

Abstract
Apolipoprotein A-I (ApoA-I) is an extracellular lipid acceptor, whose role in cholesterol efflux and high-density lipoprotein formation is mediated by ATP-binding cassette transporter A1 (ABCA1). Nevertheless, some ApoA-I variants are associated to systemic forms of amyloidosis, characterized by extracellular fibril deposition in peripheral organs. Heart amyloid fibrils were found to be mainly constituted by the 93-residue N-terminal fragment of ApoA-I, named [1–93]ApoA-I. In this paper, rat cardiomyoblasts were used as target cells to analyse binding, internalization and intracellular fate of the fibrillogenic polypeptide in comparison to full-length ApoA-I. We provide evidence that the polypeptide: (i) binds to specific sites on cell membrane (Kd= 5.90 ± 0.70 × 10−7 M), where it partially co-localizes with ABCA1, as also described for ApoA-I; (ii) is internalized mostly by chlatrin-mediated endocytosis and lipid rafts, whereas ApoA-I is internalized preferentially by chlatrin-coated pits and macropinocytosis and (iii) is rapidly degraded by proteasome and lysosomes, whereas ApoA-I partially co-localizes with recycling endosomes. Vice versa, amyloid fibrils, obtained by in vitro aggregation of [1–93]ApoA-I, were found to be unable to enter the cells. We propose that internalization and intracellular degradation of [1–93]ApoA-I may divert the polypeptide from amyloid fibril formation and contribute to the slow progression and late onset that characterize this pathology.
Keywords: apolipoprotein A-I, amyloidosis, cardiomyoblasts, binding, internalization
Introduction
Apolipoprotein A-I (ApoA-I), the major component of high-density lipoproteins (HDL), is known to play a central role in cholesterol efflux from cells and transport to the liver [1], a crucial process to prevent cellular lipid overload as well as atherosclerosis. Epidemiological studies have demonstrated that plasma levels of HDL, and their major constituent ApoA-I, are inversely correlated with the risk of atherosclerosis [2]. Nevertheless, the molecular mechanism of the atheroprotective action of ApoA-I, as well as HDL biogenesis, is not fully understood. ApoA-I may act as an extracellular receptor of cellular cholesterol and phospholipids. ApoA-I lipidation to form nascent HDL particles is a process that involves ATP-binding cassette transporter A1 (ABCA1) [3–5], that mediates cholesterol efflux to ApoA-I. ApoA-I is also known to internalize to the endosomes, an intracellular reservoir of cholesterol, to bind lipids and to be resecreted to the medium as HDL [6]. Despite intense research activity suggesting a direct association between ApoA-I and ABCA1 [7], whether or not ABCA1 is to be considered as an ApoA-I receptor is still ambiguous. ApoA-I association to cell surface might not involve direct binding to ABCA1; rather, ABCA1 would induce modifications of lipid distribution at the membrane facilitating ApoA-I docking [3].
Despite its protective role against hypercholesterolemia and cardiovascular diseases, ApoA-I is associated to systemic amyloidoses when specific mutations occur in the 243-residue sequence of ApoA-I. Amyloidoses, conformational diseases related to protein misfolding, have been associated to over 40 different peptides and proteins identified so far [8]. Mutations and/or environmental conditions able to destabilize the protein native conformation kinetically favour aggregate nucleation [9] leading to fibrils that accumulate in amyloid deposits in tissues and organs [8].
Sixteen variants of ApoA-I are responsible for systemic amyloidoses characterized by aggregate deposition in peripheral organs, such as heart, liver or kidneys [1, 10, 11]. Amyloid fibrils isolated ex vivo were found to be mainly constituted by N-terminal fragments of ApoA-I, 90–100 residue long, released by a still unidentified protease. In particular, the fragment corresponding to sequence 1–93 was found to be the main constituent of cardiac fibrils extracted from patients harbouring variant L174S ApoA-I [10] and affected by a severe systemic amyloidosis predominantly involving the heart. The 93-residue fibrillogenic domain of ApoA-I, extracted from amyloid deposits of a patient who underwent a heart transplant for end-stage heart failure, was found to be a natively unfolded protein in water at neutral pH [12]. Acidic conditions (pH 4) were able to switch on a complex fibrillogenic pathway, consisting of extensive structural rearrangements of the polypeptide, that shifts from a random coil structure to an unstable helical conformation, and then aggregates into a β-sheet based polymeric structure [12].
We produced a recombinant version of ApoA-I 1–93 fragment, denoted as [1–93]ApoA-I, as a pure and stable product, following a strategy aimed at protecting the recombinant polypeptide from intracellular degradation [13]. Conformational analyses revealed that recombinant [1–93]ApoA-I, as the native polypeptide, undergoes conformational transitions and fibrillogenesis, leading to the formation of typical amyloid fibrils on a time scale comparable with that of the natural polypeptide [13].
Nothing is known about the mechanism leading to the release of the fibrillogenic polypeptide from a full-length amyloidogenic variant of ApoA-I, or in which context the proteolytic cleavage does occur. Nevertheless, the hypothesis can be raised that the fibrillogenic polypeptide is released at the site of fibrils deposition, where it accumulates in the extracellular space of target tissues. Here, aggregation in fibrillar structures occurs favoured by molecular crowding and the unfolded structure of the polypeptide. It has been demonstrated that the N-terminal region of full-length ApoA-I is involved in lipid membrane binding [14]. Recently, we suggested that lipids have a key role in [1–93]ApoA-I aggregation [15], as cholesterol, a natural ApoA-I ligand, was found to induce and stabilize helical conformers, slowing down the aggregation process. Moreover, zwitterionic, positively and negatively charged liposomes were found to affect [1–93]ApoA-I conformation by inducing the formation of helical species [15]. Hence, it is conceivable that, although the fibrillogenic polypeptide accumulates in the extracellular space of cardiac cells, it interacts with cell membranes as does the full-length protein.
Here we report binding, internalization and intracellular fate of [1–93]ApoA-I in cultured cardiomyoblasts, in comparison to the full-length protein. Our results show for the first time that the fibrillogenic fragment of ApoA-I is able to recognize specific binding sites on cell membrane, to be internalized in target cells and to be degraded following an intracellular route only partially coincident with that of full-length ApoA-I.
Materials and methods
Proteins and reagents
All reagents, wild-type ApoA-I, fluorescein isothiocyanate (FITC)-insulin and transferrin (Tf) were from Sigma-Aldrich (St Louis, MO, USA). LysoTracker Red was from Molecular Probes (Invitrogen, Carlsbad, CA, USA). Anti-human ApoA-I polyclonal antibodies were purchased from DAKO (Glostrup, Denmark); anti-β-catenin antibody from Santa-Cruz Biotecnology (Heidelberg, Germany); anti-ABCA1 polyclonal antibodies and the chemiluminescence detection system (SuperSignal® West Pico) from Pierce Biotechnology (Rockford, IL, USA); goat anti-rabbit and antimouse antibodies, conjugated with Texas red or with Bodipy fluorescein were from Invitrogen. [1–93]ApoA-I polypeptide was expressed and purified as previously described [15], omitting the neutralization step with ammonium hydroxide. Pure [1–93]ApoA-I was lyophilized and stored at –70°C until use.
