

Abstract
Myxoma virus, a poxvirus previously considered rabbit specific, can replicate productively in a variety of human tumor cells in culture. The purpose of this study was to determine if there was efficacy or toxicities of this oncolytic virus against experimental models of human malignant gliomas in vitro, in vivo, and ex vivo in malignant glioma specimens. In vitro, the majority of glioma cell lines tested (7 of 8, 87.5%) were fully permissive for myxoma virus replication and killed by infection. In vivo, intracerebral (i.c.) myxoma virus inoculation was well tolerated and produced only minimal focal inflammatory changes at the site of viral inoculation. U87 and U251 orthotopic xenograft models were used to assess myxoma virus efficacy in vivo. A single intratumoral injection of myxoma virus dramatically prolonged median survival compared with treatment with UV-inactivated myxoma virus. Median survival was not reached in myxoma virus–treated groups versus 47.3 days (U87; P = 0.0002) and 50.7 days (U251; P = 0.0027) in UV-inactivated myxoma virus–treated groups. Most myxoma virus–treated animals (12 of 13, 92%) were alive and apparently “cured” when the experiment was finished (>130 days). Interestingly, we found a selective and long-lived myxoma virus infection in gliomas in vivo. This is the first demonstration of the oncolytic activity of myxoma virus in vivo. The nonpathogenic nature of myxoma virus outside of the rabbit host, its capacity to be genetically modified, its ability to produce a long-lived infection in human tumor cells, and the lack of preexisting antibodies in the human population suggest that myxoma virus may be an attractive oncolytic agent against human malignant glioma. (Cancer Res 2005; 65(21): 9982-90)
Introduction
Malignant gliomas continue to be a major therapeutic challenge. These are highly invasive and rapidly growing tumors that are refractory to available treatments. Long-term survivors are very rare (1) and the median survival for patients with the most malignant form of glioma, glioblastoma multiforme, is only about 1 year; this has not changed significantly in the last 30 years.
Oncolytic viruses are emerging as promising new experimental therapeutics (2–8) and several have been tested experimentally (9–13) and clinically (14–19) in gliomas. The ideal oncolytic virus should have several properties, at least some of which are obvious. These include efficacy in vitro and in vivo against a broad range of tumors and relative selectivity for tumor cells so that normal, nontransformed cells are spared. The basis of the ability of the virus to selectively infect and kill tumor cells is known for only a few oncolytic viruses. Some oncolytic viruses usurp the signaling pathways of tumor cells, such as Ras (20, 21) or p53 (7, 22), to allow a lytic infection in tumor cells to occur. Others rely on the defects in IFN signaling pathways or defects in translational control which are present in cancer cells (23–25). Other desirable properties of an oncolytic virus include the ability to engineer the virus (e.g., to enhance its beneficial properties) and a nonpathogenic profile in humans so that it is safe for patients, their contacts, and society at large. Having recently discovered the oncolytic properties of myxoma virus for a broad spectrum of human cancer cells in vitro (26), we considered myxoma virus as a promising platform in vivo because its genome has been sequenced, it is straightforward to engineer, its tropism is highly restricted (to European rabbits), and there is a lack of acquired immunity to the virus in the human population (27).
Myxoma virus is a poxvirus and has a large double-stranded DNA genome that allows for the potential insertion of large (25 kb), therapeutically relevant, eukaryotic genes (27). Myxoma virus is a rabbit-specific virus and causes a lethal disease termed myxomatosis in the European rabbit (Oryctalagus cuniculus). Its species selectivity is so narrow that it was used to control the disastrous feral rabbit population in Australia in the 1950s (28). Importantly, it is nonpathogenic for all other vertebrate species tested including humans (28, 29). Despite this extremely narrow species host range, myxoma virus can productively infect certain non-rabbit cells in vitro including immortalized baby monkey kidney fibroblasts (BGMK), primary murine cells genetically deficient in IFN responses (30), and a number of different human tumor cells in vitro (26). Cancer cells are well known to be deficient in their IFN responses (23, 24). Here, we evaluate myxoma virus as a novel oncolytic agent against experimental gliomas in vitro, in vivo, and ex vivo against human malignant glioma surgical specimens. We show for the first time that myxoma virus has oncolytic properties against human tumor cells in vivo. It infects and kills the majority of human glioma cells tested, is safe when administered intracerebrally (i.c.), and “cures” most mice when administered intratumorally in orthotopic human malignant glioma models. Further, it infects and kills all primary glioma cells tested that were obtained directly from glioma surgical specimens.
Materials and Methods
Cell Lines
Glioma cell lines and murine fibroblast NIH 3T3 cells were purchased from the American Type Culture Collection (ATCC; Manassas, VA). Cells were grown in DMEM/F12 (Hybri-care, ATCC, Manassas, VA) containing 10% fetal bovine serum (FBS) at 37°C in a humidified 5% CO2 incubator. All cells were passaged until they reached ~80% confluence, harvested by trypsin treatment, and replated in medium. Each cell line was tested routinely for Mycoplasma contamination.
Virus and Cell Infection
A derivative of myxoma virus (strain Lausanne), designated vMyxgfp (31), was used. The virus contains a green fluorescent protein insert located between open reading frames M135R and M136R of the myxoma genome. The virus was propagated and titered by plaque formation on BGMK cells. UV-inactivated dead myxoma virus was prepared by irradiating vMyxgfp with UV light for 1 hour. Cells were incubated with indicated multiplicity of infection (MOI) of live vMyxgfp or dead myxoma virus for 1 hour at 37°C, after which the virus was removed from culture and replaced with fresh culture medium and the culture resumed at 37°C, 5% CO2 until use in subsequent experiments. Phase-contrast and fluorescent images of cells were taken using a Carl Zeiss inverted microscope (Axiovert 200M) mounted with Carl Zeiss digital camera (AxioCam MRc).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazoium Bromide Assay
Cells grown to 50% to 60% confluence were infected with different doses (MOI = 0, 1, and 10) of vMyxgfp. Cell viability was measured at 72 hours postinfection by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazoium bromide (MTT) assay, as previously described (32).
