

Abstract
miR-203 is a tumour suppressor microRNA (miRNA). We studied the methylation of hsa-miR-203 in 150 samples including acute myeloid leukaemia (AML), acute lymphoblastic leukaemia (ALL), chronic myeloid leukaemia (CML), chronic lymphocytic leukaemia (CLL) and non-Hodgkin’s lymphoma (NHL) by methylation-specific PCR, and miRNA expression by stem-loop RT-qPCR. hsa-miR-203 promoter was unmethylated in normal controls but homozygously methylated in two AML and four lymphoma cell lines, in which 5-Aza-2′-deoxycytidine treatment led to promoter demethylation and miR-203 re-expression. Restoration of miR-203 expression in lymphoma cells inhibited cellular proliferation and increased cell death, suggesting an inherent tumour suppressor activity. In primary samples, hsa-miR-203 methylation was absent in CML but detected in 5.0% ALL, 10.0% AML, 42.0% CLL and 38.8% of NHL (including six [60.0%] natural killer-cell, nine [40.9%] B-cell and four [23.5%] T cell NHL). Moreover, hsa-miR-203 methylation was associated with hypermethylation of hsa-miR-34a, -124a and -196b in NHL but not CLL. In CLL, hsa-miR-203 methylation was associated with a higher presenting Hb level (P = 0.033). The projected 10 year overall survival of the CLL patients was 58.2%, which was impacted by Rai stage and high-risk karyotypes but not hsa-miR-203 methylation. hsa-miR-203 was more frequently methylated in lymphoid than myeloid malignancies (P = 0.002). In conclusion, miR-203, a tumour suppressor gene, was hypermethylated in a tumour-specific manner with gene silencing. hsa-miR-203 was more frequently hypermethylated in lymphoid than myeloid malignancies. In NHL, hsa-miR-203 methylation was associated with concomitant methylation of other tumour suppressor miRNAs. The frequent hsa-miR-203 methylation in lymphoid malignancies suggested a pathogenetic role of hsa-miR-203 methylation.
Keywords: microRNA, tumour suppressor, hypermethylation, leukaemia, lymphoma
Introduction
DNA methylation involves the addition of a methyl group to the number 5 carbon of the cytosine ring in the CpG dinucleotide, by catalysing the conversion of cytosine into methylcytosine through DNA methyltransferase [1, 2]. Cancer cells are characterized by global DNA hypomethylation but gene-specific hypermethylation of promoter-associated CpG islands of tumour suppressor genes (TSGs), resulting in transcriptional repression, and hence serve as an alternative mechanism of gene inactivation. Based on a pathway-specific approach, multiple TSGs across pathways including cell cycle regulation, Janus kinase/signal transducers and activators of transcription (JAK/STAT) signalling, WNT signalling and death-associated protein (DAP) kinase-associated intrinsic tumour suppression have been shown to be inactivated by gene hypermethylation in leukaemia, lymphoma and multiple myeloma [3–9].
MicroRNA (miRNA) is a single-stranded, non-coding RNA molecule of 22–25 nucleotides, which leads to down-regulation of target protein expression [10]. miRNAs are involved in carcinogenesis. miRNAs can be either oncogenic (oncomir) when TSGs are targeted, or tumour suppressive (tumour suppressor miRNAs) when oncogenes are targeted [10, 11]. Little is known about the role of hypermethylation of tumour suppressor miRNAs in haematological cancers.
Recently, hypermethylation of hsa-miR-203 has been reported in chronic myeloid leukaemia (CML) and hepatocellular carcinoma, conferring proliferative advantage in tumour cells [12, 13]. Specifically, restoration of miR-203 expression targets and down-regulates the oncogenic breakpoint cluster region-abelson (BCR-ABL) fusion protein, thereby inhibits cellular proliferation in CML, demonstrating the tumour suppressor role of miR-203 [12].