Fibrillar aggregates were obtained by incubating [1–93]ApoA-I for 2 weeks at 37°C at 0.3 mg/ml protein concentration in 12 mM sodium phosphate buffer, pH 6.4 containing 20% (v/v) trifluoroethanol (TFE). By centrifugation, insoluble aggregates of [1–93]ApoA-I (pellet) were separated from the unaggregated, soluble polypeptide (supernatant). To quantify aggregated [1–93]ApoA-I, the amount of the soluble polypeptide was determined spectrophotometrically and subtracted from the total amount of [1–93]ApoA-I before aggregation. The pellet was dried under N2 to remove TFE and resuspended in cell medium to reach the appropriate protein concentration [16]. The suspension of the aggregated species was tested for cytotoxicity.
Cell culture, transfection and Western blot analyses
Rat embryos heart myoblasts H9c2 and human hepatic carcinoma HepG2 cells were purchased from American Type Culture Collection (ATCC). Cells were cultured in DMEM (Sigma-Aldrich), supplemented with 10% foetal bovine serum (HyClone; Thermo Scientific, Logan, UT, USA) and antibiotics, in a 5% CO2 humidified atmosphere at 37°C. The growth medium of H9c2 cells was implemented with 2 mM L-glutamine and 2 mM sodium pyruvate. Expression vectors for enhanced green fluorescent protein (GFP) tagged Rab4 [17] and enhanced red fluorescent protein (RFP) tagged Rab5 [18] were kindly provided by Dr Marino Zerial (Max-Planck-Institute, Dresden, Germany). H9c2 cells were transiently transfected with either expression vector by the use of METAFECTENE reagent according to the manufacturer’s instructions (Biontex-USA, San Diego, CA, USA). After 24 hrs, transfected cells were incubated with the appropriate protein and analysed. To prepare cell lysates, HepG2 and H9c2 cells were scraped off in phosphate-buffered saline (PBS), centrifuged at 1000 × g for 10 min. and resuspended in lysis buffer (1 mM MgCl2, 0.25% SDS, 1% Triton X-100 in 10 mM Tris-HCl, pH 7) containing protease inhibitors. After 30 min. incubation on ice, lysates were centrifuged at 14,000 × g for 30 min. at 4°C. Supernatants were diluted in loading buffer containing 8 M urea and analysed, without boiling, by 10% polyacrylamide SDS-PAGE electrophoresis. Protein concentration was determined by Bradford assay.
Atomic force microscopy (AFM) analysis
[1–93]ApoA-I was incubated as described above to generate fibrillar aggregates. Following incubation, the whole sample was diluted 10 times using Milli-Q water; 10 μl aliquots of the diluted sample were deposited on freshly cleaved mica and dried under mild vacuum. Tapping mode AFM measurements were performed in air using a Dimension 3100 scanning probe microscope equipped with a G scanning head (maximum scan size 100 μm) and driven by a Nanoscope IIIa controller (Digital Instruments-Bruker AXS GmbH, Karlsruhe, Germany). Images were acquired in tapping mode in air using single beam uncoated silicon cantilevers (type OMCL-AC160TS, Olympus, Tokyo, Japan). The drive frequency was typically 300 kHz and the scan rate was between 0.8 and 1.0 Hz. The size of aggregates was measured from the heights in cross-section in the topographic AFM images.
Binding assays
Proteins under test (100 μg) were labelled with 1 mCi carrier-free Na125I (Amersham; GE Healthcare Bio-Sciences AB, Uppsala, Sweden) using Iodobeads (Pierce), according to the manufacturer’s instructions. Labelled proteins were desalted on PD10 columns (Amersham) equilibrated in PBS. The specific activity was about 1.5 μCi/μg. Cells were seeded in 24-well plates at a density of 5 × 104/well. After 24 hrs, 200 μl of binding buffer (25 mM Hepes, pH 7.5, 1 mg/ml bovine serum albumin in DMEM), containing increasing concentrations of the labelled protein under test, were added to the cells. Following 2 hrs incubation at 4°C, cells were washed three times with PBS containing 0.1% bovine serum albumin. Bound radioactivity (total binding) was removed by treating cells with 0.7 ml of cold 0.6 M NaCl in PBS for 2 min. on ice and measured with a γ counter (Packard Instrument Co. Inc., Meriden, CT, USA). Non-specific binding was determined by incubating the cells with the labelled protein in the presence of a 40-fold molar excess of the unlabelled protein. Specific binding was calculated by subtracting non-specific binding from total binding. Affinity constant values (Kd) were calculated according to the Scatchard equation.
Cytotoxicity assays
Cells were seeded in 96-well plates (100 μl/well) at a density of 5 × 103/well. After 24 hrs, [1–93]ApoA-I, dissolved in 12 mM sodium phosphate buffer, pH 6.4 (1 mg/ml) and centrifuged to remove insoluble material, was added to the medium to a final concentration of 5 or 10 μM. To test fibrillar aggregates, [1–93]ApoA-I was incubated as described above, and insoluble species were resuspended in cell medium at a final concentration of 5 or 10 μM and added to the cells. Cells were then grown for 72 hrs at 37°C. Cell viability was assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. MTT reagent, dissolved in DMEM without phenol red (Sigma-Aldrich), was added to the cells (100 μl/well) to a final concentration of 0.5 mg/ml. After 4 hrs at 37°C, the culture medium was removed and the resulting formazan salts were dissolved by the addition of isopropanol containing 0.1 N HCl (100 μl/well). Absorbance values of blue formazan were determined at 570 nm using an automatic plate reader (Microbeta Wallac 1420, Perkin Elmer). Cell survival was expressed as the percentage of viable cells in the presence of the protein under test, with respect to control cells grown in the absence of the protein. The occurrence of plasma membrane damage was determined by measuring the release of lactate dehydrogenase (LDH) in the culture medium [19].
Fluorescence studies
Immediately prior to be tested, proteins were dissolved or dialysed in 0.1 M sodium carbonate, pH 9.0, to a final concentration of 1 mg/ml. Proteins (500 μg) were conjugated to FITC, following the manufacturer’s protocol (Sigma-Aldrich). A Sephadex G25 column, equilibrated in PBS, was used to separate the unreacted FITC from the conjugate. The same procedure was used to label proteins with rhodamine. Fluorescent fibrillar aggregates of [1–93]ApoA-I were obtained by incubating the FITC-labelled polypeptide as described for the unlabelled polypeptide. Insoluble aggregates were resuspended in cell medium and tested as described below. Cells were seeded on glass cover slips in 24-well plates and grown to semi-confluency. Cells were incubated for the indicated times in complete medium with fluorescent proteins or compounds at the following concentrations: [1–93]ApoA-I (3 μM), ApoA-I (1 μM), dextran (5 mg/ml), Tf (0.5 mg/ml), insulin (0.1 mg/ml). Lysosomes were labelled by adding LysoTracker Red (1:500) to living cells at 37°C. After 40 min., cells were treated with 1 μg/ml Hoechst 33342 for 10 min. at 37°C and washed with PBS. When required, surface bound proteins were stripped with 1 M Hepes, pH 7.5, containing 0.5 M NaCl (Hepes/NaCl) for 5 min. Cells were then fixed for 10 min. at RT with 4% paraformaldehyde in PBS.