Immunohistochemistry
Frozen sections were fixed with 4% paraformaldehyde for 10 minutes followed by four washes with PBS. The sections were exposed to the primary antibody (M-T7, rabbit polyclonal anti-myxoma serum diluted 1:1,000 in PBS containing 2% bovine serum albumin) for 12 hours at 4°C. The biotinylated anti-rabbit immunoglobulin G (Vector Laboratories, Burlingame, CA) was used as a secondary antibody. ABC immunohistochemistry kit (Vector Laboratories) was used to carry out immunostaining. Sections were mounted and viewed with a Carl Zeiss microscope (Axiovert 200M) mounted with a Carl Zeiss digital camera (AxioCam MRc).
Western Blot
Cells were infected with 5 MOI of vMyxgfp as described above. After 12 or 24 hours, cells were washed with PBS and subsequently lysed. Protein concentrations of cell lysates were measured by bicinchoninic acid protein assay. Protein samples were separated by SDS-PAGE followed by electroblotting onto nitrocellular membrane. Polyclonal myxoma virus antibodies (M-T7 and Serp-1, recognizing early and late myxoma virus gene products, respectively) and horseradish peroxidase–conjugated secondary antibody were used to detect myxoma virus proteins. Blots were probed with a pyruvate kinase antibody for a protein loading control.
Viral Culture
Viral culture from cells
Cells were infected with 0.1 MOI of vMyxgfp. At indicated time points after infection, cells were washed with PBS and collected by trypsinization. Appropriate amount of glass beads (G8772, Sigma) was added into cell suspension (glass beads covered approximately half of the cell suspension). Cells were broken by vortexing using a yeast cell beater (Sunon SF-11580A, Sunonwealth Electric Machine Industry Co. Ltd.) for 50 seconds at 4°C. The lysates went through a freeze-thaw process thrice. Viral titer within collected cell lysates was determined on BGMK cells. Briefly, BGMK cells were incubated with serial diluted lysates at 37°C, 5% CO2, for 48 hours. Green fluorescent foci were viewed and counted under a Zeiss Axiovert microscope under a 2.5× low power lens. Viral titers based on 10,000 BGMK cells were then calculated.
Viral culture from tissues
Animals were perfused before euthanization. Tissue samples were homogenized using a Pellet Pestles Kit (VWR International, Edmonton, Canada) or homogenizer (Ultra-Turrax T25, Janke & Kunkel, Germany) and followed by a freeze-thaw protocol to release virus from cells. Supernatants were clarified by centrifugation and diluted accordingly. Myxoma virus titers were determined by focus formation on BGMK cells as described above.
Animals
CD-1 nude mice (female, 6-8 weeks old) were purchased from Charles River Canada (Constant, Canada). The animals were housed in groups of three to five in a vivarium maintained on a 12-hour light/dark schedule with a temperature of 22 ± 1°Cand a relative humidity of 50 ± 5%. Food and water were available ad libitum. All procedures were reviewed and approved by the University of Calgary Animal Care Committee.
Safety and Toxicity Study in Nude Mice
We administered myxoma virus i.c. in nude mice to test whether this approach was safe. Mice were anesthetized and burr holes drilled 1.5 to 2 mm right of the midline and 0.5 to 1 mm posterior to the coronal suture through a scalp incision. A 5-μL syringe was used to inject either 5 × 106 plaque-forming units (PFU) of vMyxgfp or dead myxoma virus (2-6 μL) at a depth of 3 mm under guidance of a stereotactic frame (Kopf Instruments, Tujanga, CA), as previously described (32–34). Animals were monitored closely for possible neurologic disorders with body weight recorded every other day. We followed these animals for different lengths of time after infection until they were sacrificed. Brains and major organs were saved and analyzed pathologically.
In vivo Studies in an Orthotopic Glioma Model in Nude Mice
Two orthotopic glioma animal models established with glioma cell lines, U87 and U251, were used to test the efficacy of myxoma virus in treating this disease. The stereotactic techniques used to implant glioma cells in the right putamen have been described (32). Briefly, mice were anesthetized, a burr hole was drilled through a scalp incision, and glioma cells (5 × 105 cells/mouse) were inoculated under guidance of a stereotactic frame (Kopf Instruments), as described above. Twelve days later, when microscopic tumors grew and animals were asymptomatic, a single intratumoral injection of 5 × 106 PFUs of either live vMyxgfp or dead myxoma virus was administered stereotactically. For experiments to assess tumor size, animals were sacrificed 35 days (an arbitrary time point) after tumor implantation. To sacrifice them, animals were anesthetized, perfused intracardially with PBS, and then fixed with 4% paraformaldehyde. Then we cut the whole brain into coronal sections, selected the section with the largest tumor, and then measured the tumor area and the area of the whole brain using ImagePro. All of the brains and major organs were examined histologically. Animals losing ≥20% of their body weight or having trouble ambulating, feeding, or grooming were sacrificed. For experiments assessing survival, animals were monitored for 130 days when we arbitrarily terminated the experiment. All the animal experiments were carried out in accordance with the Care and Use of Experimental Animals Guide issued by Canadian Council on Animal Care.
In vivo Viral Distribution Studies in an Orthotopic Glioma Model
Nude mice with established U87 xenografts in their brain were inoculated with vMyxgfp (5 × 106 PFUs) on day 20 (rather than day 12 as above) after tumor implantation, as otherwise described above. Animals were sacrificed at the following time points after infection: days 1, 7, 14, 21, 35, and 42. At the time of sacrifice, the animals were perfused with sterile PBS and the brain tumor tissues were either saved frozen for viral culture or embedded in optimum cutting temperature compound for H&E staining and immunohistochemistry for myxoma virus. Protein imaging of green fluorescent protein virus was visualized under a whole body fluorescence imaging system (32, 33, 35). Briefly, for high-magnification imaging, this system uses a Leica MZ-FLIII fluorescence stereomicroscope equipped with 100-W mercury-vapor burner and mounted with a Kodak DC 2900 digital camera (32). Images were processed and analyzed by using Photoshop 6.0 and Image-Pro Plus software.
Myxoma Virus Efficacy Studies in Bilateral Brain Tumor Model
To determine if vMyxgfp virus can target remote tumor cells in the brain, we established a dual tumor model by implanting U87 cells (3 × 105 cells/mouse/side) in both hemispheres of nude mice and injecting vMyxgfp in one tumor only (i.e., the ipsilateral tumor) by a direct single intratumoral administration. For experimental assessment of survival, animals were followed until they lost ≥20% of body weight or had trouble ambulating, feeding, or grooming. After sacrifice, brains were saved and analyzed pathologically.