In this study, we aimed to study the role of hsa-miR-203 methylation in a wide range of haematological malignancies including acute myeloid leukaemia (AML), CML, acute lymphoblastic leukaemia (ALL), chronic lymphocytic leukaemia (CLL) and non-Hodgkin’s lymphoma (NHL).
Materials and methods
Patient samples
Diagnostic bone marrow or tissue samples were obtained in 20 ALL, 20 AML, 11 Ph+ (expressing BCR-ABL) CML in chronic phase, 50 CLL and 49 NHL patients. Diagnosis of leukaemia and lymphoma were made according to the French-American-British Classification and WHO Classification of Tumours, respectively [14–17]. Of the 20 ALL patients, there were 11 males and 9 females patients with a median age of 35 years (range: 13–62). There were six common ALL, one early B precursor, ten precursor B ALL and three pre-T ALL. Of the 20 AML patients, there were 9 males and 11 females with a median age of 41.5 years (range: 20–72). The AML cases comprised three M1, fourteen M2, two M4 and one M5 FAB subtype. Of the 50 CLL patients, there were 31 (62%) patients with limited Rai stage (<stage II) and 19 (38%) with advanced Rai stage (≥ stage II) disease with a median age of 64 years (range: 37–91). Thirty-seven (74%) were male. The median presenting lymphocyte count was 17 × 109/l (range: 10–236 × 109/l). Of the 37 patients with cytogenetic data, 9 (18%) carried high-risk cytogenetic alterations [del(17p), N = 2; trisomy 12, N = 7] and 28 (56%) carried low/standard-risk cytogenetic alterations (del[13], N = 5; normal karyotype, N = 18; other karyotypic changes, N = 5). Projected 10 year overall survival (OS) of CLL patient was 58.2% for the whole group, and 65.9% in those with limited, and 47.5% in those with advanced Rai stage (P = 0.04). Projected 10 year OS of CLL patient with low/standard-risk and high-risk karyotypes were 80.6% and 13.9% (P = 0.001). Of the 49 patients with NHL, the median age was 59.6 years (range: 17–86 years). There were 17 (34.7%) patients with peripheral T cell lymphoma (two anaplastic large cell, 4 angio-immunoblastic T cell, 11 peripheral T cell, not otherwise specified), 10 (20.4%) with natural killer (NK)/T cell lymphoma, nasal type, 22 (44.9%) patients with B-cell lymphoma (9 follicular: grade 1 to 2, 9 nodal marginal zone, and two each for mantle cell and diffuse large B-cell lymphoma). The study has been approved by Institutional Review Board of Queen Mary Hospital with informed consent.
Cell lines and culture
CLL cell lines EHEB, MEC-1 and lymphoma cell lines SU-DHL-6, SU-DHL-16, GRANTA-519, JEKO-1 and MINO were purchased from Deutsche Sammlung von Mikroorganismen und Zellkulturen (Braunschweig, Germany). AML cell line SKNO-1 was purchased from Japanese Collection of Research Bioresources (Osaka, Japan). AML cell line HL-60 was kindly provided by Dr. R. Pang, Department of Medicine, HKU. CML cell lines K-562 and MEG-01 were kind gift from Dr. M. Yang, Department of Paediatrics, HKU. Cell lines were maintained in 90% RPMI 1640 + 10% FBS (HL-60, K-562, MEG-01, EHEB), 90% RPMI 1640 + 10% FBS + 10 ng/ml GM-CFS (SKNO-1), 90% IMDM + 10% FBS (MEC-1) and 85% RPMI 1640 + 15% FBS (SU-DHL-6, SU-DHL-16, GRANTA-519, JEKO-1 and MINO). Culture media were supplemented with 50 U/ml penicillin and 50 μg/ml streptomycin (Invitrogen, Carlsbad, CA, USA), and maintained in a humidified atmosphere of 5% CO2 at 37°C.