To inhibit clathrin-dependent endocytosis, cells were pre-incubated with either 100 μM monodansylcadaverine (MDC) for 30 min. or with 300 μM sucrose for 15 min. As a control, cells were incubated under the same conditions but in the absence of the inhibitors. Cells were then incubated for 6 hrs with the fluorescent protein under test. For immunofluorescence analyses, cells were permeabilized with 0.5% Triton X-100 in PBS (5 min.). Cells were then incubated for 30 min. with 3% goat serum in PBS to saturate non-specific binding sites. Afterwards, cells were incubated overnight at 4°C with anti-ABCA1 antibodies (1:200), or anti-β-catenin antibody (1:200), and then rinsed with 0.1% Triton X-100 in PBS. Finally, cells were incubated 1 hr in the darkness with fluorescent goat anti-rabbit or antimouse IgG (1:500). Slides were washed with 0.1% Triton X-100 in PBS and then with PBS, and mounted in 50% glycerol in PBS. Samples were examined using a Leica 6000 UV microscope and a Leica TCS SP5 confocal microscope, equipped with a Leica application suite software (Leica Microsystems GmbH, Wetzlar, Germany). All images were taken under identical conditions.
Protein degradation analyses
To inhibit proteasome activity, cells were pre-treated for 4 hrs at 37°C with 2.5 μM Z-Leu-Leu-Leu-al (MG132), or with 10 μM N-Ac-Leu-Leu-norleucinal (ALLN). Cells were then incubated with the fluorescent protein for the indicated times. Intralysosomal catabolism was inhibited by treating cells with 20 mM ammonium chloride or 100 μM chloroquine. As a control, cells were incubated under the same conditions but in the absence of the inhibitors.
Results
[1–93]ApoA-I binding to cardiomyoblasts
The ability of [1–93]ApoA-I to recognize specific binding sites on cell surface was investigated by performing binding assays of 125I-labelled [1–93]ApoA-I to rat cardiomyoblasts (H9c2) and human hepatocytes (HepG2). These cell lines are of interest, as in ApoA-I associated amyloidoses the heart is a natural target for aggregate deposition in vivo, whereas the liver is the major source of ApoA-I. The binding curves shown in Fig. 1A and B were obtained incubating cardiomyoblasts or hepatocytes, respectively, for 2 hrs at 4°C with increasing concentrations of the iodinated polypeptide, in the absence (total binding) or presence (non-specific binding) of a 40-fold molar excess of the unlabelled polypeptide. Specific binding was calculated by subtracting the values relative to non-specific binding from total binding. The data points represent the average of three independent experiments carried out in triplicate determinations. The linearization of the binding data (specific binding) was performed according to the Scatchard equation to obtain the linear plots shown in the insets of Fig. 1. The apparent affinity constants (Kd) were determined and found to be 5.90 ± 0.70 × 10−7 M and 1.78 ± 0.26 × 10−7 M for H9c2 and HepG2, respectively. These results indicate that the fibrillogenic fragment is able to bind with high affinity to specific sites on cell surface of both cell lines.
Fig 1.
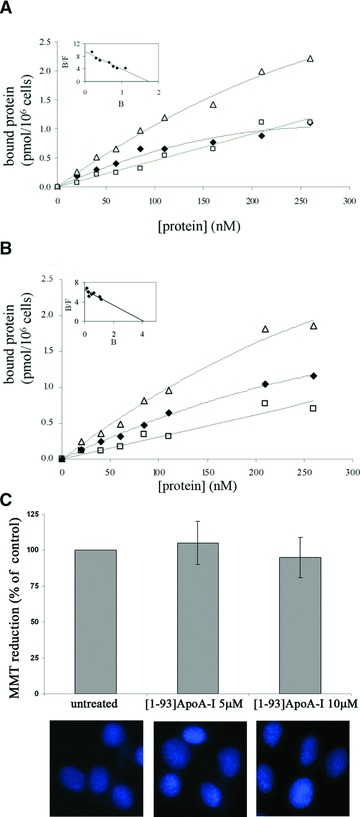
Binding of [1–93]ApoA-I to cultured cells and its effects on cell viability. Binding curves were obtained incubating H9c2 cells (A) or HepG2 cells (B) for 2 hrs at 4°C with increasing concentrations of iodinated [1–93]ApoA-I, in the absence (Δ, total binding) or in the presence (□, non-specific binding) of a 40-fold molar excess of the unlabelled polypeptide. Specific binding values (♦) were obtained by subtracting the values relative to non-specific binding from those of total binding. The linearization of specific binding curves was obtained according to the Scatchard equation (insets of A and B). B: pmoles of protein bound to 1 × 106 cells; F: concentration of the unbound protein. (C) MTT reduction assay and Hoechst staining of H9c2 cells untreated or treated with 5 μM or 10 μM [1–93]ApoA-I. Error bars indicate standard deviations obtained from four independent experiments. All images have been acquired at the same magnification.
To test the effects of the fibrillogenic polypeptide on cell viability, cardiomyoblasts were incubated for 72 hrs in the presence of 5 or 10 μM freshly prepared [1–93]ApoA-I. MTT reduction assays were performed to test metabolically active cells. No inhibition of cell viability was observed in treated cells with respect to untreated cells (Fig. 1C). Furthermore, we tested membrane destabilization of treated cells by measuring LDH release into the culture medium. No significant LDH release was observed upon treatment with [1–93]ApoA-I (data not shown). Finally, the absence of apoptotic nuclei in treated cells (Fig. 1C) confirmed that the fibrillogenic polypeptide, at least in our experimental conditions, is not cytotoxic for cardiomyoblasts.
Endocytosis of [1–93]ApoA-I in cardiomyoblasts
To test whether the fibrillogenic polypeptide of ApoA-I undergoes endocytosis upon interaction with cardiomyoblasts plasma membrane, we labelled [1–93]ApoA-I, as well as full-length ApoA-I, with FITC. H9c2 cells were incubated for different lengths of time either with the polypeptide or with the full-length protein. Cells were then treated with high salt buffer (Hepes/NaCl) to remove proteins specifically bound to the extracellular side of plasma membrane. Following membrane permeabilization with Triton X-100, anti-β-catenin antibody was added to the cells to label the membrane compartment. As shown in Fig. 2A, after 2 hrs incubation the polypeptide (green) mostly co-localizes with β-catenin (red), whereas after 6 hrs the polypeptide fluorescent signal was found to be mostly intracellular (Fig. 2B). Hence, the polypeptide binds to the membrane first and then is internalized in target cells. Similar results were obtained with labelled ApoA-I (Fig. 2C and D). Immunostaining was specific, as no fluorescent signals were observed in the absence of primary antibody (data not shown).
Fig 2.
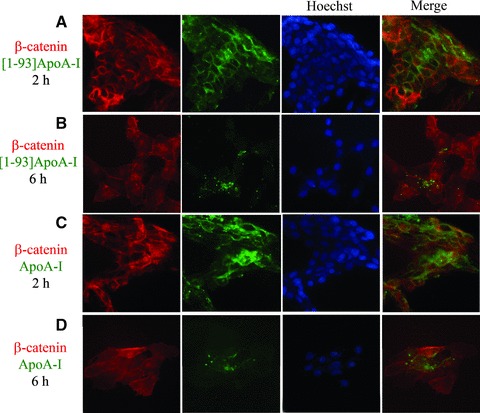
Endocytosis of [1–93]ApoA-I and full-length ApoA-I in H9c2 cells. Cells were grown on cover slips, incubated 2 hrs (A) or 6 hrs (B) with 3 μM FITC-[1–93]ApoA-I (green) and immunofluorescently stained for β-catenin (red). (C) and (D), cells incubated 2 or 6 hrs, respectively, with 1 μM FITC-ApoA-I (green) and immunostained for β-catenin (red). Nuclei were stained with Hoechst (blue). Cells were analysed by epifluorescence microscopy.