Primary Human Glioma Cultures
Patients samples from human gliomas were obtained following brain tumor surgery from the operating room of the Foothills Hospital (Calgary) and short-term cultures were established. This study was approved by the Conjoint Medical Ethics Committee. A neuropathologist confirmed the histopathologic diagnosis. Briefly, the specimen was split in half and fixed in 10% formalin (to confirm its identity) or placed in DMEM/F12 with 10% FBS. Tissue was then washed several times, transferred to a 35-mm tissue culture dish, cut into small pieces (~1 mm in diameter), and dissociated with trypsin (0.25%) and 50 μg/mL DNase for 30 minutes at 37°C. After filtering and washing with DMEM/F12 (20% FBS), cells were resuspended in 20% FBS in DMEM/F12 and plated (10,000-200,000 cells/well) in 96-well plates. Cells were infected the following day with vMyxgfp virus, both live and UV inactivated, at MOIs of 0.1, 1, and 10. Cell viability was measured at 48 hours postinfection by MTT assay and cytopathic effect and green fluorescent protein virus internalization were obvious 40 hours after infection as previously described.
Statistical Analyses
Statistical Analysis Software (SAS Institute, Inc., Cary, NC) and GraphPad Prism (version 4, GraphPad Software, Inc., San Diego, CA) were used for statistical analyses. Survival curves were generated by the Kaplan-Meier method. The log-rank and Mann-Whitney tests were used to compare the distributions of survival times and tumor sizes, respectively. All reported P values were two-sided and were considered to be statistically significant at <0.05.
Results
Myxoma virus productively infects and kills most human malignant glioma cell lines
We analyzed the permissiveness of infection and cell killing by vMyxgfp of eight established malignant glioma cell lines [six human (U87, U118, U251, U373, U343, and A172) and two racine (RG2 and 9L)]. The majority of these (7 of 8, 87.5%) were permissive to infection although to different degrees (Fig. 1A); U87 was the most permissive whereas U251 and U118 were scored as semipermissive. A cell line was defined as permissive to myxoma virus infection when visible green fluorescent protein–expressing foci were formed. The permissive cell line BGMK was used as a reference line. We next examined susceptibility of these lines to cell killing (Fig. 1B) and found similar results. The malignant glioma cell lines tested were susceptible to killing with myxoma virus whereas U118 was the least susceptible. Seven of eight (87.5%) cell lines were considered fully susceptible (>80% killed and lysed by 10 MOI myxoma virus) to live vMyxgfp 72 hours after infection (Fig. 1B) whereas it took 96 hours for >80% of U118 cells to be killed (data not show). Cells receiving either dead myxoma virus or no virus remained fully viable.
Figure 1.

Myxoma virus infects and kills most glioma cells in vitro. A, glioma cells were infected with live vMyxgfp (MOI = 10, left and center column) or dead virus (dead myxoma virus, i.e., UV-inactivated vMyxgfp, right column). Cytopathic effect and green fluorescent protein expression were evident in all live vMyxgfp–infected cell lines, but not in the dead myxoma virus-infected cell lines, 72 hours after infection (magnification, ×100). B, cells infected with different MOIs of vMyxgfp were evaluated for viability by MTT assay at 72 hours postinfection. U87 and RG2 cell lines were very susceptible. The U118 cell line was much less susceptible and a significant proportion of cells were killed only at 96 hours and an MOI of 10 (data not shown). BGMK and NIH 3T3 were used as positive and negative controls, respectively. C, myxoma viral protein synthesis was evident after virus infection. An increase in both early (M-T7) and late (Serp-1, an indicator of virus replication) viral gene products was detected in glioma lines and BGMK by Western blot analysis, 12 to 24 hours after myxoma virus infection (MOI = 5). Pyruvate kinase was used as a protein loading control. D, myxoma virus replicates within glioma cells. Cells were infected with vMyxgfp (MOI = 0.1) followed by collection of cell lysates at the indicated times after infection. Viral titers were determined by plaque titration on BGMK cells. Viral titers increased in cell lysates of all glioma cell lines although only minimally in U118 and U251. —◆–, U118; —○—, U87; —▲—, BGMK; —□—, U251; —△—, 3T3.
We then confirmed that glioma cells supported a productive infection by examining early/late viral gene expression and postinfection viral titers. We selected three glioma cell lines which spanned the spectrum of susceptibility to myxoma virus (the highly susceptible U87, less susceptible U251, and least susceptible U118), the fully permissive control BGMK, and the semipermissive rodent cell line NIH 3T3. These five cell lines were infected at an MOI of 5, samples were collected at 12 and 24 hours after infection, and cell lysates were prepared for Western blot analysis. To examine viral gene expression, early and late gene products were analyzed by Western blot. Both M-T7, a 35 kDa myxoma virus protein expressed and secreted early in infection (Fig. 1C), and Serp-1, a 55 kDa myxoma virus protein secreted late in infection, were seen in all cell lines tested and roughly correlated with susceptibility to infection. For example, the most susceptible line U87 (and the positive control line BGMK) expressed the highest levels of early and late viral gene products whereas the least susceptible lines (U118 and NIH 3T3) had the lowest levels of expression. We then quantified the virus titers on the indicated cell lines (Fig. 1D) and found that viral titers varied significantly between cell lines and mirrored their susceptibility. Remarkably, viral titers obtained in U87 cells were as high as BGMK cells, suggesting that this malignant glioma cell line was very efficiently infected. Viral titers in the less susceptible lines NIH 3T3, U251, and U118 were significantly lower. Hence, myxoma kills human malignant glioma cell lines and they support varying degrees of productive myxoma virus infection.