Methylation-specific polymerase chain reaction (MSP)
DNA was extracted from bone marrow samples of ALL, AML, CML and CLL at diagnosis, diagnostic tissues (either lymph node or nasal biopsy in nasal NK-cell lymphoma) in patients with NHL, and cell lines by standard method. MSP for aberrant gene promoter methylation was performed as previously described [18]. Treatment of DNA with bisulphite for conversion of unmethylated cytosine to uracil (but unaffecting methylated cytosine) was performed with a commercially available kit (EpiTect Bisulphite Kit, QIAGEN, Hilden, Germany). Methylated MSP (M-MSP) primers were forward: 5′-GAG TAT TTT CGG TTT AGA CGA GAC-3′; reverse: 5′-CCT TTT ATA CGA CGC AAC CG-3’. Unmethylated MSP (U-MSP) primers were forward: 5′-TTT GAG TAT TTT TGG TTT AGA TGA GAT-3′; reverse: 5′-AAC ACC TTT TAT ACA ACA CAA CCA-3′. DNA from normal bone marrow donors (N = 8) was used as negative control, whereas enzymatically methylated control DNA (CpGenome Universal Methylated DNA, Chemicon, Temecula, CA, USA) was used as positive control in all the experiments. MSP was performed in a thermal cycler (9700, Applied Biosystems, Foster City, CA, USA) with the following cycling conditions: 95°C for 5 min., 35 cycles of 95°C for 30 sec., 58°C for 30 sec., 72°C for 30 sec. and a final extension of 10 min. at 72°C. The MSP mixture contained 50 ng of bisulphite-treated DNA, 0.2 mM deoxyribonucleotide triphosphates (dNTPs), 1.5 mM MgCl2, 10 pmol of each primer, 1 × PCR buffer and 2.5 units of AmpliTaq Gold DNA Polymerase (Applied Biosystems) in a final volume of 25 μl. Ten microlitres of PCR products were loaded onto 6% non-denaturing polyacrylamide gels, electrophoresed and visualized under ultraviolet light after staining with ethidium bromide.
5-Aza-2′-deoxycytidine (5-AzadC) treatment
For treatment with 5-AzadC (Sigma-Aldrich, St. Louis, MO, USA), cells were seeded in six-well plates at a density of 1 × 106 cells/ml, and cultured with 1 μM of 5-AzadC for 3 days. Cells on day 0 and day 3 of 5-AzadC treatment were harvested.
RNA isolation and stem-loop reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated using mirVana™ miRNA Isolation Kit (Ambion, Austin, TX, USA), according to the manufacturer’s instructions. RT was performed with Taqman® MicroRNA RT Kit and Taqman® MicroRNA Assay Kit (Applied Biosystems), according to the manufacturer’s instructions. Total RNA was reverse transcribed in 1 mM dNTPs, 50 U MultiScribe™ Reverse Transcriptase, 1 × RT Buffer, 3.8 U RNase Inhibitor and 1 × stem-loop RT primer at following thermal cycling condition: 16°C for 30 min., 42°C for 30 min. and 85°C for 5 min. RT-qPCR of miR-203 was performed in triplicate using 1.33 μl of 1:15 diluted RT product in 1 × Taqman® Universal PCR Master Mix and 1 × Taqman® Assay at 95°C for 10 min., followed by 40 cycles of 95°C for 15 sec. and 60°C for 1 min. RNU48 was used as reference for data analysis using the 2−ΔΔCt method [19].
Mature miR-203 overexpression
Mature miR-203 RNA oligonucleotide mimic (100 nM) (Ambion) was transfected into 1 × 106 cells using X-tremeGENE siRNA Transfection Reagent (Roche, Basel, Switzerland), according to manufacturers’ instructions. Control experiments were performed with non-targeting precursor mimic, negative control #1.
MTT assay
Cell proliferation was determined by colorimetric quantification of purple formazan formed from the reduction of yellow tetrazolium MTT (3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide) by proliferative cells. In brief, cells were seeded in a 96-well microtitre plate at 2.5 × 104 /well in 100 μl of medium. At the assay time-point, each well was added with 10 μl of 5 mg/ml MTT reagent and incubated for a further 4 hrs. Each well was added with 100 μl of dimethyl sulfoxide (DMSO). The absorbance reading at 550 nm with reference to 650 nm was recorded.