It is known that the plasma membrane is the main platform where lipidation of ApoA-I occurs [4], mediated by the ATP-binding cassette transporter A1 (ABCA1). This allows cellular free cholesterol and phospholipids to be transferred to ApoA-I leading to the biogenesis of nascent HDL. It has been demonstrated that ApoA-I is internalized in an ABCA1-dependent manner, because no internalization was observed in cells ABCA1–/–[20]. To test whether ABCA1 is expressed in cardiomyoblasts, cell lysates were analysed by Western blotting with anti-ABCA1 antibodies. HepG2 lysates were used as a positive control, as high levels of this transporter have been found in the liver. As shown in Fig. 3A, an immuno-positive species, with an apparent molecular mass corresponding to that expected for ABCA1 (about 210 kDa), was present in cell extracts prepared from H9c2 (lane 2) and HepG2 (lane 1) cells. These results were confirmed by immuno-fluorescence analyses of H9c2 and HepG2 cells with anti-ABCA1 antibodies, which revealed immuno-positive signals in both cell lines (Fig. 3B and C). We observed that in H9c2 cells ABCA1 is localized both on the plasma membrane and in intracellular compartments, whereas in HepG2 cells it is mainly located on the plasma membrane. Although data reported in the literature indicate that the transporter is mostly localized on cell plasma membrane [21, 22], consistently with its role in cellular lipid efflux, ABCA1 has been also observed in intracellular compartments [5, 23, 24].
Fig 3.
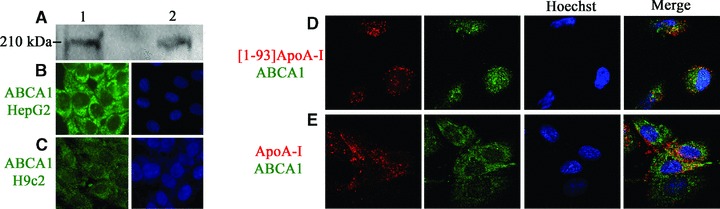
ABCA1 expression and co-localization with [1–93]ApoA-I and ApoA-I. (A) Western blot analysis with anti-ABCA1 antibodies of cell lysates prepared from HepG2 cells (25 μg total proteins, lane 1) and from H9c2 cells (50 μg, lane 2). Immunostaining for ABCA1 (green) of HepG2 cells (B) and H9c2 cells (C). Nuclei were stained with Hoechst (blue). (D) and (E), co-localization of [1–93]ApoA-I and ApoA-I with ABCA1. H9c2 cells were incubated for 2 hrs either with 3 μM rhodamine-[1–93]ApoA-I (D), or with 1 μM rhodamine-ApoA-I (E), and immunostained for ABCA1 (green). Nuclei were stained with Hoechst (blue). Cells were observed by confocal microscopy.
Furthermore, to test whether the fibrillogenic polypeptide co-localizes with ABCA1, we incubated H9c2 cells with rhodamine-[1–93]ApoA-I for 2 hrs at 37°C. Cells were then fixed and incubated with anti-ABCA1 antibodies to label the transporter. We found little co-localization between [1–93]ApoA-I (red) and ABCA1 (green). Representative images are shown in Fig. 3D. Similar results were obtained with rhodamine-ApoA-I (Fig. 3E), in agreement with recent reports showing that the majority of cell-associated ApoA-I does not co-localize with ABCA1 [25]. Immunostaining was specific as no signals were detected in the absence of primary antibody (data not shown).
Internalization pathway of [1–93]ApoA-I
To learn about the mechanism of [1–93]ApoA-I uptake in cardiomyoblasts, we analysed different routes of endocytosis. First, the involvement of chlathrin-coated pits was evaluated using Rab5 as a marker, as this protein regulates vesicular transport from the plasma membrane to the endosomes. We transiently expressed Rab5 fused to the RFP in H9c2 cells. Twenty-four hours after transfection, cells were incubated with the FITC protein under test for 6 hrs at 37°C, to allow internalization. As indicated in Fig. 4A, a significant, albeit partial, co-localization of internalized [1–93]ApoA-I (green) with RFP-Rab5 (red) was observed, indicating that a fraction of the internalized polypeptide is associated to early endosomes. Similar results were obtained when ApoA-I was tested (Fig. 4B), in line with recent findings [4, 5]. Additional experiments were performed with labelled transferrin (FITC-Tf), as a marker of the endocytic pathway. For both proteins we confirmed the results obtained with Rab5, because after 6 hrs incubation partial co-localization with FITC-Tf was observed (data not shown). To further confirm that both [1–93]ApoA-I and ApoA-I are internalized in H9c2 cells by chlatrin-mediated endocytosis, we used specific inhibitors of this internalization pathway, such as MDC and sucrose. Upon pre-incubation of H9c2 cells with either MDC or sucrose, we observed that the amount of internalized polypeptide, and that of the full-length protein, appeared to be reduced (data not shown). Hence, endocytosis of both proteins is slowed down, although not fully blocked, by inhibitors of chlatrin-mediated endocytosis. This indicates either that in our experimental conditions the endocytic pathway still functions, albeit less efficiently, or that endocytosis of [1–93]ApoA-I and ApoA-I does not occur solely via chlathrin-coated pits.
Fig 4.
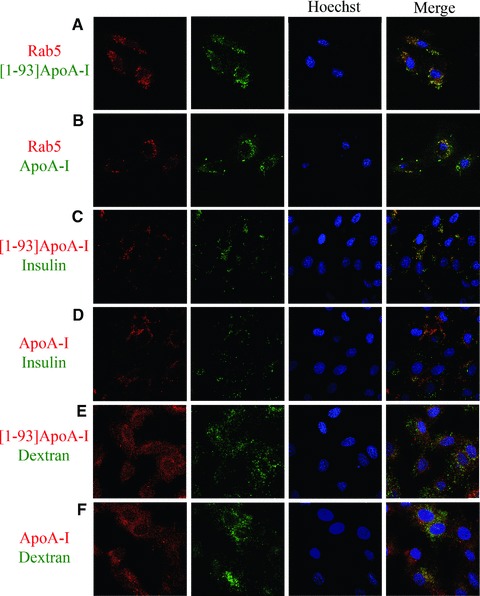
Analysis of the route of [1–93]ApoA-I and ApoA-I endocytosis in H9c2 cells by confocal microscopy. (A) and (B), clathrin-mediated endocytosis. H9c2 cells were transiently transfected with an expression vector for RFP-Rab5. After 24 hrs, cells were incubated 6 hrs at 37°C either with 3 μM FITC-[1–93]ApoA-I (A) or with 1 μM FITC-ApoA-I (B). (C) and (D), lipid rafts-mediated internalization. Cells were incubated 4 hrs at 37°C either with 3 μM rhodamine-[1–93]ApoA-I (C), or with 1 μM rhodamine-ApoA-I (D), in the presence of FITC insulin (0.1 mg/ml). (E) and (F), macropinocytosis. Cells were incubated 4 hrs at 37°C either with 3 μM rhodamine-[1–93]ApoA-I (E), or with 1 μM rhodamine-ApoA-I (F), in the presence of FITC dextran (5 mg/ml). Nuclei were stained with Hoechst (blue).