The intracerebral administration of myxoma virus is safe in nude mice
To determine if myxoma is safe when administered i.c. in immunocompromised (nude) mice, we tested the highest dose (5 × 106 PFUs) that we delivered in this study. Others have reported that myxoma virus is safe when administered into the brains of immunocompetent mice (30). There was a slight trend for live vMyxgfp–treated animals to lose some body weight within 2 weeks of viral administration but this difference was not statistically significant (compared with dead myxoma virus–treated animals; two way ANOVA, P = 0.6360; Fig. 2A). Histologic examination of the brains showed minimal inflammation (lymphocytes, microglia, and macrophage infiltration; Fig. 2B) and no evidence of a diffuse meningoencephalitis. Similarly, the subarachnoid space, subependymal regions, and ventricles were normal. Therefore, because this dose was well tolerated i.c., we used this dose in our therapeutic experiments.
Figure 2.

i.c. administration of myxoma virus is safe in nude mice. A, nude mice receiving live vMyxgfp (live vMyxgfp, 12 mice) i.c. showed a slight lose in body weight the first week after virus administration although this was not significantly different than dead myxoma virus–treated (4 mice) animals (two-way ANOVA, P = 0.6360). The mice appeared normal throughout the experiment and none died. B, histologic changes in the brain of live vMyxgfp–treated mice showed mild focal inflammation and some minor lymphocytic and microglial infiltration at the site of viral inoculation (right two columns; top, ×25; bottom, ×400) but not in the dead myxoma virus–treated animals (left column; top, ×25; bottom, ×400). There was no diffuse meningoencephalitis, hydrocephalus, or other significant abnormalities. Casing represents regions that are magnified.
Myxoma virus produces long-term survival in two animal models of human malignant glioma
We compared the in vivo effectiveness of myxoma virus against a highly susceptible cell line in vitro (i.e., U87) to a line which was somewhat less susceptible in vitro (i.e., U251). Accordingly, we implanted U87 malignant glioma cells into the putamen of nude mice and, 12 days later, administered a single dose of 5 × 106 PFUs/mouse vMyxgfp intratumorally. Thirty-five days after tumor implantation, the animals were sacrificed and tumor size measured. All dead myxoma virus–treated animals had large tumors (occupied, on average, 9.16% of coronal sections of the brain) whereas macroscopic tumor was found in only two live vMyxgfp–treated animals (occupied, on average, 0.359% of coronal sections of the brain; Mann-Whitney, P = 0.0029; Fig. 3A). We then repeated these procedures but measured survival. We found that a single myxoma virus intratumoral administration dramatically prolonged survival and 87.5% of live vMyxgfp–treated animals were long-term survivors (Fig. 3B) and were apparently cured. The median survival of live vMyxgfp–treated animals was not reached because only one of eight (12.5%) died (from progressive tumor). In contrast, all dead myxoma virus–treated animals died (100%) with a significantly shorter median survival of 47.3 days (log-rank, P = 0.0002). This survival experiment was repeated with similar results.
Figure 3.

A single intratumoral administration of myxoma virus produced smaller tumors and prolonged survival in both U87 and U251 orthotopic human glioma animal models. U87 and U251 human glioma xenografts were established in brains of CD-1 nude mice. Twelve days after tumor implantation, animals were injected intratumorally with myxoma virus (5 × 106 PFUs/mouse) or dead myxoma virus (control). A, animals were sacrificed on day 35 and tumor size was measured. U87 tumors were significantly smaller in the live vMyxgfp–treated group than in the dead myxoma virus–treated group (tumors occupied 0.359% versus 9.16% of coronal sections of the brain, respectively; Mann-Whitney, P = 0.0029); most live vMyxgfp–treated animals had no visible microscopic tumors. B, live vMyxgfp–treated nude mice with orthotopic U87 xenografts survived significantly longer after a single administration of vMyxgfp (5 × 106 PFUs/mouse) delivered intratumorally. Live vMyxgfp–treated mice had a significantly longer survival (median survival not reached) than dead myxoma virus–treated animals (median survival, 47.3 days; log-rank, P = 0.0002). Only one of eight (12.5%) live vMyxgfp–treated animals died whereas all (100%) dead myxoma virus–treated animals died. The experiment was arbitrarily terminated on day 130. A similar result was found using the U251 orthotopic human glioma animal model. C, the U251 tumors were significantly smaller in the live vMyxgfp–treated group than in the dead myxoma virus–treated group (tumors occupied 3.77% versus 31.66% of coronal sections of the brain, respectively; Mann-Whitney, P = 0.0419). D, median survival was also significantly longer in the live vMyxgfp–treated (none of the animals died) than the dead myxoma virus–treated group (live vMyxgfp not reached versus 50.7 days; log-rank, P = 0.0027). E, histologic examination of the U87 orthotopic glioma model showed large tumors in the putamen of dead myxoma virus–treated mice (left column; top, ×25; bottom, ×400) but showed only microscopic residual tumor cells and focal calcification in live vMyxgfp–treated mice. Most live vMyxgfp–treated mice also had slightly dilated ventricles (middle and right columns; top, ×25; bottom, ×400). F, myxoma virus protein was detected within the microscopic residual U87 tumor cells in the brain of live vMyxgfp–treated animals (right column; top, ×25; bottom, ×400) 130 days after infections, but not in the dead myxoma virus–treated animals (left column; top, ×25; bottom, ×400).
We next repeated the above experiments with U251 orthotopic animal model and found similar results. Tumor size was compared 35 days after tumor implantation (and 23 days after viral inoculation). Tumor size was significantly smaller in live vMyxgfp–treated (occupied, on average, 3.77% of coronal sections of the brain) than dead myxoma virus–treated animals (occupied, on average, 31.66% of coronal sections of the brain; Mann-Whitney, P = 0.0419; Fig. 3C). In U251-bearing mice, the survival was also significantly longer in live vMyxgfp–treated mice and none died. The median survival of dead myxoma virus–treated mice was 50.7 days (log-rank, P = 0.0027; Fig. 3D). This survival experiment was repeated with similar results.
Histologic examination of U87-bearing mice showed all dead myxoma virus–treated animals died with large tumors at the site of implantation (Fig. 3E). In the live vMyxgfp–treated group, six mice (75%) had no microscopic tumor and the others (25%) had residual tumors which were usually very small (<1 mm; Fig. 3E). Myxoma virus protein was detected by immunohistochemistry in microscopic residual tumor cells in live vMyxgfp–treated mice (Fig. 3F). Similar results were seen in the U251 orthotopic model (data not shown).