Trypan blue exclusion assay
Dead cells were measured by trypan blue exclusion method under microscope. Dead cells (%) = (total number of dead cells per microscopic field/ total number of cells per microscopic field) × 100. Five random microscopic fields were counted in each sample.
Statistical analysis
The frequency of hsa-miR-203 methylation in different types of haematological cancers was computed by chi-square or Fisher’s exact test. In CLL, correlation between hsa-miR-203 methylation status with continuous (mean age, mean diagnostic haemoglobin, lymphocyte and platelet counts) and categorical variables (gender, Rai staging, high-risk cytogenetic aberrations and methylation of hsa-miR-34a, -124a and -196b) were studied by Student’s t-test and chi-square test (or Fisher’s exact test), respectively. OS is measured from the date of diagnosis to the date of last follow-up or death. In CLL, OS of patients with limited Rai stage (stages 0, I and II) were compared to those with advanced stage (stages III and IV). Survival is plotted by the Kaplan–Meier method and compared by the log-rank test. All P-values were two sided. In NHL, correlation between hsa-miR-203 methylation with continuous (mean age) and categorical variables (gender, lineage [B, T or NK/T], nodal/extranodal presentation and methylation of hsa-miR-34a, -124a and -196b) were studied by Student’s t-test and chi-square test (or Fisher’s exact test), respectively.
Results
MSP
Controls
None of the eight normal control marrows showed aberrant methylation of hsa-miR-203 by MSP (Fig. 1A). The positive and negative controls showed the expected MSP results (normal DNA: U-MSP+/M-MSP–; methylated DNA: U-MSP–/M-MSP+).
Fig 1.
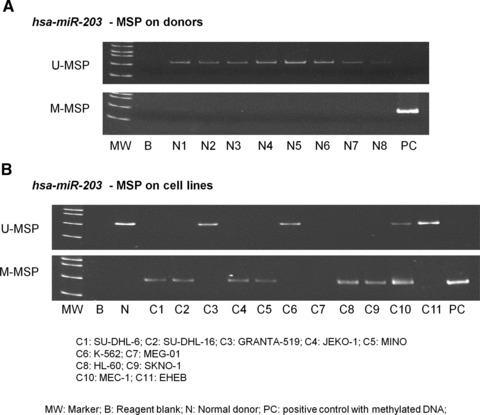
Methylation of hsa-miR-203. (A) U-MSP showed that the methylated control [M] was totally methylated, and all eight normal controls (N1–N8) were unmethylated. In the M-MSP, the methylated control was positive (methylated) but all normal controls were negative (unmethylated). (B) For the cell lines, SU-DHL-6, SU-DHL-16, JEKO-1, MINO, HL-60 and SKNO-1 were completely methylated of hsa-miR-203.
Leukaemia and lymphoma cell lines
The profile of methylation of hsa-miR-203 of leukaemia and lymphoma cell lines was shown in Fig. 1B. SU-DHL-6, SU-DHL-16, JEKO-1, MINO, HL-60 and SKNO-1 were homozygously methylated for hsa-miR-203.