Experiments were then performed to test internalization by lipid rafts. To do so, FITC insulin was used as a marker. H9c2 cells were incubated with rhodamine-[1–93]ApoA-I in the presence of FITC insulin for 4 hrs at 37°C. As shown in Fig. 4C, strong signals of co-localization of [1–93]ApoA-I (red) with insulin (green) were detected, indicating that [1–93]ApoA-I uptake occurs also by lipid rafts. On the other hand, when the same experiment was performed with rhodamine-ApoA-I, little co-localization was observed with FITC insulin (Fig. 4D).
As it has been demonstrated that ApoA-I is also internalized by macropinocytosis [4], we tested this internalization route for [1–93]ApoA-I. H9c2 cells were incubated with rhodamine-[1–93]ApoA-I in the presence of FITC dextran, a macropinocytosis marker, for 4 hrs at 37°C. As shown in Fig. 4E, little co-localization was observed between [1–93]ApoA-I (red) and dextran (green). On the contrary, clear signals of co-localization were detected for ApoA-I (Fig. 4F).
According to recent reports, ApoA-I, once internalized, is recycled back to the cell surface [3–5]. To investigate the retroendocytosis pathway, Rab4 was used as a marker, as it is located in vesicles directing protein recycling from early endosomes to the plasma membrane. We transiently transfected H9c2 cardiomyoblasts with a vector encoding Rab4 fused to the GFP. Twenty-four hours after transfection, cells were incubated with rhodamine-[1–93]ApoA-I, or ApoA-I, for 6 hrs at 37°C. As shown in Fig. 5A, [1–93]ApoA-I (red) does not co-localize with Rab4+ endosomal compartments (green), whereas significant signals of co-localization were observed for ApoA-I (Fig. 5B). Taken together, these results clearly indicate that the fibrillogenic polypeptide, once internalized in cardiomyoblasts, is not recycled to the cell membrane, whereas ApoA-I is shuttled back to the plasma membrane to be resecreted, as described for other cell lines [3–5].
Fig 5.
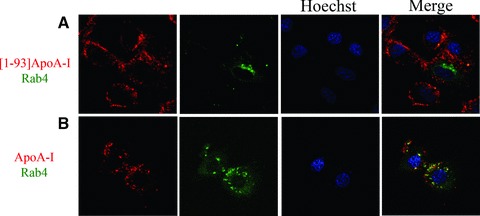
Co-localization of [1–93]ApoA-I and ApoA-I with Rab4. H9c2 cells were transiently transfected with an expression vector for GFP-Rab4. After 24 hrs, cells were incubated 6 hrs at 37°C either with 3 μM rhodamine-[1–93]ApoA-I (A) or with 1 μM rhodamine-ApoA-I (B). Nuclei were stained with Hoechst (blue). Cells were observed by confocal microscopy.
Intracellular fate of [1–93]ApoA-I
Next, we analysed the fate of the internalized polypeptide in cardiomyoblasts. After a prolonged exposure to FITC-[1–93]ApoA-I (24 hrs), the complete disappearance of intracellular fluorescent signals associated to the polypeptide was observed (Fig. 6A and C), suggestive of polypeptide massive degradation. To investigate the degradation pathway, we used specific inhibitors of proteasomal and lysosomal activities. When cells were pre-incubated with the proteasome inhibitor MG132, we observed the persistence of [1–93]ApoA-I intracellular fluorescence at 24 hrs (Fig. 6B), indicative of proteasome involvement in [1–93]ApoA-I degradation. To test whether [1–93]ApoA-I is targeted to lysosomes, we incubated cells with FITC-[1–93]ApoA-I in the presence of ammonium chloride, an inhibitor of intralysosomal catabolism. Following incubation, lysosomes were labelled with LysoTracker red. As shown in Fig. 6D, a strong fluorescence signal associated to the polypeptide (green) was found to co-localize with lysosomes (red), suggesting that lysosomes play a role in [1–93]ApoA-I catabolism. Different results were obtained instead with full-length ApoA-I, as even at 24 hrs incubation with FITC-ApoA-I, the persistency of protein fluorescence within the cells was observed (Fig. 6E and G). This is in agreement with recent reports indicating that in different cell types ApoA-I is not significantly degraded [4]. Moreover, as shown in Fig. 6G, ApoA-I was found to co-localize with lysosomes, in line with reports indicating that lysosomes are an intracellular station of ApoA-I [3, 4]. In the presence of either MG132 or ammonium chloride, fluorescence signals associated to ApoA-I did not significantly increase with respect to cells untreated with the inhibitors, as shown in Fig. 6F and H, respectively. Furthermore, by using different inhibitors, such as ALLN for the proteasome, and chloroquine for the lysosomes, we confirmed the data reported above (data not shown). Taken together, our results indicate that the inhibition of lysosomal or proteasomal activity does not significantly alter the amount of intracellular ApoA-I, whereas both pathways seem to be involved in the degradation of the fibrillogenic polypeptide.
Fig 6.
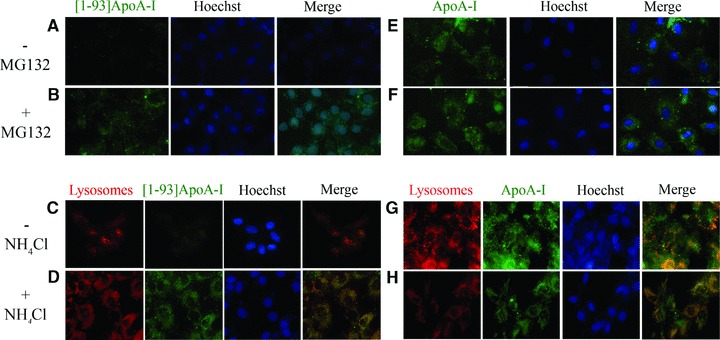
Analysis of the degradation pathway of [1–93]ApoA-I and ApoA-I in H9c2 cells by epifluorescence microscopy. (A)–(D) [1–93]ApoA-I degradation. Cells were incubated 24 hrs at 37°C with 3 μM FITC-[1–93]ApoA-I in the absence (A, C) or in the presence of MG132 (2.5 μM) (B), or ammonium chloride (100 μM) (D). (E)–(H), ApoA-I degradation. Cells were incubated for 24 hrs at 37°C with 1 μM FITC-ApoA-I, in the absence (E and G) or in the presence of MG132 (F), or ammonium chloride (H). Lysosomes were stained with LysoTracker red. Nuclei were stained with Hoechst (blue).
