta222aR 5'-gttaagggagtgcactcgagtgaagccacgttttt-3', DraI.dmdtm90c: tm90cF5'-actgcagaacatgtataaggactccagcgctt-3' and tm90cR5'-aattaccttgacatctgcattttggcc-3', DdeI.
Comparison of human and zebrafish dystrophin. Numbered boxes represent exons of the human dystrophin isoform Dp427m with amino acid numbers indicated. The conservation of each exon in comparison to the zebrafish dystrophin sequence is written inside each box. Blue boxes represent exons that have the same number of amino acids in human and zebrafish and yellow ones have a different number of amino acids, as indicated below each yellow box.
Variation of exon skipping in individual larvae.At 3 dpf, the exon-skipping efficiency was analysed in individuallarvae administered with an equal mixture of Z32E(+133+157) andZ32E(+83+107) at a concentration of 12 μM each. The variation inexon-skipping efficiency is relatively low, allowing pooling of twolarvae for subsequent analysis in further experiments.