Primary samples at diagnosis
Direct sequencing of the M-MSP products from the methylated primary samples showed the expected nucleotide changes after bisulphite treatment, therefore confirming complete bisulphite conversion and specificity of MSP (Fig. 2A). hsa-miR-203 hypermethylation was not detected in any of the CML. On the other hand, hsa-miR-203 methylation was preferentially found in NHL patients with methylation occurring in 1 (5%) ALL, 2 (10%) AML, 21 (42%) CLL and 19 (38.8%) NHL samples (P < 0.0001) (Fig. 2B). hsa-miR-203 was more frequently hypermethylated in lymphoid than myeloid malignancies (P = 0.002). In CLL patients, the mean survival for the whole group was 86.97 months (89.28 months and 76.71 months for those with or without hsa-miR-203 methylation). Moreover, hsa-miR-203 methylation was associated with a higher mean Hb level than the unmethylated patients (12.2 g/dl versus 10.4 g/dl, P = 0.03) but not diagnostic lymphocyte count (P = 0.98) and platelet count (P = 0.55). Moreover, there was no correlation between hsa-miR-203 methylation and age (P = 0.38), gender (P = 0.99), advanced Rai stage (≥stage 2) (P = 0.14), high-risk karyotypic aberrations (P = 0.99) or death (P = 0.76). The projected OS in CLL patients with and without hsa-miR-203 methylation were 69% and 62% (P = 0.76). Amongst lymphoma samples, hsa-miR-203 was methylated in 9 (40.9%) B-cell NHL, 4 (23.5%) T cell and 6 (60%) NK/T cell NHL (P = 0.165). However, hsa-miR-203 methylation did not correlate with age (P = 0.74), gender (P = 0.24), nodal/extranodal presentation (P = 0.383) or Ann Arbor stage (P = 0.118) of the lymphoma patients.
Fig 2.
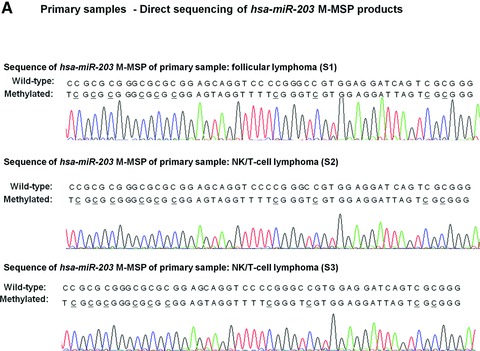

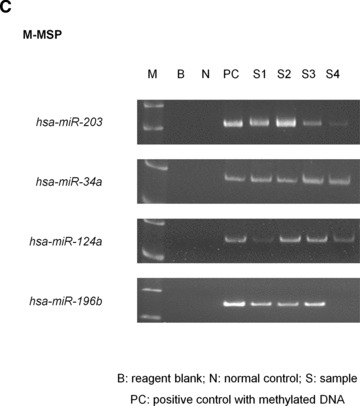
Methylation of hsa-miR-203 in primary samples. (A) Sequence analysis of the M-MSP product from bisulphite-treated DNA showed that the cytosine [C] residues of CpG dinucleotides were methylated and remained unchanged, whereas all the other C residues were unmethylated and were converted to thymidine [T], indicating complete bisulphite conversion and specificity of MSP. (B) M-/U-MSP analysis of hsa-miR-203 methylation in primary samples of haematological malignancies. (C) Correlation of hsa-miR-203, -34a, -124a and -196b methylation in samples as shown by M-MSP analysis from four representative NHL patients (B: reagent blank; S: primary sample; N: normal control; PC: positive control with methylated DNA).
Associations of hsa-miR-203 methylation with hsa-miR-34a, -124a and -196b methylation
In the 50 cases of CLL, there were 2 (4%) with hsa-miR-34a, 7 (14%) with hsa-miR-124a and 3 (6%) with hsa-miR-196b methylation. There was no association of hsa-miR-203 methylation with hsa-miR-34a (P = 0.171), -124a (P = 0.99), -196b methylation (P = 0.565). In patients with NHL, 31 (63.3%) had concomitant study of the methylation status of hsa-miR-34a, -124a and -196b in addition to hsa-miR-203. There were 5 (16.13%) with hsa-miR-34a, 10 (32.26%) with hsa-miR-124a and 12 (38.7%) with hsa-miR-196b methylation. hsa-miR-203 methylation was associated with hsa-miR-34a (P = 0.027), hsa-miR-124a (P = 0.004) and hsa-miR-196b methylation (P = 0.021) (Fig. 2C).