Analysis of the uptake of [1–93]ApoA-I fibrils in cardiomyoblasts and the effects on cell viability
As it is conceivable that the accumulation of the fibrillogenic polypeptide in the extracellular space leads to fibrils deposition, [1–93]ApoA-I fibrils were obtained by incubating the polypeptide for 2 weeks at pH 6.4 in the presence of the co-solvent TFE. AFM analysis of the incubated sample showed the presence of fibrils with height of 2.4 ± 0.1 nm and length between 0.4 and 1.5 μm (Fig. 7A). Fibrils coexist with prefibrillar aggregates, including annular protofibrils, which form a network in the image background; spheroidal aggregates of variable size (height between 3 and 15 nm) are also present.
Fig 7.
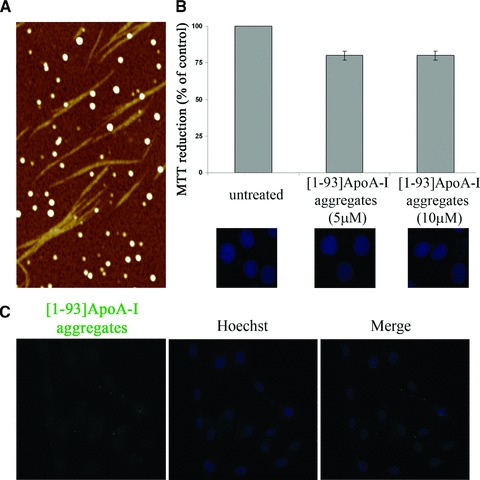
Analysis of [1–93]ApoA-I fibrils. (A) Tapping mode AFM image (height data) of aggregated [1–93]ApoA-I. Upon incubation in the aggregating conditions, the whole sample was observed. Fibrils coexist with prefibrillar aggregates; spheroidal aggregates are also found. Scan size 3.0 μm, Z range 10 nm. (B) Effects of [1–93]ApoA-I fibrils on cell viability. MTT reduction assay and Hoechst staining of H9c2 cells, untreated or treated with 5 μM or 10 μM [1–93]ApoA-I fibrils, are shown. Error bars indicate standard deviations obtained from three independent experiments. Nuclei images have been acquired at the same magnification. (C) Analysis of internalization of [1–93]ApoA-I fibrils in H9c2 cells. Cells were incubated for 6 hrs with 3 μM FITC-labelled [1–93]ApoA-I fibrils and analysed by epifluorescence microscopy. Nuclei were stained with Hoechst (blue).
To test the effects of fibrils on cell viability, cardiomyoblasts were incubated for 72 hrs in the presence of 5 or 10 μM aggregated [1–93]ApoA-I (insoluble species). No inhibition of cell viability was observed by MTT assays in treated cells with respect to untreated cells (Fig. 7B). This was confirmed by the absence of apoptotic nuclei in treated cells (Fig. 7B).
To verify whether the fibrillar material is able to enter the cells, we produced fluorescent fibrils by incubating FITC-labelled [1–93]ApoA-I under the conditions previously described. H9c2 cells were incubated for 6 hrs with fluorescent fibrils (insoluble species) and then treated with Hepes/NaCl buffer to remove polypeptide molecules specifically bound to the extracellular side of the plasma membrane. No fluorescent signals associated to [1–93]ApoA-I fibrils were observed by epifluorescence microscopy analysis, demonstrating that no significant internalization of fibrils occurs in cardiomyoblasts (Fig. 7C).
Discussion
ApoA-I represents the intriguing case of a protein which, in its native form, plays a key role in cholesterol homeostasis, as it acts as an extracellular acceptor of lipids. Nevertheless, by specific mutations ApoA-I is converted into the precursor of natively unfolded pathogenic fragments associated with familial systemic amyloidoses [1]. N-terminal fragments of ApoA-I, 90–100 residue long, accumulate in tissues and organs of patients carrying one of the 16 amyloidogenic mutations identified so far in ApoA-I gene [1, 10, 11]. The molecular mechanism responsible for ApoA-I associated amyloid diseases remains largely unknown. However, recent findings allowed us to raise the hypothesis that the mutations located in ApoA-I N-terminal region are amyloidogenic as they favour the proteolytic cleavage responsible for the release of the fibrillogenic polypeptide [26].
As the heart is one of the targets of ApoA-I amyloid aggregate deposition in patients affected by the disease, rat cardiomyoblasts (H9c2 cell line) were selected for in vitro analyses. In this study, we demonstrated that the fibrillogenic polypeptide is able to specifically bind to cardiomyoblasts, as well as to human hepatocytes. The apparent affinity constants (Kd= 5.90 ± 0.70 × 10−7 M and 1.78 ± 0.26 × 10−7 M for H9c2 and HepG2, respectively) were found to be comparable to those previously reported for lipid-free ApoA-I binding to HepG2 cells (Kd= 0.84 × 10−7 M) [27], and to aortic endothelial cells (Kd= 0.8 × 10−7 M) [28]. These results are also consistent with the finding that region 62–77 of ApoA-I is a membrane binding domain of lipid-free ApoA-I, because the corresponding synthetic peptide binds with high affinity (Kd∼ 10−7 M) to HepG2 cells [29]. Our data are also reinforced by the finding that region 1–43 is involved in ApoA-I lipid binding [14].
As ABCA1 transporter plays a central role in ApoA-I membrane binding and lipidation [3–5], we analysed the presence of this transporter in H9c2 cells. Immunofluorescence analyses demonstrated that ABCA1 is expressed in cardiomyoblasts, where it was found to partially co-localize with both full-length ApoA-I and its fibrillogenic polypeptide, supporting the hypothesis that ApoA-I and [1–93]ApoA-I share common determinants for membrane association. Low co-localization between ApoA-I and ABCA1 is in agreement with the general view that only a small fraction of membrane bound ApoA-I appears to co-localize with ABCA1 [25]. To explain these observations, a model was recently proposed [4, 30], in which the interaction of a small fraction of lipid-free ApoA-I to ABCA1 is sufficient to activate ABCA1 lipid translocase activity, which in turn promotes the formation of specialized lipid domains, acting as high affinity binding sites for ApoA-I [30]. Therefore, although ApoA-I binding occurs in an ABCA1-dependent manner, most of ApoA-I molecules bind to lipids rather than to ABCA1 itself [4].
We demonstrated that ApoA-I fibrillogenic polypeptide is able to enter cardiac cells, as shown by fluorescence analyses. Our results support roles for both chlathrin- and lipid raft-mediated pathways in [1–93]ApoA-I internalization, with very little contribution of macropinocytosis. Upon internalization, no involvement of the retroendocytosis pathway was observed; rather, the fibrillogenic fragment is targeted to proteasomal and lysosomal stations for degradation, as at 24 hrs no intracellular fluorescent signals were detected. This is not surprising, considering that the largely unfolded structure of the fibrillogenic polypeptide is responsible for its susceptibility to proteolytic cleavages, as previously demonstrated by experiments of limited proteolysis [13]. Accordingly, to produce the recombinant form of the polypeptide in a prokaryotic expression system, it has been necessary to transiently fuse the polypeptide to a stable bacterial protein to avoid intracellular degradation [13]. The rapid degradation of the polypeptide is also in agreement with the absence of cytotoxic effects on cardiomyoblasts, at least in the experimental conditions tested.