Myxoma virus produces a selective, productive and long-lived infection in human gliomas in vivo
We implanted U87 cells into the brain of nude mice and, 20 days later, inoculated vMyxgfp intratumorally as described above. We chose 20 rather than 12 days (used above) because these tumors were slightly larger and easier to image. Animals were sacrificed at different times after infection and tumor-containing brain regions and regions of the brain without visible tumor were examined using immunohistochemistry for myxoma virus antigens and fluorescent imaging of green fluorescent protein-labeled myxoma virus and viral culture. We found green fluorescent protein expression only at the site of viral inoculation and limited to within the tumor and not within other areas of the brain. Green fluorescent protein expression lasted at least 42 days although the tumors were very small (Fig. 4A). There was no green fluorescent protein expression in the dead myxoma virus–treated animals. Similarly, results were confirmed by immunohistochemistry for myxoma virus protein (Fig. 4A). To further examine whether the virus was viable, we isolated tissue from both the site of tumor/viral inoculation and the opposite hemisphere of the brain at each time point and did virus culture on BGMK cells in vitro. We found an increase in viral titers of more than 1 log, which peaked on day 14 and persisted at least 35 days (Fig. 4B). Importantly, we did not find any infectious myxoma virus in the contralateral, noninoculated, brain.
Figure 4.
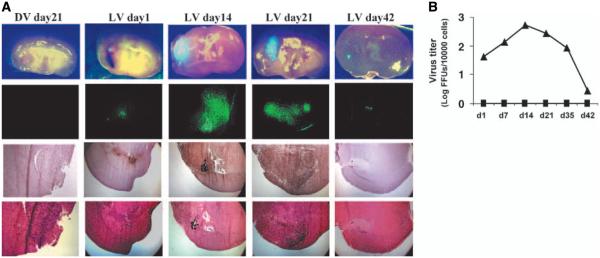
Myxoma virus infection is selective, productive, and produces a long-lived infection in human gliomas in vivo. A, vMyxgfp expression was limited to the tumor and was not found in any normal brain areas under bright- and dark-field imaging (first and second rows, respectively). Fourth row, low-magnification images of H&E-stained sections. U87 allografts were established followed by intratumoral vMyxgfp inoculation. Animals were sacrificed at different times postinfection and tumor-containing brain regions and regions of the brain without visible tumor were examined using immunohistochemistry for myxoma virus antigens (third row) and fluorescent imaging of green fluorescent protein expression (second row). Green fluorescent protein expression and viral antigens were visualized only at the site of viral inoculation in the live vMyxgfp–treated animals. No evidence of virus replication was detected in the dead myxoma virus–treated animals. B, quantification of intratumoral virus replication in virus inoculation side (—▲—) and contralateral side (—■—) for both live vMyxgfp– and dead myxoma virus–treated animals. Virus titers (log PFUs/10,000 cells) at days 1, 7, 14, 21, and 42 postinfection [points, mean (n = 3/time point); bars, SD]. Virus titers increased by >1 log and peaked 14 days after inoculation in virus inoculation (tumor) side. Both dead myxoma virus–treated tumors (not shown) and normal brain regions (contralateral) had no viable virus recovered. Viral cultures were done on BGMK cells by plaque assay.
Myxoma virus inhibited tumor growth in the ipsilateral inoculated tumor but not in the noninoculated tumor in the contralateral hemisphere
We implanted both hemispheres of the brain with U87 cells and, 12 days later, administered intratumorally a single dose of 5 × 106 PFUs/mouse (or dead myxoma virus) only in the ipsilateral tumor. We found that a single ipsilateral myxoma virus inoculation did not prolong survival (log-rank, P = 0.1386; Fig. 5A). Histologic examination showed all live vMyxgfp–treated animals had large tumors arising from the contralateral, noninoculated, hemisphere. The ipsilateral tumors which were inoculated with virus were very small (see arrow) or not detectable (Fig. 5B).
Figure 5.
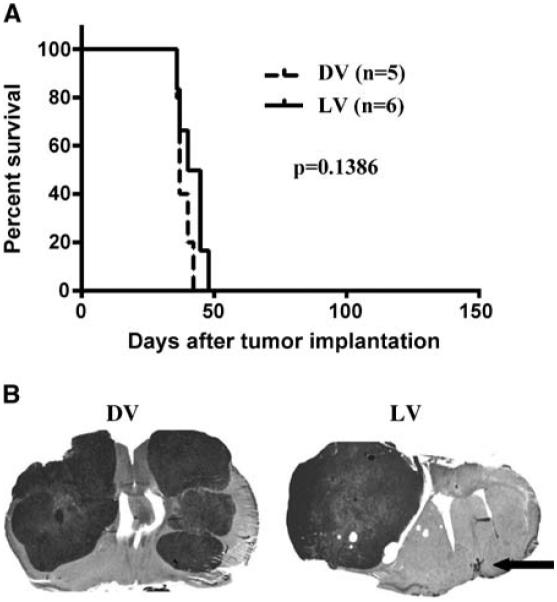
Myxoma virus inhibited tumor growth following a single intratumoral administration but had no effect on tumors in the contralateral hemisphere. A, myxoma virus did not increase survival in nude mice with bilateral U87 tumors in which only the ipsilateral tumor was injected with 5 × 106 PFUs vMyxgfp. B, representative H&E-stained sections show the inoculated tumor was very small (arrow) or absent whereas the tumor in the contralateral hemisphere was very large (right). Dead myxoma virus–treated animals had large tumors in both hemispheres (left).
Myxoma virus infects and kills human gliomas obtained from surgical specimens
To determine whether myxoma oncolysis also occurred in primary cultures from brain tumor surgical specimens, we tested 10 ex vivo brain tumor specimens derived from 2 glioblastoma multiforme, 1 anaplastic oligoastrocytoma, 1 anaplastic oligodendroglioma, 1 oligoastrocytoma, 1 low-grade oligodendroglioma, 2 mixed oligoastrocytoma, and 2 meningiomas. vMyxgfp infected and killed all 8 (100%) primary glioma cultures but had no effect on the 2 meningiomas cultured. Viral proteins were detected in live vMyxgfp–treated glioma cells using indirect immunofluorescent microscopy (Fig. 6A). Dead myxoma virus–treated tumor specimens remained healthy and continued to proliferate. MTT-assay showed that all glioma samples were susceptible to vMyxgfp infection (Fig. 6B). A172 glioma cell line was used as a positive control for myxoma virus susceptibility. The number of specimens was small but these data suggest that myxoma oncolysis could be effective in a substantial portion of primary human gliomas.