5-AzadC treatment of lymphoma cells
JEKO-1 cells were completely methylated for hsa-miR-203. Upon 5-AzadC demethylation treatment, hsa-miR-203 U-MSP signal emerged on day 3 (Fig. 3A), with re-expression of mature miR-203 as shown by Taqman stem-loop RT-qPCR (Fig. 3B).
Fig 3.

Effect of 5-AzadC treatment on JEKO-1 lymphoma cells. (A) M-/U-MSP analysis of hsa-miR-203 promoter methylation status and (B) stem-loop RT-qPCR analysis of the mature miR-203 expression. 5-AzadC treatment resulted in progressive demethylation of hsa-miR-203 promoter, and re-expression of the mature miR-203 in JEKO-1 cells.
Effect of miR-203 overexpression in lymphoma cells
Upon transfection of mature miR-203 mimic which led to overexpression mature miR-203 (Fig. 4A), JEKO-1 showed 20% reduction in cell proliferation as compared with the negative control at 24th hr by MTT assay (Fig. 4B), and 2-fold increase of dead cells as measured by trypan blue exclusion assay (Fig. 4C), thereby suggesting the tumour suppressor role of miR-203 in lymphoma cells.
Fig 4.

Effect of miR-203 in lymphoma cells. JEKO-1 cells, homozygously methylated for hsa-miR-203, were transfected with mature miR-203 mimic oligo as compared with non-targeting precursor control. (A) Stem-loop RT-qPCR analysis of mature miR-203 expression at 24 hrs after transfection. (B) Cellular proliferation of lymphoma cells in response to overexpression of miR-203 was measured by MTT assay. (C) Dead cells were measured by trypan blue exclusion assay. Error bars represent standard deviation.
Discussion
In this study, we showed that hsa-miR-203 is hypermethylated and silenced in lymphoma cell lines but not normal blood or marrow cells, which is consistent with a tumour-specific pattern of miRNA-methylation. By contrast, some miRNAs may be hypermethylated in both normal and tumour cells, and hence is consistent with a tissue-specific but not tumour-specific pattern of gene methylation. For example, miR-127 and miR-373 are hypermethylated in both the normal and cancer cells [20, 21].
Furthermore, if miR-203 is a TSG, restoration of miR-203 expression is expected to inhibit cellular proliferation or induce cell death. Indeed, in this study, restoration of miR-203 expression in JEKO-1 lymphoma cells led to inhibition of cellular proliferation and enhancement of cell death, which is consistent with previous studies showing inhibition of cell proliferation upon re-expression of miR-203 in cells carrying homozygous hsa-miR-203 hypermethylation [12]. Therefore, these findings, together with the pattern of tumour-specific methylation and miRNA silencing, suggested that miR-203 possesses tumour suppressor property.
In a previous study, hsa-miR-203 was shown to be preferentially hypermethylated in Ph+ leukaemias (with expression of BCR-ABL) but less commonly, in Ph– acute leukaemias or Ph– chronic myeloproliferative diseases [12]. By contrast, we showed that miR-203 was unmethylated in a Ph+ CML blastic cell lines, K-562, and also 11 patients with Ph+ CML at chronic phase. However, the difference might be due to the small number of Ph+ leukaemia patients in our study. On the other hand, hsa-miR-203 was preferentially hypermethylated in lymphoid than myeloid malignancies. Moreover, hsa-miR-203 methylation has been shown to be frequently methylated in T cell lymphoma [12]. In this study, although hsa-miR-203 methylation was moderately frequent in T cell lymphoma, it was also prevalent in B-cell or NK-cell lymphoma. Therefore, the spectrum of hsa-miR-203 methylation may be more extensive than Ph+ neoplasms.
NK/T cell lymphoma is an Epstein–Barr virus-associated, aggressive extranodal lymphoma more frequently encountered in Asia, and Central and South America [22]. As yet, only few TSGs have been shown to be hypermethylated in NK/T cell lymphoma including p73, CDKN2A, CDKN2B, hMLH1 and RARβ[23]. On the other hand, as yet, miR-34a was the only miRNA reported to be hypermethylated in NK/T cell lymphoma [24]. Therefore, frequent methylation of hsa-miR-203 in NK/T cell lymphoma may be an important biomarker or pathogenetic event.