Furthermore, in parallel experiments we provided evidence that full-length ApoA-I is internalized in cardiomyoblasts via chlatrin-dependent endocytosis and macropinocytosis as predominant internalization routes, with very low signals of co-localization with lipid rafts. To our knowledge, this is the first evidence of ApoA-I internalization in cardiac cells. Moreover, although ApoA-I is known to interact with plasma membrane lipid rafts to control cholesterol export [31], this is the first time that lipid rafts involvement in ApoA-I internalization has been analysed. Once internalized, ApoA-I associates to Rab4-labelled endosomal compartment, a station involved in ApoA-I recycling to the cell membrane in other cell lines [3–5, 23]. At 24 hrs, ApoA-I associated fluorescent signals are still observed in cardiomyoblasts and co-localize with lysosomes. It is noteworthy that lysosomes are best known for their role in degradation, although recent studies have shown that they may also fuse to the plasma membrane and release their content to the extracellular medium [32]. The question of the physiological role of ApoA-I in lysosomes remains controversial. In fact, some authors demonstrated that ApoA-I reaches lysosomes to be degraded [3, 4], but studies support the idea that ABCA1-bound ApoA-I traffics to late endosomal vesicles and/or to lysosomes. Being these stations intracellular cholesterol reservoirs, here nascent lipoprotein particles are formed and then secreted from the cell [33–35].
In conclusion, the data reported here reveal that ApoA-I fibrillogenic fragment, the main constituent of amyloid fibrils, binds to target cells, is internalized and rapidly degraded. The internalization routes, intracellular pathways and degradation mechanisms of full-length ApoA-I and its fibrillogenic polypeptide are not fully coincident, as schematically depicted in Scheme 1.
Scheme 1.
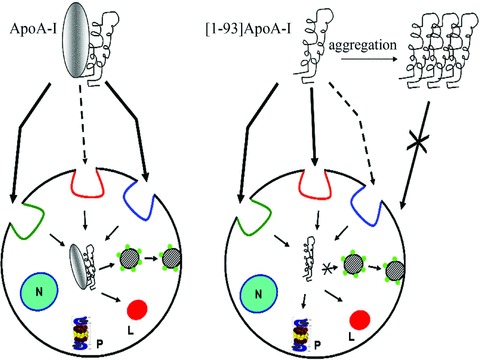
Schematic representation of internalization routes and intracellular fates of [1–93]ApoA-I and ApoA-I in H9c2 cells. Lipid rafts are coloured in red; chlatrin-coated pits in green; macropinocytosis in blue. Rab4 vesicles are represented by green circles. N: nucleus; P: proteasome; L: lysosomes.
We surmise that the intracellular fate of the polypeptide may be relevant in the development of the pathology. The continuous accumulation of the natively unfolded polypeptide in the cardiac tissue leads to a progressive, massive occupancy of the extracellular space by amyloid deposits, as observed in pathological hearts, from which the natural fibrillogenic polypeptide can be isolated [10]. During the fibrillogenic process, a dynamic equilibrium between monomeric species and aggregated states has been proposed [36]. Here we provided evidence that, besides aggregation in the extracellular space, an alternative fate is available to the polypeptide, i.e. the interaction with target cells, internalization and subsequent degradation. This would subtract unaggregated molecules of [1–93]ApoA-I from the equilibrium directing the polypeptide towards a non-pathological route. Relevant results were obtained when the analyses were extended to the polypeptide in the fibrillar state. Typical amyloid fibrils were obtained in vitro, as demonstrated by AFM analysis. Evidence was provided that, unlike the unaggregated polypeptide, fibrils have no access to the intracellular compartment. Thus, the intracellular degradation pathway is precluded to fibrillar aggregates. Hence, the hypothesis can be raised that internalization and subsequent degradation of the unaggregated fibrillogenic polypeptide represent a protective mechanism against fibrillogenesis, able to balance [1–93]ApoA-I progressive aggregation and to slow down the fibrillogenic process. This phenomenon may be relevant in the slow progression and late onset of ApoA-I-associated amyloid pathology.
Acknowledgments
We thank Prof. Chiara Campanella for helpful discussions, Dr. Marino Zerial (Max-Planck-Institute, Germany) for generously providing fluorescent Rab proteins constructs and Dr. Amanda Penco for help in AFM measurements. Confocal microscopy investigations were carried out at the CISME (Interdepartmental Centre of Electronic Microscopy) of the University of Naples Federico II. This work was supported by MIUR, Ministero dell’Università e della Ricerca Scientifica, Italy (PRIN 2007, Project N. 2007XY59ZJ_003; PRIN 2008, Project N. 20083ERXWS_002).
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Obici L, Franceschini G, Calabresi L, et al. Structure, function and amyloidogenic propensity of apolipoprotein A-I. Amyloid. 2006;13:1–15. doi: 10.1080/13506120600960288. [DOI] [PubMed] [Google Scholar]
- 2.Gordon DJ, Rifkind BM. High-density lipoprotein–the clinical implications of recent studies. N Engl J Med. 1989;321:1311–6. doi: 10.1056/NEJM198911093211907. [DOI] [PubMed] [Google Scholar]
- 3.Cavelier C, Lorenzi I, Rohrer L, et al. Lipid efflux by the ATP-binding cassette transporters ABCA1 and ABCG1. Biochim Biophys Acta. 2006;1761:655–66. doi: 10.1016/j.bbalip.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 4.Denis M, Landry YD, Zha X. ATP-binding cassette A1-mediated lipidation of apolipoprotein A-I occurs at the plasma membrane and not in the endocytic compartments. J Biol Chem. 2008;283:16178–86. doi: 10.1074/jbc.M709597200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Azuma Y, Takada M, Shin HW, et al. Retroendocytosis pathway of ABCA1/apoA-I contributes to HDL formation. Genes to Cells. 2009;14:191–204. doi: 10.1111/j.1365-2443.2008.01261.x. [DOI] [PubMed] [Google Scholar]
- 6.Mukherjee S, Zha X, Tabas I, et al. Cholesterol distribution in living cells: fluorescence imaging using dehydroergosterol as a fluorescent cholesterol analog. Biophys J. 1998;75:1915–25. doi: 10.1016/S0006-3495(98)77632-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chroni A, Liu T, Fitzgerald ML, et al. Cross-linking and lipid efflux properties of apoA-I mutants suggest direct association between apoA-I helices and ABCA1. Biochemistry. 2004;43:2126–39. doi: 10.1021/bi035813p. [DOI] [PubMed] [Google Scholar]