Figure 6.
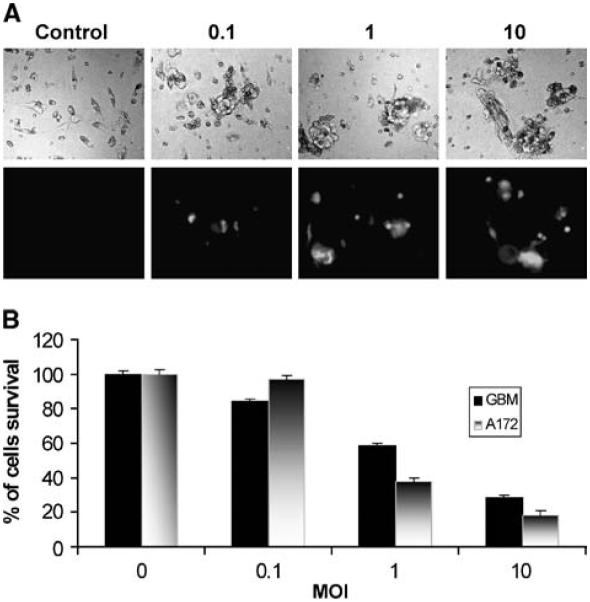
Myxoma virus infects and kills glioma cells grown ex vivo . Primary glioma cultures derived from patient surgical specimens were infected with vMyxgfp (MOI = 0, 0.1, 1, and 10). A, a representative line derived from a patient with a glioblastoma multiforme. Forty hours after viral infection, cultures were visualized for cell killing by cytopathic effect (phase-contrast; top) and viral production (visualized by green fluorescent protein fluorescence; bottom); magnification, ×100. B, cell killing was quantified by MTT assay 48 hours after cells were infected with vMyxgfp. A172 glioma cell line was used as a positive control for myxoma susceptibility.
Discussion
This is the first demonstration of the potential usefulness and minimal toxicity of replication competent myxoma virus against experimental models of human cancer in vivo. What makes this particularly surprising is that, until recently (30), myxoma infection was considered to be a rabbit-specific phenomenon and myxoma virus was thought to be incapable of infecting cells from any other species (including humans). Here, we made several observations: First, myxoma virus infects and kills most glioma cell lines in vitro. Second, it has marked efficacy without significant toxicities in vivo in the immunocompromised nude mouse host. Third, it produces a remarkably long-lived persistent infection in tumor cells in vivo that could potentially be very important therapeutically. These observations, when coupled with several other properties of myxoma virus (26), suggest that myxoma virus warrants further evaluation as an oncolytic agent against malignant glioma and other cancers.
Until recently, myxoma virus was regarded as a rabbit-specific virus that causes the fatal disease termed myxomatosis in European rabbits (28). However, species-specific host cell surface receptors for poxviruses have never been identified and it is now believed that intracellular events downstream of poxvirus binding and entry determine whether a poxvirus infection will be “permissive” or “nonpermissive” (36). Despite this very narrow host range in nature, myxoma virus can productively infect certain non-rabbit cells in vitro such as immortalized BGMK and primary human dermal fibroblasts (depending on how long they have been passaged; ref. 37). And it is now becoming clear that the intracellular milieu following virus binding and entry is critical in determining if a productive myxoma virus infection will occur (30, 31, 37). In untransformed cells, kinase-mediated signal transduction pathways seem to be important (31). As well, type 1 IFN responses are key determinants of poxvirus infection (37). Hence, the host range and apparent species specificity of myxoma virus infection are linked directly to the intracellular environment and IFN responsiveness of the host cell (36).
The mechanism underlying the tropism of myxoma for human cancer cells remains to be better elucidated. The first report of the ability of myxoma to infect human tumor cell lines in vitro was recently published (26). In that report, the myxoma gene product M-T5 (an ankyrin repeat protein) was critical for late viral gene expression in tumor cells but this observation did not explain the mechanism of action. Other viruses which are sensitive to IFN and are also oncolytic include vesicular stomatitis virus (23, 24), Newcastle disease virus (38), and, to a lesser degree, the poxvirus vaccinia (39, 40). Whether the oncolytic properties of myxoma are dependent on defective IFN signaling in human tumor cells or other intracellular events is the subject of ongoing work in our laboratories.
In this report, we did not exhaustively compare the specificity of myxoma virus for tumor cells with untransformed cells in vitro but rather relied on the specificity we observed for human gliomas in vivo. We took this approach because the results of in vitro testing can be somewhat misleading because the susceptibility of primary human fibroblasts to myxoma virus infection in vitro depends exquisitely on the number of times these cells are passaged (37). For example, human dermal fibroblasts are permissive to infection by myxoma virus at low passage number whereas high passage fibroblasts can lose the ability to support a productive infection (37). Hence, until the proper conditions are identified to test the specificity of myxoma in vitro, we regard in vivo testing as the most relevant demonstration of specificity and lack of toxicity. Furthermore, from a practical perspective, testing in non-human primates and ultimately in clinical trials will definitively determine the safety and specificity of myxoma. The only time that myxoma virus was tested in humans, over 50 years ago, there was no infectivity or pathogenicity of any kind (29). And there has never been a report of myxoma virus infection in humans despite widespread release of the virus to control feral rabbit populations (41).
One of the most surprising findings of our study was the long-lived myxoma virus infection we found in tumor tissues in vivo. Intratumoral viral titers increased for at least 14 days following viral inoculation and persisted for at least 35 days. This is in contrast to our experience with reovirus (another oncolytic virus) in which glioma cell killing occurs rapidly in vivo and of which viral titers begin to decrease after 24 hours (34). Whether this property will be beneficial or limiting in a clinical setting is unknown but suggests the possibility that myxoma virus may be particularly well suited to the approach of augmenting the oncolytic effect of the virus with the sustained delivery of therapeutic gene products. The addition of therapeutic genes [such as TRAIL (42), tumor necrosis factor (43), or the prodrug cytosine deaminase (44)] may enhance the therapeutic effect of oncolytic viruses. Replication competent vectors show superior delivery of therapeutic gene products when compared with replication defective viral vectors (45). Hence, in future studies, we will add a variety of therapeutic genes using myxoma virus as a platform to combine its oncolytic effect with a cytotoxic approach.