In our cohort of CLL, despite that it was a retrospective study, survival analysis confirmed that this is a representative CLL with an indolent clinical course, similar to Caucasian CLL patients. Moreover, Rai stage and high-risk karyotypes were important risk factors for survival, thereby testifying that CLL in Chinese runs a similar clinical course as Caucasian CLL patients. However, apart from association with a higher diagnostic Hb level, hsa-miR-203 methylation did not impact survival.
Finally, although hsa-miR-203 was frequently methylated in both CLL and NHL, in contrast to CLL in which hsa-miR-203 was not associated with methylation of other miRNAs, hsa-miR-203 methylation was associated with methylation of hsa-miR-34a, -124a and -196b. This finding suggested that different haematological cancers may carry disease-specific miRNA-methylation profiles, which might be important in the pathogenesis of disease.
In summary, hsa-miR-203 hypermethylation is tumour-specific, associated with gene silencing, which could be reversed by 5-AzadC hypomethylating treatment. miR-203 possesses tumour suppressor activity. Amongst haematological malignancies, hsa-miR-203 is more frequently hypermethylated in lymphoid instead of myeloid malignancies. CLL in Chinese patients runs a similarly indolent clinical course, which is impacted by Rai stage and karyotype. Finally, hsa-miR-203 methylation was associated with concomitant methylation of other miRNAs in NHL.
Acknowledgments
We thank Dr Curtis C. Harris M.D., Chief, Laboratory of Human Carcinogenesis, National Cancer Inst., USA for his expert comments. This work is supported by The University of Hong Kong Seed Funding Programme for Basic Research, and Hong Kong Research Grants Council General Research Fund to Dr. C.S.C.
Financial disclosures
There is no financial disclosure.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Chim CS, Liang R, Kwong YL. Hypermethylation of gene promoters in hematological neoplasia. Hematol Oncol. 2002;20:167–76. doi: 10.1002/hon.694. [DOI] [PubMed] [Google Scholar]
- 2.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–54. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 3.Chim CS, Fung TK, Liang R. Disruption of INK4/CDK/Rb cell cycle pathway by gene hypermethylation in multiple myeloma and MGUS. Leukemia. 2003;17:2533–5. doi: 10.1038/sj.leu.2403133. [DOI] [PubMed] [Google Scholar]
- 4.Chim CS, Wong ASY, Kwong YL. Epigenetic inactivation of the CIP/KIP cell-cycle control pathway in acute leukemias. Am J Hematol. 2005;80:282–7. doi: 10.1002/ajh.20503. [DOI] [PubMed] [Google Scholar]
- 5.Chim CS, Fung TK, Cheung WC, et al. SOCS1 and SHP1 hypermethylation in multiple myeloma: implications for epigenetic activation of the Jak/STAT pathway. Blood. 2004;103:4630–5. doi: 10.1182/blood-2003-06-2007. [DOI] [PubMed] [Google Scholar]
- 6.Chim CS, Wong ASY, Kwong YL. Epigenetic dysregulation of the Jak/STAT pathway by frequent aberrant methylation of SHP1 but not SOCS1 in acute leukaemias. Ann Hematol. 2004;83:527–32. doi: 10.1007/s00277-004-0843-1. [DOI] [PubMed] [Google Scholar]
- 7.Chim CS, Kwong YL, Liang R. Gene hypermethylation in multiple myeloma: lessons from a cancer pathway approach. Clin Lymphoma Myelom. 2008;8:331–9. doi: 10.3816/CLM.2008.n.048. [DOI] [PubMed] [Google Scholar]
- 8.Chim CS, Pang R, Fung TK, et al. Epigenetic dysregulation of Wnt signaling pathway in multiple myeloma. Leukemia. 2007;21:2527–36. doi: 10.1038/sj.leu.2404939. [DOI] [PubMed] [Google Scholar]