- 8.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–66. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 9.Eckman CB, Eckman EA. An update on the amyloid hypothesis. Neurol Clin. 2007;25:669–82. doi: 10.1016/j.ncl.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Obici L, Bellotti V, Mangione P, et al. The new apolipoprotein A-I variant Leu174→Ser causes chardiac amyloidosis, and the fibrils are constituted by the 93-residue N-terminal polypeptide. Am J Pathol. 1999;155:695–702. doi: 10.1016/S0002-9440(10)65167-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eriksson M, Schönland S, Yumlu S, et al. Hereditary apolipoprotein AI-associated amyloidosis in surgical pathology specimens: identification of three novel mutations in the APOA1 gene. J Mol Diagn. 2009;11:257–62. doi: 10.2353/jmoldx.2009.080161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andreola A, Bellotti V, Giorgetti S, et al. Conformational switching and fibrillogenesis in the amyloidogenic fragment of apolipoprotein A-I. J Biol Chem. 2003;278:2444–51. doi: 10.1074/jbc.M204801200. [DOI] [PubMed] [Google Scholar]
- 13.Di Gaetano S, Guglielmi F, Arciello A, et al. Recombinant amyloidogenic domain of ApoA-I: analysis of its fibrillogenic potential. Biochem Biophys Res Commun. 2006;351:223–8. doi: 10.1016/j.bbrc.2006.10.026. [DOI] [PubMed] [Google Scholar]
- 14.Tanaka M, Dhanasekaran P, Nguyen D, et al. Contributions of the N- and C-terminal helical segments to the lipid-free structure and lipid interaction of apolipoprotein A-I. Biochemistry. 2006;45:10351–8. doi: 10.1021/bi060726t. [DOI] [PubMed] [Google Scholar]
- 15.Monti DM, Guglielmi F, Monti M, et al. Effects of a lipid environment on the fibrillogenic pathway of the N-terminal polypeptide of human apolipoprotein A-I, responsible for in vivo amyloid fibril formation. Eur Biophys J. 2010;39:1289–99. doi: 10.1007/s00249-010-0582-2. [DOI] [PubMed] [Google Scholar]
- 16.Campioni S, Mannini B, Zampagni M, et al. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat Chem Biol. 2010;6:140–7. doi: 10.1038/nchembio.283. [DOI] [PubMed] [Google Scholar]
- 17.Sönnichsen B, De Renzis S, Nielsen E, et al. Distinct membrane domains on endosomes in the recycling pathway visualized by multicolor imaging of Rab4, Rab5, and Rab11. J Cell Biol. 2000;149:901–14. doi: 10.1083/jcb.149.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rink J, Ghigo E, Kalaidzidis Y, et al. Rab conversion as a mechanism of progression from early to late endosomes. Cell. 2005;122:735–49. doi: 10.1016/j.cell.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 19.Gharibyan AL, Zamotin V, Yanamandra K, et al. Lysozyme amyloid oligomers and fibrils induce cellular death via different apoptotic/necrotic pathways. J Mol Biol. 2007;365:1337–49. doi: 10.1016/j.jmb.2006.10.101. [DOI] [PubMed] [Google Scholar]
- 20.Zha X, Genest J, Jr, McPherson R. Endocytosis is enhanced in Tangier fibroblasts: possible role of ATP-binding cassette protein A1 in endosomal vesicular transport. J Biol Chem. 2001;276:39476–83. doi: 10.1074/jbc.M105067200. [DOI] [PubMed] [Google Scholar]
- 21.Fitzgerald ML, Mendez AJ, Moore KJ, et al. ATP-binding cassette transporter A1 contains an NH2-terminal signal anchor sequence that translocates the protein’s first hydrophilic domain to the exoplasmic space. J Biol Chem. 2001;276:15137–45. doi: 10.1074/jbc.m100474200. [DOI] [PubMed] [Google Scholar]
- 22.Orsó E, Broccardo C, Kaminski WE, et al. Transport of lipids from golgi to plasma membrane is defective in tangier disease patients and Abc1-deficient mice. Nat Genet. 2000;24:192–6. doi: 10.1038/72869. [DOI] [PubMed] [Google Scholar]
- 23.Neufeld EB, Stonik JA, Demosky SJ, Jr, et al. The ABCA1 transporter modulates late endocytic trafficking: insights from the correction of the genetic defect in Tangier disease. J Biol Chem. 2004;279:15571–8. doi: 10.1074/jbc.M314160200. [DOI] [PubMed] [Google Scholar]
- 24.Neufeld EB, Remaley AT, Demosky SJ, et al. Cellular localization and trafficking of the human ABCA1 transporter. J Biol Chem. 2001;276:27584–90. doi: 10.1074/jbc.M103264200. [DOI] [PubMed] [Google Scholar]
- 25.Vedhachalam C, Ghering AB, Davidson WS, et al. ABCA1-induced cell surface binding sites for ApoA-I. Arterioscler Thromb Vasc Biol. 2007;27:1603–9. doi: 10.1161/ATVBAHA.107.145789. [DOI] [PubMed] [Google Scholar]
- 26.Raimondi S, Guglielmi F, Giorgetti S, et al. Effects of the known pathogenic mutations on the aggregation pathway of the amyloidogenic peptide of Apolipoprotein A-I. J Mol Biol. 2011 doi: 10.1016/j.jmb.2011.01.044. 10.1016/j.jmb.2011.01.044. [DOI] [PubMed] [Google Scholar]
- 27.Barbaras R, Collet X, Chap H, et al. Specific binding of free apolipoprotein A-I to a high-affinity binding site on HepG2 cells: characterization of two high-density lipoprotein sites. Biochemistry. 1994;33:2335–40. doi: 10.1021/bi00174a047. [DOI] [PubMed] [Google Scholar]
- 28.Rohrer L, Cavelier C, Fuchs S, et al. Binding, internalization and transport of apolipoprotein A-I by vascular endothelial cells. Biochim Biophys Acta. 2006;1761:186–94. doi: 10.1016/j.bbalip.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 29.Georgeaud V, Garcia A, Cachot D, et al. Identification of an ApoA-I ligand domain that interacts with high-affinity binding sites on HepG2 cells. Biochem Biophys Res Commun. 2000;267:541–5. doi: 10.1006/bbrc.1999.1990. [DOI] [PubMed] [Google Scholar]
- 30.Vedhachalam C, Duong PT, Nickel M, et al. Mechanism of ATP-binding cassette transporter A1-mediated cellular lipid efflux to apolipoprotein A-I and formation of high density lipoprotein particles. J Biol Chem. 2007;282:25123–30. doi: 10.1074/jbc.M704590200. [DOI] [PubMed] [Google Scholar]
- 31.Gaus K, Kritharides L, Schmitz G, et al. Apolipoprotein A-1 interaction with plasma membrane lipid rafts controls cholesterol export from macrophages. FASEB J. 2004;18:574–6. doi: 10.1096/fj.03-0486fje. [DOI] [PubMed] [Google Scholar]
- 32.Luzio JP, Pryor PR, Bright NA. Lysosomes: fusion and function. Nat Rev Mol Cell Biol. 2007;8:622–32. doi: 10.1038/nrm2217. [DOI] [PubMed] [Google Scholar]
- 33.Oram JF. The ins and outs of ABCA. J Lipid Res. 2008;49:1150–1. doi: 10.1194/jlr.E800006-JLR200. [DOI] [PubMed] [Google Scholar]
- 34.Chen W, Sun Y, Welch C, et al. Preferential ATP-binding cassette transporter A1-mediated cholesterol efflux from late endosomes/lysosomes. J Biol Chem. 2001;276:43564–9. doi: 10.1074/jbc.M107938200. [DOI] [PubMed] [Google Scholar]
- 35.Chen W, Wang N, Tall AR. A PEST deletion mutant of ABCA1 shows impaired internalization and defective cholesterol efflux from late endosomes. J Biol Chem. 2005;280:29277–81. doi: 10.1074/jbc.M505566200. [DOI] [PubMed] [Google Scholar]
- 36.Carulla N, Caddy GL, Hall DR, et al. Molecular recycling within amyloid fibrils. Nature. 2005;436:554–8. doi: 10.1038/nature03986. [DOI] [PubMed] [Google Scholar]