Our study has several limitations. First, our in vivo model is immunocompromised and this limits the generalizability of our findings. Second, the ability of an oncolytic virus to infect and kill remote (invasive or metastatic) tumor cells is critically important in the treatment of cancer in general and gliomas in particular. Myxoma virus infection did not cause regression of glioma tumors in the contralateral, noninoculated, tumors. We are currently determining strategies to facilitate infection and killing of remote glioma cells by manipulating the immune response and by using alternative strategies of delivery (e.g., blood-brain barrier breakdown or intra-arterial delivery).
Acknowledgments
Grant support: National Cancer Institute of Canada with funds raised by the Canadian Cancer Society (P.A. Forsyth) and the Terry Fox Foundation (J. Bell, G. McFadden, and P.A. Forsyth). G. McFadden holds a Canada Research Chair in Molecular Virology.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
References
- 1.Scott JN, Rewcastle NB, Brasher PM, et al. Which glioblastoma multiforme patient will become a long-term survivor? A population-based study. Ann Neurol. 1999;46:183–8. [PubMed] [Google Scholar]
- 2.Thorne SH, Kirn DH. Future directions for the field of oncolytic virotherapy: a perspective on the use of vaccinia virus. Expert Opin Biol Ther. 2004;4:1307–21. doi: 10.1517/14712598.4.8.1307. [DOI] [PubMed] [Google Scholar]
- 3.Lichty BD, Power AT, Stojdl DF, Bell JC. Vesicular stomatitis virus: re-inventing the bullet. Trends Mol Med. 2004;10:210–6. doi: 10.1016/j.molmed.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Bell JC, Lichty B, Stojdl D. Getting oncolytic virus therapies off the ground. Cancer Cell. 2003;4:7–11. doi: 10.1016/s1535-6108(03)00170-3. [DOI] [PubMed] [Google Scholar]
- 5.Shah AC, Benos D, Gillespie GY, Markert JM. Oncolytic viruses: clinical applications as vectors for the treatment of gliomas. J Neurooncol. 2003;65:203–26. doi: 10.1023/b:neon.0000003651.97832.6c. [DOI] [PubMed] [Google Scholar]
- 6.Chiocca EA. Oncolytic viruses. Nat Rev Cancer. 2002;2:938–50. doi: 10.1038/nrc948. [DOI] [PubMed] [Google Scholar]
- 7.Kirn D, Martuza RL, Zwiebel J. Replication-selective virotherapy for cancer: Biological principles, risk management and future directions. Nat Med. 2001;7:781–7. doi: 10.1038/89901. [DOI] [PubMed] [Google Scholar]
- 8.Norman KL, Farassati F, Lee PW. Oncolytic viruses and cancer therapy. Cytokine Growth Factor Rev. 2001;12:271–82. doi: 10.1016/s1359-6101(00)00024-1. [DOI] [PubMed] [Google Scholar]
- 9.Wilcox ME, Yang W, Senger D, et al. Reovirus as an Oncolytic Agent Against Experimental Human Malignant Gliomas. J Natl Cancer Inst. 2001;93:903–12. doi: 10.1093/jnci/93.12.903. [DOI] [PubMed] [Google Scholar]
- 10.Fueyo J, Gomez-Manzano C, Alemany R, et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo . Oncogene. 2000;19:2–12. doi: 10.1038/sj.onc.1203251. [DOI] [PubMed] [Google Scholar]
- 11.Gromeier M, Lachmann S, Rosenfeld MR, Gutin PH, Wimmer E. Intergeneric poliovirus recombinants for the treatment of malignant glioma. Proc Natl Acad Sci USA. 2000;97:6803–8. doi: 10.1073/pnas.97.12.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ikeda K, Ichikawa T, Wakimoto H, et al. Oncolytic virus therapy of multiple tumors in the brain requires suppression of innate and elicited antivirual responses. Nat Med. 1999;5:881–7. doi: 10.1038/11320. [DOI] [PubMed] [Google Scholar]
- 13.Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science. 1991;252:854–6. doi: 10.1126/science.1851332. [DOI] [PubMed] [Google Scholar]
- 14.Chiocca EA, Abbed KM, Tatter S, et al. A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Mol Ther. 2004;10:958–66. doi: 10.1016/j.ymthe.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 15.Csatary LK, Gosztonyi G, Szeberenyi J, et al. MTH-68/H oncolytic viral treatment in human high-grade gliomas. J Neurooncol. 2004;67:83–93. doi: 10.1023/b:neon.0000021735.85511.05. [DOI] [PubMed] [Google Scholar]
- 16.Freeman A, Gomori JM, Linetsky E, et al. Phase I/II trial of intravenous OV001 oncolytic virus in resistant glioblastoma multiforme (GBM); Journal of Clinical Oncology 2004 ASCO Annual Meeting Proceedings (Post-Meeting Edition); 2004. July 15 Supplement. [Google Scholar]
- 17.Papanastassiou V, Rampling R, Fraser M, et al. The potential for efficacy of the modified (ICP 34.5()) herpes simplex virus HSV1716 following intratumoural injection into human malignant glioma: a proof of principle study. Gene Ther. 2002;9:398–406. doi: 10.1038/sj.gt.3301664. [DOI] [PubMed] [Google Scholar]
- 18.Rampling R, Cruickshank G, Papanastassiou V, et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 2000;7:859–66. doi: 10.1038/sj.gt.3301184. [DOI] [PubMed] [Google Scholar]
- 19.Markert JM, Medlock MD, Rabkin SD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7:867–74. doi: 10.1038/sj.gt.3301205. [DOI] [PubMed] [Google Scholar]
- 20.Coffey MC, Strong JE, Forsyth PA, Lee PW. Reovirus therapy of tumors with activated Ras pathway. Science. 1998;282:1332–4. doi: 10.1126/science.282.5392.1332. [DOI] [PubMed] [Google Scholar]