- 9.Chim CS, Liang R, Fung TK, et al. Epigenetic dysregulation of the death-associated protein kinase/p14/HDM2/ p53/Apaf-1 apoptosis pathway in multiple myeloma. J Clin Pathol. 2007;60:664–9. doi: 10.1136/jcp.2006.038331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Esquela-Kerscher A, Slack FJ. Oncomirs – microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–69. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 11.Chen CZ. MicroRNAs as oncogenes and tumor suppressors. N Engl J Med. 2005;353:1768–71. doi: 10.1056/NEJMp058190. [DOI] [PubMed] [Google Scholar]
- 12.Bueno MJ, Perez de Castro I, Gomez de Cedron M, et al. Genetic and Epigenetic Silencing of MicroRNA-203 Enhances ABL1 and BCR-ABL1 Oncogene Expression. Cancer Cell. 2008;13:496–506. doi: 10.1016/j.ccr.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 13.Furuta M, Kozaki KI, Tanaka S, et al. miR-124 and miR-203 are epigenetically silenced tumor-suppressive microRNAs in hepatocellular carcinoma. Carcinogenesis. 2010;31:766–76. doi: 10.1093/carcin/bgp250. [DOI] [PubMed] [Google Scholar]
- 14.Bennett JM, Catovsky D, Marie-Theregse D, et al. Proposals for the Classification of the Acute Leukaemias French-American-British (FAB) Co-operative Group. Br J Haematol. 1976;33:451–8. doi: 10.1111/j.1365-2141.1976.tb03563.x. [DOI] [PubMed] [Google Scholar]
- 15.Van den Berghe H. Morphologic, immunologic and cytogenetic (MIC) working classification of the acute myeloid leukaemias. Br J Haematol. 1988;68:487–94. doi: 10.1111/j.1365-2141.1988.tb04242.x. [DOI] [PubMed] [Google Scholar]
- 16.Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111:5446–56. doi: 10.1182/blood-2007-06-093906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaffe ES, Harris NL, Stein H, et al. World Health Organization classification of tumours: pathology and genetics of tumours of haematopoietic and lymphoid tissues. Lyon: IARC Press; 2001. [Google Scholar]
- 18.Chim CS, Liang R, Fung TK, et al. Infrequent epigenetic dysregulation of CIP/KIP family of cyclin-dependent kinase inhibitors in multiple myeloma. Leukemia. 2005;19:2352–5. doi: 10.1038/sj.leu.2403904. [DOI] [PubMed] [Google Scholar]
- 19.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-[delta][delta]CT method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 20.Lujambio A, Esteller M. CpG island hypermethylation of tumor suppressor microRNAs in human cancer. Cell Cycle. 2007;6:1455–9. [PubMed] [Google Scholar]
- 21.Lujambio A, Calin GA, Villanueva A, et al. A microRNA DNA methylation signature for human cancer metastasis. PNAS. 2008;105:13556–61. doi: 10.1073/pnas.0803055105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chim CS, Ma SY, Au WY, et al. Primary nasal natural killer cell lymphoma: long-term treatment outcome and relationship with the International Prognostic Index. Blood. 2004;103:216–21. doi: 10.1182/blood-2003-05-1401. [DOI] [PubMed] [Google Scholar]
- 23.Siu LL, Chan JK, Wong KF, et al. Aberrant promoter CpG methylation as a molecular marker for disease monitoring in natural killer cell lymphomas. Br J Haematol. 2003;122:70–7. doi: 10.1046/j.1365-2141.2003.04396.x. [DOI] [PubMed] [Google Scholar]
- 24.Chim CS, Wong KY, Qi Y, et al. Epigenetic inactivation of the miR-34a in hematological malignancies. Carcinogenesis. 2010;31:745–50. doi: 10.1093/carcin/bgq033. [DOI] [PubMed] [Google Scholar]