- 21.Norman KL, Hirasawa K, Yang AD, Shields MA, Lee PW. Reovirus oncolysis: the Ras/RalGEF/p38 pathway dictates host cell permissiveness to reovirus infection. Proc Natl Acad Sci U S A. 2004;101:11099–104. doi: 10.1073/pnas.0404310101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nemunaitis J, Edelman J. Selectively replicating viral vectors. Cancer Gene Ther. 2002;9:978–1000. doi: 10.1038/sj.cgt.7700547. [DOI] [PubMed] [Google Scholar]
- 23.Stojdl DF, Lichty BD, tenOever BR, et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell. 2003;4:263–75. doi: 10.1016/s1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- 24.Stojdl DF, Lichty B, Knowles S, et al. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med. 2000;6:821–5. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- 25.Balachandran S, Barber GN. Defective translational control facilitates vesicular stomatitis virus oncolysis. Cancer Cell. 2004;5:51–65. doi: 10.1016/s1535-6108(03)00330-1. [DOI] [PubMed] [Google Scholar]
- 26.Sypula J, Wang F, Ma Y, Bell J, McFadden G. Myxoma virus tropism in human tumor cells. Gene Ther Mol Biol. 2004;8:103–14. [Google Scholar]
- 27.Kerr P, McFadden G. Immune responses to myxoma virus [review] Viral Immunol. 2002;15:229–46. doi: 10.1089/08828240260066198. [DOI] [PubMed] [Google Scholar]
- 28.Fenner F, Ross J. Myxomatosis. In: Thompson GV, King CM, editors. The European rabbit, the history and biology of a successful colonizer. Oxford University Press; Oxford: 1994. pp. 205–39. [Google Scholar]
- 29.Burnet FM. Changing Patterns: An Atypical Autobiography. W. Heinemann; Melbourne (Australia): 1968. pp. 105–20. [Google Scholar]
- 30.Wang F, Ma Y, Barrett JW, et al. Disruption of Erk-dependent type I interferon induction breaks the myxoma virus species barrier. Nat Immunol. 2004;5:1266–74. doi: 10.1038/ni1132. [DOI] [PubMed] [Google Scholar]
- 31.Johnston JB, Barrett JW, Chang W, et al. Role of the serine-threonine kinase PAK-1 in myxoma virus replication. J Virol. 2003;77:5877–88. doi: 10.1128/JVI.77.10.5877-5888.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang W, Senger D, Muzik H, et al. Reovirus prolongs survival and reduces the frequency of spinal and leptomeningeal metastases from medulloblastoma. Cancer Res. 2003;63:3162–72. [PubMed] [Google Scholar]
- 33.Yang WQ, Senger DL, Lun XQ, et al. Reovirus as an experimental therapeutic for brain and leptomeningeal metastases from breast cancer. Gene Ther. 2004a;11:1579–89. doi: 10.1038/sj.gt.3302319. [DOI] [PubMed] [Google Scholar]
- 34.Yang WQ, Lun XQ, Palmer CA, et al. Efficacy and safety evaluation of human reovirus type 3 in immunocompetent animals: racine and non-human primates. Clin Cancer Res. 2004b;10:8561–76. doi: 10.1158/1078-0432.CCR-04-0940. [DOI] [PubMed] [Google Scholar]
- 35.Yang M, Baranov E, Jiang P, et al. Whole-body optical imaging of green flourescent protein-expressing tumors and metastases. Proc Natl Acad Sci U S A. 2000;97:1206–11. doi: 10.1073/pnas.97.3.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McFadden G. Poxvirus tropism [review] Nat Rev Microbiol. 2005;3:201–13. doi: 10.1038/nrmicro1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnston JB, Nazarian SH, Natale R, McFadden G. Myxoma virus infection of primary human fibroblasts varies with cellular age and is regulated by host interferon responses. Virology. 2005;332:235–48. doi: 10.1016/j.virol.2004.11.030. [DOI] [PubMed] [Google Scholar]
- 38.Pecora AL, Rizvi N, Cohen GI, et al. Phase I trial of intravenous administration of PV701, an oncolytic virus, in patients with advanced solid cancers. J Clin Oncol. 2002;20:2251–66. doi: 10.1200/JCO.2002.08.042. [DOI] [PubMed] [Google Scholar]
- 39.Lindenmann J, Gifford GE. Studies on vaccinia virus plaque formation and its inhibition by interferon. III. A simplified plaque inhibition assay of interferon. Virology. 1963;19:302–9. doi: 10.1016/0042-6822(63)90068-0. [DOI] [PubMed] [Google Scholar]
- 40.Gifford GE, Toy ST, Lindenmann J. Studies on vaccinia virus plaque formation and its inhibition by interferon. II. Dynamics of plaque formation by vaccinia virus in the presence of interferon. Virology. 1963;19:294–301. doi: 10.1016/0042-6822(63)90067-9. [DOI] [PubMed] [Google Scholar]
- 41.Fenner F, Ratcliffe FN. Myxomatosis. Cambridge University Press; Cambridge (UK): 1965. [Google Scholar]
- 42.Lin T, Gu J, Zhang L, et al. Targeted expression of green fluorescent protein/tumor necrosis factor-related apoptosis-inducing ligand fusion protein from human telomerase reverse transcriptase promoter elicits antitumor activity without toxic effects on primary human hepatocytes. Cancer Res. 2002;62:3620–5. [PubMed] [Google Scholar]
- 43.Rasmussen H, Rasmussen C, Lempicki M, et al. TNFerade Biologic: preclinical toxicology of a novel adenovector with a radiation-inducible promoter, carrying the human tumor necrosis factor α gene. Cancer Gene Ther. 2002;9:951–7. doi: 10.1038/sj.cgt.7700518. [DOI] [PubMed] [Google Scholar]
- 44.Nakamura H, Mullen JT, Chandrasekhar S, Pawlik TM, Yoon SS, Tanabe KK. Multimodality therapy with a replication-conditional herpes simplex virus 1 mutant that expresses yeast cytosine deaminase for intratumoral conversion of 5-fluorocytosine to 5-fluorouracil. Cancer Res. 2001;61:5447–52. [PubMed] [Google Scholar]
- 45.Ichikawa T, Chioca EA. Comparative analyses of transgene delivery and expression in tumors inoculated with a replication-conditional or -defective viral vector. Cancer Res. 2001;61:5336–9. [PubMed] [Google Scholar]